
Abstract
Agonists and antagonists of the canonical Wnt signaling pathway are modulators of pathological aspects of rheumatoid arthritis (RA). Their activity is primarily modifying bone loss and bone formation, as shown in animal models of RA. More recently, modulation of Wnt signaling by the antagonist Sclerostin has also been shown to influence soft-tissue-associated inflammatory aspects of the disease pointing towards a role of Wnt signaling in soft-tissue inflammation as well. Yet, nothing is known experimentally about the role of Wnt ligands in RA. Here we provide evidence that altering Wnt signaling at the level of a ligand affects all aspects of the rheumatoid arthritic disease. WNT9a levels are increased in the pannus tissue of RA patients, and stimulation of synovial fibroblasts (SFB) with tumor necrosis factor (TNF) leads to increased transcription of Wnt9a. Loss of Wnt9a in a chronic TNF-dependent RA mouse model results in an aggravation of disease progression with enhanced pannus formation and joint destruction. Yet, loss of its activity in the acute K/BxN serum-transfer induced arthritis (STIA) mouse model, which is independent of TNF signaling, has no effect on disease severity or progression. Thus, suggesting a specific role for WNT9a in TNF-triggered RA. In synovial fibroblasts, WNT9a can activate the canonical Wnt/β-catenin pathway, but it can also activate P38- and downregulate NFκB signaling. Based on in vitro data, we propose that loss of Wnt9a creates a slight proinflammatory and procatabolic environment that boosts the TNF-mediated inflammatory response.
Similar content being viewed by others

Introduction
Inflammation, together with increased pannus formation, as well as progressive joint and bone destruction, are hallmarks of rheumatoid arthritis (RA), leading to joint malformations. These joints lack any signs of cartilage or bone repair, with the observed bone loss being accredited to dysregulated osteoclastogenesis. Clinically, RA presents a symmetric polyarthritis affecting mostly diarthrodial joints1. Due to infiltration of inflammatory cells and enhanced local cellular proliferation, the synovium expands, forming the pannus. The inflamed tissue covers and invades the articular cartilage and adjacent bone and produces proinflammatory cytokines, such as TNF and IL-1β. TNF stimulates, among others, the differentiation of osteoclasts that are responsible for the focal bone erosions in different RA mouse models2,3,4. In mice, transgenic expression of a stabilized mRNA encoding human TNF (hTNFtg) leads to joint alterations, essentially mimicking those found in human RA patients5. IL-1β is the key-cytokine in the serum-transfer-induced K/BxN RA (STIA) mouse model6,7,8,9.
Among others, Wnt signaling is modulated in the joint tissue of human RA patients and hTNFtg mice and has been implicated to play a role in RA10,11,12,13,14,15,16. Wnt signaling is well known for its roles in bone and cartilage formation as well as in bone homeostasis17,18,19. The WNT protein family encompasses 19 distinct ligands that regulate morphogenesis processes during embryogenesis, stem cell maintenance, tissue homeostasis, and proliferation during embryogenesis and in cancer20,21,22,23,24,25,26. WNTs can interact with different receptor complexes and signal through diverse pathways, among which the canonical Wnt/β-catenin pathway is best understood27,28. Canonical Wnt signaling is characterized by stabilization and nuclear translocation of β-catenin and can be visualized by increased expression of target genes, such as Axin229. Noncanonical Wnt signaling results in the activation of very diverse signaling pathways27. Wnt signaling is modulated and fine-tuned extracellularly through secreted agonists and antagonists. One of these antagonists, Dickkopf 1 (DKK1), is upregulated in the serum of RA patients and the serum and synovial tissue of hTNFtg mice and has been proposed to be a master regulator in joint remodeling blocking bone formation and promoting bone destruction11. Yet, its inhibition in hTNFtg mice did not affect pannus-associated inflammatory aspects of RA11. Another antagonist, Wnt inhibitory factor 1 (WIF1), expressed in chondrocytes, is downregulated in experimental arthritis and articular cartilage of RA patients30,31,32. Its loss partially protected hTNFtg mice against bone but not cartilage erosion, the latter being even aggravated, but soft-tissue inflammatory disease aspects were unaffected32. Both antagonists are Wnt/β-catenin pathway targets33. Treatment of hTNFtg mice with the agonist, R-Spondin 1, protected against bone and cartilage loss, but again soft-tissue inflammation was unaffected34. On the other hand, loss of yet another antagonist, Sclerostin (SOST), which is also upregulated in RA biopsies and synovial tissue in the hTNFtg RA mouse model, led to worsening of all disease aspects, including soft-tissue inflammation35.
Wnt9a is expressed in the developing synovial joints, and Wnt9a-deficient embryos are smaller in size, display temporally delayed chondrocyte maturation and synovial chondromatosis in their elbow joints36. Here, we show that WNT9a levels are increased in the pannus of RA patients and that its expression can be induced in mouse SFBs by TNF treatment. Hence, WNT9a may either be involved in disease progression or act as a protective factor. To distinguish between these possibilities, hTNFtg mice lacking Wnt9a in the limb mesenchyme were generated, and the STIA model was applied to Wnt9a-deficient mice.
Material and methods
Human material
Research with human material was performed in accordance with the Declaration of Helsinki. Human synovial tissue was dissected during surgery of RA (n = 4) and OA (n = 4) patients, fixed in freshly prepared 4% paraformaldehyde/PBS at 4 °C for 24 h, embedded in paraffin, and sectioned at 4 μm. Human synovial fibroblasts were isolated from synovial tissue of RA (n = 6) and OA (n = 6) patients undergoing joint replacement surgery. RA patients met the American College of Rheumatology criteria. Isolated fibroblasts were cultured using high glucose DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C, 5% CO2 for maximal eight passages before RNA isolation using the Qiagen RNeasy Midi Kit. The Invitrogen SuperScript™ III system was used for first-strand synthesis.
The study was approved by the Institutional Ethical Review Board of the Faculty of Medicine of the Otto-von-Guericke University (IRB No 73/18). Informed consent was obtained from the patients prior to surgery. We included 20 patients undergoing either surgery for TKA implantation due to osteoarthritis or rheumatoid arthritis.
Animal models
The following mouse strains were used: R26CreERT237, Prx1Cre38, Wnt9a floxed and Wnt9a germline deleted (Wnt9a+/−)36, Ctnnb1 (exon 3) floxed39, and hTNFtg (strain Tg197)5 with a mixed 129 Sv/J;C57Bl/6 J background. For the hTNFtg arthritis model, Wnt9a+/−;Prx1Cre;hTNFtg/+ male mice were bred with Wnt9afl/fl females to generate hTNFtg/+;Wnt9afl/−;Prx1Cre (hTNFtg/+;Wnt9a∆Prx/−) mice. hTNFtg/+;Wnt9afl/+ or hTNFtg/+;Wnt9a∆Prx/+ littermates correspond to hTNFtg/+ specimens and Wnt9afl/+ or Wnt9a∆Prx/+ littermates to controls. From the age of 5 weeks onward, mice of both genders were scored in a blinded fashion once a week for weight, grip strength, and semi-quantitatively for paw swelling, and finally sacrificed at the age of 6 and 8 weeks, or 12 weeks in the case of hTNFtg/+ mice. Detailed information of the gender of the mice is provided in the Supplementary data and Supplementary Table S1. For the STIA model, 8-week-old female littermates of the genotypes Wnt9a∆Prx1/− and Wnt9a∆Prx1/+ (control) were housed together and injected two times (on days 0 and 2) intraperitoneal with 150 µl of arthritogenic serum collected from K/BxN mice6. Mice were scored three times a week for weight, grip strength, and using calibers for ankle width in a blinded fashion, and finally sacrificed 14 or 28 days later. Grip strength was determined in a blinded fashion using a grip strength device (Bioseb) following manufacturers’ instructions. Sample sizes in both models (n ≥ 8) were based on previous studies using these models. For cell culture experiments, 7-day-old homozygous Wnt9afl/fl, Wnt9afl/fl;R26CreER, and Ctnnb1fl/fl;R26CreER mice were used. Adult mice were sacrificed by cervical dislocation and juveniles by decapitation. Genotyping was performed using previously published PCR protocols. As animals of specific genotypes were allocated to the different groups, no randomization was performed, and no animals were excluded from the analysis. Animal experiments were performed in accordance with the 10 Essentials of the ARRIVE guideline and relevant regulations under the licenses 84-02.04.2014.A056 and 81-02.04.2017.A409 approved by the national and local authorities.
Micro-Computed tomography (microCT), histomorphometric analysis, and scoring
Mouse limbs were fixed for 48 h in 4% paraformaldehyde at RT, washed in 70% ethanol, and scanned using the SkyScan 1176 microCT (Bruker) with an 0.5 mm aluminum filter at 50 kV, 500 μA, 8.52 μm image pixel size, and 955 ms exposure. Sections were reconstructed using the NRecon v1.7.4.6 software (SkyScan; Bruker) with beam hardening correction set to 40%. The CT Analyzer v1.18.9.0 software was used for trabecular bone analysis of the 426–2131 μm region below the growth plate. MicroCT images of mouse paws were semi-quantitatively scored for affected areas (0 = none, 1 = few small & localized, 2 = multiple small to medium, 3 = multiple medium to large), degree of erosion (0 = none, 1 = roughness, 2 = pitting, 3 = holes), and bone formation (0 = none, 1 = small osteophytes, 2 = spurs, 3 = bone deformity or fusion), with the total score of 0–9 being the sum of the individual scores. Scanning, reconstruction, and the final scoring was performed in a semi-blinded fashion, as recording was based on the identification-number (ear marking) of the individual animal without direct association to the genotype.
Beta-galactosidase staining, histology, trap, and immunostainings
For beta-galactosidase staining, hind paws were fixed in 0.2% glutaraldehyde for 18 h at 4 °C and stained for 48 h at 37 °C with X-Gal solution (1 mg/ml X-Gal, 2 mM MgCl, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 0.2% NP-40), followed by decalcification with 10% EDTA/tris-buffered saline before dehydration, embedding in paraffin, sectioning at 5 µm, and counterstaining with nuclear fast red.
For histological analysis and tartrate-resistant-alkaline-phosphatase (TRAP) staining, decalcified and paraffin-embedded hind paws were sectioned at 5 µm. Following deparaffinization and rehydration, sections were stained sequentially for 90 s in 0.02% Fast Green FCF and 30 min in 0.1% Safranin O to assess cartilage defects.
For TRAP, sections were incubated for 20 min with 600 µg/ml Fast Red Violet LB Salt and 100 µg/ml Naphthol ASMX Phosphate in dimethylformamide, buffered with 40 mM Sodium acetate/10 mM Sodium tartrate at pH 5.0, and counterstained with Mayer’s Hematoxylin.
For immunohistochemistry, sections were enzymatically pretreated, incubated in 3% H2O2 for 30 min, blocked with 10% serum, and incubated with primary antibodies (see Supplementary Table S2). Sections were developed using the appropriate species-specific biotinylated secondary antibody (Vector Laboratories) diluted 1:250 in combination with the Vectastain Elite ABC Kit and Diaminobenzidine, and counterstained with methyl green.
Isolation and cultivation of primary murine cells
Wild-type deep layer chondrocytes (DLCs) were isolated from the knees (femoral and tibial head) and SFBs from the paws of 7-day-old pups. For this, pups were skinned, and the required parts cleared of soft tissue. To isolate DLCs, knees were predigested with 0.3% trypsin for 1 h. Afterwards, the remaining tissue was treated with 0.5 mg/ml collagenase II for 18 h at 37 °C while shaking. DLCs were cultured in DMEM/F-12 with 10% FCS. For SFB isolation, tarsal and metatarsal elements were dissected and digested with 1 mg/ml collagenase IV (310 u/mg dw) for about 20 min under heavy stirring. Cells were cultured in high glucose DMEM with 10% FCS at 37 °C, 5% CO2 up to passage 4/5.
The floxed allele in Wnt9afl/fl cells was recombined using a Cre-expressing lentivirus. Controls were infected with a Cre-less lentivirus. In Wnt9afl/fl;R26CreER, and Ctnnb1fl/fl;R26CreER SFBs, recombination was achieved by 4-hydroxytamoxifen (80 ng/ml) treatment for 48 h. For cytokine treatment, SFBs were exposed either for 6, 8, and 12 h to analyze short-term effects, or 48 h to analyze longer-term effects to the following concentrations: hTNFα (10 ng/ml), mTNFα (10 ng/ml), or mIL-1β (10 ng/ml). For WNT9a gain-of-function experiments, SFBs were stimulated for 5 days or as for the β-catenin-dependent Axin2 assay for 48 h with rWNT9a (300 ng/ml) or rWNT3a (100 ng/ml). To assess the rWNT9a effect on hTNF treatment, cells were stimulated with rWNT9a for 2 h before hTNF application.
Lentiviral preparation
Calcium-phosphate transfection was performed in HEK-293T cells with the following plasmid mixture: 3.5 µg pMD2-VSVG (Addgene #12259), 6.5 µg psPAx2 (Addgene #12260), and either 10 µg p156RRL-nlsCre (Addgene #12106) or p156RRL-nlsCre− (for control). After 6–8 h, the medium was exchanged with medium of the respective target cell type. Two days later, supernatant was collected, filtrated (0.45 µm), and mixed 1:1 with target cell medium, supplemented with 10 µg/ml polybrene, and applied to target cells. Cells were lysed for RNA isolation 24 h later. Deletion efficiency was detected by qPCR.
Immunoblots
Whole protein lysates from SFBs were prepared as previously described40. 12.5 µg lysate was run on a 10% SDS-PAGE and transferred to a 0.45-μm PVDF-membrane by semi-dry transfer (PerfectBlue™, PeqLab). Membranes were blocked using 5% milk or 4% BSA in TBST, incubated with the appropriate primary (see Supplementary Table S3) followed by the respective species-specific HRP-coupled secondary antibodies, and developed using ECL substrate and hyperfilm film.
RNA isolation, cDNA synthesis, and qPCR analysis
Total RNA was isolated from cultivated DLCs or SFBs using the RNAqueous Kit. First-strand cDNA synthesis was performed using PrimeScript RT Reagent Kit. For qPCR, cDNA was mixed with PerfeCTa SYBR Green FastMix and respective primers (see Supplementary Table S4) in 25 µl total volume. Gene expression was monitored using a BioRad CFX96 cycler. Reaction conditions were 95 °C 30 s, 45× (95 °C 15 s, 60 °C 30 s, 72 °C 20 s + plate read), 72 °C 5 min, melting curve (55–99 °C), in 0.5 °C increments for 5 s + plate read. Values were calculated using the comparative ∆C(t) method and normalized to two housekeeping genes (HPRT, B2M, Hprt, Sdha).
In vitro osteoclastogenesis assay
For osteoclast cultures, monocytes were isolated by flushing the bone marrow from 8-week-old wild-type mice, incubated for 1 day with minimum essential medium eagle, 10% FCS, and primed towards osteoclastic differentiation using 50 ng/ml M-CSF. The next day, cells in the supernatant were collected, transferred to 96-well plates (2 × 105 cells/well), and cultured for 4 days in the presence of 50 ng/ml M-CSF, 20 ng/ml RANKL, and 20% conditioned medium (CM). CM was collected after three days of culture from Wnt9a-deficient or control SFBs and filtered before use. Osteoclasts were detected using a Leukocyte Acid Phosphatase (TRAP) Kit, according to the manufacturer’s instructions.
Image acquisition
Histological bright-field images were acquired on the Zeiss AxioImager.M2 or Zeiss Observer.Z1 using the AxioCam MRc 6.45 µm color camera and the Zen 2 or Zen 3 Zeiss software (Jena, Germany).
Statistical analysis
Statistical analysis was performed by two-tailed, unpaired Student’s t-test using the GraphPad Prism software 6.0. Data are displayed as mean values ± standard deviation. P-values less than 0.05 were considered significant. The minimal number of biological replicates for each experiment was n = 3.
Detailed information regarding the vendors and order numbers can be found in Supplementary Table S5.
Results
WNT9a levels in synovial fibroblasts are elevated under inflammatory conditions
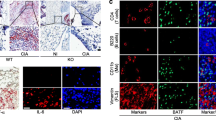
Biopsies of human RA patients displayed increased WNT9a levels compared to OA patients (Fig. 1a). Accordingly, WNT9a expression was slightly elevated in cultured SFBs of RA relative to OA patients. The expression of other Wnts, which are either expressed in the developing murine synovial joint or upregulated in human RA joints10,16,31,36,41, was tendentially increased in cultured RA relative to OA SFBs, as were the AXIN2 and Cyclin D1 (CCND1) levels (Fig. 1b).

a Immunohistological analysis of WNT9a in human OA and RA tissue samples (n = 4). b qPCR analysis of WNT pathway-related genes in RA relative to OA SFB samples displayed as a dot plot with P-values noted above (n = 6). c qPCR analysis of murine Wnt9a, Wnt4, and Wnt16 in cultured deep layered chondrocytes (DLC; n = 3) and synovial fibroblasts (SFB; n = 6) after treatment with murine TNF for 48 h, displayed as relative fold change to untreated cells. d qPCR analysis of Wnt and Wnt pathway-related genes in SFBs treated with recombinant murine TNF (mTNF) or human TNF (hTNF) for 48 h, displayed as relative fold change to untreated cells (n ≥ 3). P-values: *<0.05, **<0.01, and ***<0.001.
Next, we assessed the effects of exogenous TNF on the expression of Wnt pathway components. For this, mouse SFBs and DLCs were treated with either murine (m) or human (h) TNF for 48 h. In SFBs, but not DLCs, Wnt9a expression increased, while Wnt4 and Wnt16 decreased in DLCs and SFBs upon stimulation with mTNF (Fig. 1c,d). In SFBs, the Wnt9a increase in response to hTNF was lower, while all other Wnt genes analyzed either decreased or, as Wnt1, remained nearly unchanged after m/hTNF treatment (Fig. 1d). Yet, in shorter-term treatments, Wnt9a expression was only significantly increased after mTNF treatment for 12 h (Supplementary Fig. 1a), suggesting a secondary mechanism for Wnt9a regulation.
The transcription of Wnt pathway components/regulators was primarily downregulated in mSFBs in response to m/hTNF, with two exceptions, Wisp1 (Wnt1-inducible signaling pathway protein 1) and Dkk1. Wisp1 expression was unaltered, while Dkk1 was elevated (Fig. 1d).
In SFBs, either Wnt9a-deficient or treated with recombinant Wnt9a (rWNT9a), Wnt pathway components/regulators were changed as following: Wnt4 and Wnt5a increased slightly in Wnt9a-deficient SFBs (Supplementary Fig. 1b). On the other hand, Wnt4, Wnt11, and Sfrp2 decreased, while Axin2 and Wif1 expression increased in response to rWNT9a treatment (Supplementary Fig. 1b). While Axin2 expression was not altered in Wnt9a-deficient SFBs, it decreased in Wnt9a-deficient DLCs (Supplementary Fig. 1b, c). In certain cells, including chondrocytes, WNT9a can activate the canonical Wnt/β-catenin pathway, but it may also activate noncanonical Wnt pathways36,42,43,44. To analyze whether WNT9a utilizes the Wnt/β-catenin pathway in SFBs, Axin2 levels were examined in Ctnnb1-deficient SFBs treated with rWNT9a or rWNT3a (as a control). This resulted in a 10-fold reduction of Axin2 expression in response to either rWNT3a or rWNT9a treatment (Supplementary Fig. 1d), suggesting that Axin2 upregulation in response to rWNT9a is β-catenin-dependent. In RA, according to the literature, opposite effects on canonical Wnt signaling have been reported: β-catenin levels are elevated in the synovium of RA patients, suggesting increased signaling45, while the high DKK1 levels in the hTNFtg mouse model imply an overall decrease in signaling. Yet, anti-DKK1 antibody treatment did not affect pannus size11. Hence, canonical Wnt signaling in the pannus may be unaffected to a certain extent. Concordantly, BAT-gal activity was increased in the pannus of hTNFtg/+;BAT-gal mice (Supplementary Fig. 1e), and AXIN2 levels were increased in the hTNF tg/+ synovium (Supplementary Fig. 1f). This is somewhat contradictory to the hTNF-induced Axin2 downregulation in SFBs in vitro, yet, cells in the pannus of hTNFtg mice are exposed to TNF signaling for a much longer time.
Loss of Wnt9a aggravates the hTNF-transgene-driven inflammation
To address disease progression of the hTNFtg model in a Wnt9a-deficient background, we used mice in which the conditional Wnt9a allele was deleted in the limb mesenchyme using the Prx1-Cre line combined with a germline-deleted Wnt9a allele (hTNFtg/+;Wnt9a∆Prx/−)38. Loss of Wnt9a in the limb mesenchyme worsened the clinical signs of inflammation, such as paw swelling and grip strength loss, in the hTNFtg/+ background (Fig. 2a). Although hTNFtg/+;Wnt9a∆Prx/− mice had a reduced weight compared to hTNFtg/+ mice, their net weight gain was similar to hTNFtg/+ mice (Fig. 2a). Radiographic examination revealed significantly increased bone destruction in the hind paws of 6- and 8-week-old hTNFtg/+;Wnt9a∆Prx/− mice compared to hTNFtg/+ littermates, as quantified in different regions (Fig. 2b–d and Supplementary Fig. 2a, b). Bone destruction in the 8-week-old hTNFtg/+;Wnt9a∆Prx/− mice was even more severe than in 12-week-old hTNFtg/+ mice (Fig. 2e). Chronic inflammation is associated with a general trabecular bone loss46, which also occurred in 8-week-old hTNFtg/+ mice and was even more pronounced in hTNFtg/+;Wnt9a∆Prx/− mice (Supplementary Fig. 2c). Histopathological examination of the paws revealed an increased inflamed pannus area, being statistically significant at the 8-week timepoint (Fig. 3a, b). Increased cartilage destaining, although not statistically significant, and significant reduction in cartilage thickness were detected in 6- and 8-week-old hTNFtg/+;Wnt9a∆Prx/− mice (Fig. 3b). The paws of hTNFtg/+;Wnt9a∆Prx/− mice also contained more TRAP-positive osteoclasts and neutrophils (Fig. 3c, d). CD45R+ B cells were only sparsely detected in some pannus areas of hTNFtg/+ and hTNFtg/+;Wnt9a∆Prx/− mice (Supplementary Fig. 3a, c). Yet, more and bigger follicular-like B cell clusters were present in the bone marrow of hTNFtg/+;Wnt9a∆Prx/− hind paws (Supplementary Fig. 3a–c).

a Assessment of disease progression by measurement of paw swelling, grip strength, and weight in control (ctrl), hTNFtg/+, and hTNFtg/+;Wnt9a∆Prx/− mice over 5–8 weeks. The net weight gain in the hTNFtg/+and hTNFtg/+;Wnt9a∆Prx/− mice was similar. Data are presented as the mean ± SD (ctrl: n = 19; hTNFtg/+: n = 34; hTNFtg/+;Wnt9a∆Prx/−: n = 21 for the 5- and 6-week timepoints, and ctrl: n = 9; hTNFtg/+: n = 25; hTNFtg/+;Wnt9a∆Prx/−: n = 12, for the 7- and 8-week timepoints). b Representative three-dimensional microCT images of ctrl, hTNFtg/+, and hTNFtg/+;Wnt9a∆Prx/− hind paw regions at the 6- and 8-week timepoint (n = 9). c Scoring of the three-dimensional microCT images with regard to the extent of bone destruction (arbitrary units) (n = 9). d Two-dimensional representative microCT images of ctrl, hTNFtg/+, and hTNFtg/+;Wnt9a∆Prx/− hind paw regions at the 6- and 8-week timepoint (n = 9). e For comparison, a two-dimensional representative microCT image of the hTNFtg/+ hind paw region at a 12-week timepoint (n = 3). P-values: *<0.05, **<0.01.

a Representative images of Safranin O stained tarsal joint sections from 6- and 8-week-old hTNFtg/+ and hTNFtg/+;Wnt9a∆Prx/−, showing increased pannus formation in the hTNFtg/+;Wnt9a∆Prx/− hind paw regions (n = 9). b Quantitative histomorphometric analysis of destaining of cartilage, cartilage thickness, and pannus area in 6- and 8-week-old hTNFtg/+ and hTNFtg/+;Wnt9a∆Prx/− hind paws (n = 9). c Representative images of tartrate-resistant acid phosphatase (TRAP) staining (red color) of tarsal joint sections from 6- and 8-week-old hTNFtg/+and hTNFtg/+;Wnt9a∆Prx/− mice (n ≥ 4). Quantification of TRAP-positive osteoclasts on sections of 8-week-old hTNFtg/+ (n = 7) and hTNFtg/+;Wnt9a∆Prx/− (n = 4) within the tarsal region (as in a). d Representative images of immunohistochemically stained sections of 6- and 8-week-old hTNFtg/+ and hTNFtg/+;Wnt9a∆Prx/− hind paws, showing the increased presence of Ly6B.2+ neutrophils in the Wnt9a-deficient background (n = 9). Data in b and c are presented as mean ± SD, P-values: *<0.05.
Alterations of RA-relevant factors in response to altered Wnt9a signaling
Activated synovial fibroblasts are considered to be the main culprits in RA, and TNF levels are particularly high in these cells in the hTNFtg RA mouse model. Therefore, we addressed whether the expression of RA-relevant factors is altered in Wnt9a-deficient SFBs. Regarding proinflammatory cytokines, Tnf expression was decreased, while transcriptional levels of Il1b, Il6, Il15, and Cxcl16 were slightly increased (Fig. 4a). Of the catabolic enzymes, the expression of Adamts4 was unchanged, Adamts5 and Mmp13 slightly increased, while Mmp9 was significantly decreased (Fig. 4b). Interestingly, rWNT9a treatment of wild-type SFBs increased Tnf, Rankl, Il15, and Cxcl16 expression (Fig. 4a). Concerning the catabolic enzymes, rWNT9a treatment decreased Adamts5, while increasing Mmp9 and Mmp13 expression (Fig. 4b). The transcriptional levels of two other cytokines, Il17a and Il9, were very low in SFBs and did not change in response to either deletion of Wnt9a or stimulation with rWNT9a (data not shown). Similar alterations in the expression levels of Il1b, Il6, Adamts5, and Mmp13 were also observed in Ctnnb1-deficient SFBs independent of the presence or absence of WNT9a stimulation (Supplementary Fig. S4a). Consequently, loss of WNT9a or β-catenin activity in SFBs fosters a slight proinflammatory and procatabolic environment. Next, we asked if hTNF treatment would lead to differential effects in Wnt9afl/fl versus Wnt9a∆/∆ SFBs. Except for a decreased Tnf expression and slight increases in Tnfr1, Il1b, Adamts5, and Mmp13 levels, the expression of the analyzed genes did not change (Fig. 4c, d). As more osteoclasts are present in the hTNFtg/+;Wnt9a∆Prx/− hind paws, we asked whether Wnt9a-deficient SFBs produce a secreted factor stimulating osteoclast differentiation. Conditioned medium from Wnt9a∆/∆ SFBs in presence of 20 ng/ml RANKL led to an increase in the area covered by osteoclasts, while the average number of osteoclasts per well did not change (Fig. 4e).

a–d qPCR analysis of proinflammatory cytokines, TNF receptors 1 and 2, and Rankl in Wnt9a-deficient and wild-type (Wnt9afl/fl) SFBs (a, c), and of catabolic enzymes involved in cartilage degradation (b, d) stimulated with rWNT9a for 5 days (a, b) or treated with hTNF for 48 h (c, d). Data are displayed as fold expression changes relative to untreated SFBs (n = 3). e Images of osteoclast differentiation cultures treated for 4 days with conditioned medium (CM) from control (Wnt9afl/fl) or Wnt9a-deficient SFBs in the presence of 20 ng/ml RANKL; quantified for OC.A (average area covered by a single osteoclast), OC.A/A in % (osteoclast area per total area), and OC.N (osteoclast number per well). f Immunoblot for p-P38, P38, IκBα, and GAPDH using extracts from Wnt9afl/fl and Wnt9a∆/∆ hTNF-treated SFBs, and Wnt9afl/fl SFBs rWNT9a pretreated (2 h) followed by hTNF treatment for the indicated time; below the quantification of IκBα relative to GAPDH levels. g Immunoblot for p-P38 and P38 using extracts from rWNT9a-treated SFBs; below the quantification of the p-P38 relative to P38 levels. h Immunoblot for IκBα, pIκBα, and GAPDH (loading control) using extracts from rWNT9a-treated SFBs. (a–h) n = 3. P-values: *<0.05, **<0.01.
Next, we asked if loss of Wnt9a or treatment with rWNT9a would alter TNF activated signaling pathways, such as P38 and NFκB47. Loss of Wnt9a did not affect P38 activation upon TNF stimulation (Fig. 4f). Yet interestingly, phospho-P38 (p-P38) levels increased in response to rWNT9a application (Fig. 4g). Nevertheless, rWNT9a pretreatment of SFBs for 2 h before hTNF application did not alter p-P38 levels (Fig. 4f). TNF-induced degradation of the NFκB inhibitor IκBα was also not affected by the loss of Wnt9a (Fig. 4f). Yet, pretreatment with rWNT9a delayed IκBα degradation (Fig. 4f), while rWNT9a treatment by itself did not affect IκBα or pIκBα levels in SFBs (Fig. 4h). Thus, WNT9a can, like WNT4, interfere with NFκB signaling and like other WNTs it can activate p3848,49,50.
Loss of Wnt9a does not alter disease severity in the acute serum-transfer RA model
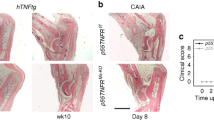
Next, we asked whether the loss of Wnt9a affects the phenotype of the STIA model, driven primarily by autoantibodies and IL-1β as the main cytokine. Here, two timepoints were analyzed, 14 days (peak of the arthritic-like phenotype) and 28 days (regression of the arthritic-like phenotype) after serum transfer35. Based on clinical parameters, loss of Wnt9a in the mesenchymal limb cells did neither affect the severity of bone destruction in the STIA mouse model, nor disease regression. Net weight gain, grip strength, and paw swelling were very similar between the arthritic Wnt9afl/− control and Wnt9a∆Prx1/− mice (Fig. 5a). Concurrently, the extent of bone destruction and reversal thereof was also very similar according to the radiographic images of hind paws from arthritic Wnt9afl/− control and Wnt9a∆Prx1/− mice at the 14- and 28-day timepoint (Fig. 5b). This was confirmed by the analysis and quantification of the bone volume (based on µCT-sections of hind paws at the two timepoints) (Fig. 5c,d). Also histologically, no differences were detected between the K/BxN-treated Wnt9afl/− control and K/BxN-treated Wnt9a∆Prx1/− mice at the 14-day timepoint (Fig. 6a). Next, we asked whether Wnt9a expression in SFBs was altered by a 48 h IL-1β treatment. By qPCR, Wnt9a was upregulated in SFBs upon treatment with 10 ng IL-1β (Fig. 6b). Yet, short-term IL-1β treatment led to a transient downregulation of Wnt9a at the 6 h timepoint (Fig. 6c). Of the other Wnt pathway-related genes, Wnt16 and Wnt5a were both upregulated in response to IL-1β treatment (Fig. 6b). All other Wnt pathway-related genes behaved similarly to TNF treatment except for Wisp1, which was downregulated upon IL-1β treatment (Fig. 6b). Except for Axin2, these expression changes were all statistically significant. Interestingly, in Wnt9a-deficient SFBs only Wisp1 expression was altered after IL-1β treatment; here, the downregulation in response to IL-1β was diminished (Fig. 6b). Consistent with reports on autoregulation and regulation of Il6 and Rankl, IL-1β treatment of SFBs upregulated its own expression as well as the expression of Il6 and Rankl (Fig. 6c, d)51,52,53. Furthermore, IL-1β upregulated Tnf expression at the early timepoints, but not after 48 h of exogenous treatment (Fig. 6c, d). Consistent with the literature, IL-1β treatment increased the expression of catabolic proteolytic enzymes, such as Mmp9, Mmp13, Adamts4, and Adamts5 in wild-type SFBs (Fig. 6d)54,55. In the Wnt9a-deficient SFBs, the expression of these genes was not different, except for Mmp9, which was slightly, but not statistically significantly downregulated (Fig. 6d).

a Assessment of net weight gain, grip strength, and ankle width in K/BxN-treated control (Wnt9afl/−) and Wnt9a-deficient mice over 28 days. b Three- and c two-dimensional microCT images of Wnt9afl/− (control) and Wnt9a-deficient hind paws, 14 (n = 19) and 28 days (n = 9) after K/BxN serum transfer. d Quantification of the bone volume in the area outlined by the white dashed line in c, encompassing the navicular, intermediate, and lateral cuneiform bones, at the 14- (n = 10) and 28-day (n = 9) timepoint.

a Representative images of Safranin O stained tarsal joint sections of the 14-day timepoint (n = 3). b qPCR analysis of Wnt genes and Wnt pathway-related genes in Wnt9afl/fl and Wnt9a-deficient SFBs after treatment with recombinant murine IL-1β. c qPCR analysis of short-term effects on the expression of Wnt9a, Il1b, and Tnf in response to stimulation with recombinant IL-1β. d qPCR analysis of long-term effects on the expression of cytokines and catabolic enzymes in Wnt9afl/fl (wild-type) and Wnt9a-deficient SFBs in response to stimulation with recombinant IL-1β for 48 h.
Discussion
In contrast to previous investigations regarding the role of Wnt signaling in the experimental hTNFtg mouse model, such as the inhibition of the antagonist DKK1, the loss-of the antagonist WIF1, or treatment with the agonist R-Spondin 1, where soft-tissue inflammatory aspects remained unaffected11,32,34, all aspects of the RA-like symptoms were affected by the loss of Wnt9a. Furthermore, disease progression and severity were augmented in the hTNFtg/+;Wnt9a∆Prx/− mice. This phenotype is similar to the alterations observed following the loss of the Wnt inhibitor sclerostin35. Yet, sclerostin levels remained unaltered in Wnt9a-deficient SFBs with or without TNF, or SFBs treated with rWNT9a, and in hTNFtg/+;Wnt9a∆Prx/− mice (Supplementary Fig. 1g, and data not shown).
As shown in previous studies, the mild TNF-induced inflammatory response can be dramatically boosted by co-administration of IL-1β56. Thus, the modestly increased transcriptional levels of the cytokines IL-1β and IL-6, as observed in Wnt9a- and Ctnnb1-deficient SFBs, may provide a sensitized proinflammatory environment that could markedly enhance the inflammatory effect of the hTNF transgene and thereby accelerate disease progression (Fig. 7a). Joints of 3-month-old Wnt9a∆Prx1/− mice do not show any inflammatory alterations (data not shown), which could partly be due to the observed downregulation of endogenous Tnf expression in the Wnt9a-deficient SFBs. We also noted a slight upregulation of the expression of the catabolic enzymes ADAMTS5 and MMP13 in the Wnt9a- as well as Ctnnb1-deficient SFBs. Both enzymes degrade cartilage matrix and, based on genetic experiments, are essential for cartilage degradation in murine osteoarthritis and rheumatoid arthritis models54,57,58. Hence, the in vitro observations are pointing to the presence of a procatabolic environment in the Wnt9a∆Prx1/− mice. In combination with the proinflammatory environment, this may predisposition the articular cartilage even further for destruction in the presence of the hTNF transgene. In support of this notion, in 6-month old Wnt9a∆Prx/− specimens ADAMTS5- and MMP13-positive cells were observed in the synovium (Supplementary Fig. 4b, c), and 9-month and older Wnt9a∆Prx/− mice develop osteoarthritis-like changes in their joints (C.H. and S.T. manuscript in preparation). Our in vitro results suggest that this deregulation of the procatabolic/proinflammatory environment is dependent on β-catenin (Fig. 7b).

a Compared to the situation in the wild-type, Wnt9a-deficient joints display a mild proinflammatory (slightly increased transcriptional levels of Il1b and Il6) and procatabolic (slightly increased transcriptional levels of Mmp13 and Adamts5) environment. In the hTNFtg/+;Wnt9a∆Prx/− joints, this is converted into a highly inflamed catabolic environment through the activity of TNF. Due to the predispositioning in the Wnt9a-deficient joints, the effects of the TNF transgene are more intense compared to the alteration in hTNFtg/+ joints. b WNT9a acts on the one hand through the Wnt/β-catenin pathway to maintain low transcriptional levels of the proinflammatory factors Il1b and Il6, and of the procatabolic factors Adamts5 and Mmp13 in synovial fibroblasts. On the other hand, WNT9a dampens the activity of the transcription factor NFκB, which is required for osteoclastogenesis downstream of TNF and RANKL.
Interestingly, these potential predispositioning parameters also exist in the Wnt9a∆Prx1/− STIA RA model. Yet surprisingly, the loss of Wnt9a did not influence disease progression or severity in this model. In contrast to the hTNFtg model, STIA is a transient RA model being driven by autoantibodies against the glucose-6-phosphate isomerase and mimicking primarily the effector phase of RA59. IL-1β signaling is crucial in this model, while TNF plays only a partial, not yet fully understood role7. In vitro in SFBs, we observed no upregulation of Tnf expression in response to long-term IL-1β treatment, and in Wnt9a-deficient SFBs, the endogenous Tnf expression levels were even reduced. Hence, one could speculate that this may in part be responsible for the observation that the absence of WNT9a-activity in the STIA model does not lead to an aggravation of the phenotype, as the TNF levels may be too low. Alternatively, the time the Wnt9a-deficient cells are exposed to inflammatory cytokines may be too short in the acute STIA model, which only exhibits a temporary RA-phenotype. The latter, could be addressed in the future by establishing the chronic STIA model in Wnt9a∆Prx/− mice, similarly to what has been observed regarding ApoE-deficiency in combination with the STIA model60. Furthermore, in vitro, we detected differences between IL-1β- and TNF-treated SFBs concerning other Wnts, and this may also contribute to the observed differences between the two models.
Based on the results from the hTNFtg/+;Wnt9a∆Prx mice our data suggest that the observed WNT9a upregulation in the synovium of human RA biopsies occurs likely as a protective response. As shown here, exogenous WNT9a can interfere with NFκB signaling in vitro. Hence, one possible mechanism how the in vivo observed upregulation of WNT9a may act in a protective fashion is by negatively influencing TNF-NFκB-driven osteoclastogenesis (Fig. 7b). Along this line, we noted that the number of osteoclasts was increased in the hTNFtg/+;Wnt9a∆Prx mice, and that conditioned medium from Wnt9a-deficient SFBs altered the spreading of osteoclasts in vitro in an M-CSF/RANKL mediated osteoclastogenesis. Interestingly, TNF can, in contrast to IL-1β, stimulate osteoclastogenesis even in the absence of Rankl61.
Nevertheless there are still some limitations to this study: (a) the expression changes were only analyzed in Wnt9a in-vitro-deleted SFBs, which were exogenously treated with TNF, instead of primary cells from the animals, which have a different history, and (b) WNT9a is produced in vivo by other cell-types besides SFBs, such as chondrocytes and osteoblasts, which are also targeted by the Prx1-Cre line and may contribute to the phenotype.
Supplementary information is available at Cell Death & Disease’s website.
Data availability
Representative data of datasets generated or analyzed during this study are included in this published article and its supplementary information files. All individual datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Walsh, N. C., Crotti, T. N., Goldring, S. R. & Gravallese, E. M. Rheumatic diseases: the effects of inflammation on bone. Immunol. Rev. 208, 228–251 (2005).
Lam, J. et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 106, 1481–1488 (2000).
Redlich, K. et al. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Invest. 110, 1419–1427 (2002).
Pettit, A. R. et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159, 1689–1699 (2001).
Keffer, J. et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031 (1991).
Monach, P. A., Mathis, D. & Benoist, C. The K/BxN arthritis model. Curr. Protoc. Immunol. https://doi.org/10.1002/0471142735.im1522s81 (2008).
Ji, H. et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J. Exp. Med. 196, 77–85 (2002).
Choe, J. Y., Crain, B., Wu, S. R. & Corr, M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J. Exp. Med. 197, 537–542 (2003).
Corr, M. et al. Interleukin 1 receptor antagonist mediates the beneficial effects of systemic interferon beta in mice: implications for rheumatoid arthritis. Ann. Rheum. Dis. 70, 858–863 (2011).
Sen, M. Wnt signalling in rheumatoid arthritis. Rheumatology 44, 708–713 (2005).
Diarra, D. et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 13, 156–163 (2007).
Schett, G., Zwerina, J. & David, J. P. The role of Wnt proteins in arthritis. Nat. Clin. Pract. Rheumatol. 4, 473–480 (2008).
Rabelo Fde, S. et al. The Wnt signaling pathway and rheumatoid arthritis. Autoimmun. Rev. 9, 207–210 (2010).
Cici, D., Corrado, A., Rotondo, C. & Cantatore, F. P. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int. J. Mol. Sci. 20, 5552 (2019).
Imai, K. et al. Differential expression of WNTs and FRPs in the synovium of rheumatoid arthritis and osteoarthritis. Biochem. Biophys. Res. Commun. 345, 1615–1620 (2006).
Sen, M. et al. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl Acad. Sci. USA 97, 2791–2796 (2000).
Teufel, S. & Hartmann, C. Wnt-signaling in skeletal development. Curr. Top. Dev. Biol. 133, 235–279 (2019).
Hartmann, C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol. 16, 151–158 (2006).
Baron, R. & Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192 (2013).
Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Clevers, H., Loh, K. M. & Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346, 1248012 (2014).
Grigoryan, T., Wend, P., Klaus, A. & Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 22, 2308–2341 (2008).
Klaus, A. & Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 8, 387–398 (2008).
Holland, J. D., Klaus, A., Garratt, A. N. & Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 25, 254–264 (2013).
Steinhart Z. & Angers S. Wnt signaling in development and tissue homeostasis. Development https://doi.org/10.1242/dev.146589 (2018).
Yang, Y. Wnt signaling in development and disease. Cell Biosci. 2, 14 (2012).
van Amerongen R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a007914 (2012).
Clevers, H. & Nusse, R. Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Yan, D. et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta -catenin signaling is activated in human colon tumors. Proc. Natl Acad. Sci. USA 98, 14973–14978 (2001).
Surmann-Schmitt, C. et al. Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J. Cell Sci. 122, 3627–3637 (2009).
Witte, F., Dokas, J., Neuendorf, F., Mundlos, S. & Stricker, S. Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr. Patterns 9, 215–223 (2009).
Stock, M. et al. Wif-1-deficiency uncouples cartilage and bone destruction in TNFalpha-mediated experimental arthritis. Arthritis Rheum. 65, 2310–2322 (2013).
Niida, A. et al. DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene 23, 8520–8526 (2004).
Kronke, G. et al. R-spondin 1 protects against inflammatory bone damage during murine arthritis by modulating the Wnt pathway. Arthritis Rheum. 62, 2303–2312 (2010).
Wehmeyer, C. et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 8, 330ra335 (2016).
Spater, D. et al. Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development 133, 3039–3049 (2006).
Seibler, J. et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 31, e12 (2003).
Logan, M. et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
Harada, N. et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 (1999).
Amara, C. S., Fabritius, C., Houben, A., Wolff, L. I. & Hartmann, C. CaMKII signaling stimulates Mef2c activity In vitro but only minimally affects murine long bone development in vivo. Front. Cell Dev. Biol. 5, 20 (2017).
Kan, A. & Tabin, C. J. c-Jun is required for the specification of joint cell fates. Genes Dev. 27, 514–524 (2013).
Guo, X. et al. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 18, 2404–2417 (2004).
Matsumoto, K., Miki, R., Nakayama, M., Tatsumi, N. & Yokouchi, Y. Wnt9a secreted from the walls of hepatic sinusoids is essential for morphogenesis, proliferation, and glycogen accumulation of chick hepatic epithelium. Dev. Biol. 319, 234–247 (2008).
Ali, I., Medegan, B. & Braun, D. P. Wnt9A induction linked to suppression of human colorectal cancer cell proliferation. Int. J. Mol. Sci. 17, 495 (2016).
Xiao, C. Y. et al. Expression of beta-catenin in rheumatoid arthritis fibroblast-like synoviocytes. Scand. J. Rheumatol. 40, 26–33 (2011).
Hardy, R. & Cooper, M. S. Bone loss in inflammatory disorders. J. Endocrinol. 201, 309–320 (2009).
Varfolomeev, E. E. & Ashkenazi, A. Tumor necrosis factor: an apoptosis JuNKie? Cell 116, 491–497 (2004).
Caverzasio, J. & Manen, D. Essential role of Wnt3a-mediated activation of mitogen-activated protein kinase p38 for the stimulation of alkaline phosphatase activity and matrix mineralization in C3H10T1/2 mesenchymal cells. Endocrinology 148, 5323–5330 (2007).
Chang, J. et al. Noncanonical Wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J. Biol. Chem. 282, 30938–30948 (2007).
Yu, B. et al. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat. Med. 20, 1009–1017 (2014).
Toda, Y. et al. Autocrine induction of the human pro-IL-1beta gene promoter by IL-1beta in monocytes. J. Immunol. 168, 1984–1991 (2002).
Tsukihara, S. et al. Interleukin-1beta-induced expression of IL-6 and production of human chorionic gonadotropin in human trophoblast cells via nuclear factor-kappaB activation. Am. J. Reprod. Immunol. 52, 218–223 (2004).
Deschner, J. et al. Regulation of RANKL by biomechanical loading in fibrochondrocytes of meniscus. J. Biomech. 39, 1796–1803 (2006).
Stanton, H. et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 434, 648–652 (2005).
Bondeson, J., Wainwright, S. D., Lauder, S., Amos, N. & Hughes, C. E. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res. Ther. 8, R187 (2006).
Henderson, B. & Pettipher, E. R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor alpha in the rabbit: evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 75, 306–310 (1989).
Singh, A. et al. Collagenase-3 (MMP-13) deficiency protects C57BL/6 mice from antibody-induced arthritis. Arthritis Res. Ther. 15, R222 (2013).
Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 (2013).
Christensen, A. D., Haase, C., Cook, A. D. & Hamilton, J. A. K/BxN serum-transfer arthritis as model for human inflammatory arthritis. Front. Immunol. 7, 213 (2016).
Archer, A. M. et al. ApoE deficiency exacerbates the development and sustainment of a semi-chronic K/BxN serum transfer-induced arthritis model. J. Transl. Med. 14, 170 (2016).
Wang, Y. et al. IL1beta and TNFalpha promote RANKL-dependent adseverin expression and osteoclastogenesis. J. Cell Sci. 131, jcs213967 (2018).
Acknowledgements
We thank Ulrich König for the preparation of the lentiviruses, Berno Dankbar for reagents and his valuable comments on the manuscript, and Rene Gronewald for taking care of the hTNF colony and producing the K/BxN serum.
Funding
This work was supported by grants from the Interdisciplinary Center of Clinical Research (IZKF, Har2/002/14) and the DFG (HA 4767/5-1). L.I.W. was supported by a grant of the Federal Ministry of Education and Science (01EC1408F), as part of the OVERLOAD-PrevOP consortium. The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
S.T.: Investigation; Formal Analysis; Project administration; Supervision; Visualization; Writing—original draft; Writing—review and editing, P.K.: Investigation, C.F.: Investigation, L.I.W.: Investigation; Writing—review and editing, J.B. and T.P.: Resources; Writing—review and editing, C.H.: Conceptualization; Funding acquisition; Resources; Project administration; Supervision; Visualization; Writing—original draft; Writing—review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics statement
Animal experiments in these studies were performed in accordance with the EU guidelines and approved by the North Rhine-Westphalia State Agency for Nature, Environment and Consumer Protection (LANUV). The collection of human material used in this study was approved by the Institutional Ethical Review Board of the Faculty of Medicine of the Otto-von-Guericke University (IRB No 73/18). Informed consent was obtained from the patients prior to surgery.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Agostini
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teufel, S., Köckemann, P., Fabritius, C. et al. Loss of the WNT9a ligand aggravates the rheumatoid arthritis-like symptoms in hTNF transgenic mice. Cell Death Dis 12, 494 (2021). https://doi.org/10.1038/s41419-021-03786-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03786-6
- Springer Nature Limited
This article is cited by
-
Genetic polymorphism of WNT9A is functionally associated with thumb osteoarthritis in the Chinese population
Advances in Rheumatology (2024)
-
RelA/MicroRNA-30a/NLRP3 signal axis is involved in rheumatoid arthritis via regulating NLRP3 inflammasome in macrophages
Cell Death & Disease (2021)