
Abstract
Atherosclerosis is a chronic inflammatory disease characterized by the accumulation of fatty deposits in the inner walls of vessels. These plaques restrict blood flow and lead to complications such as heart attack or stroke. The development of atherosclerosis is influenced by a variety of factors, including age, genetics, lifestyle, and underlying health conditions such as high blood pressure or diabetes. Atherosclerotic plaques in stable form are characterized by slow growth, which leads to luminal stenosis, with low embolic potential or in unstable form, which contributes to high risk for thrombotic and embolic complications with rapid clinical onset. In this complex scenario of atherosclerosis, macrophages participate in the whole process, including the initiation, growth and eventually rupture and wound healing stages of artery plaque formation. Macrophages in plaques exhibit high heterogeneity and plasticity, which affect the evolving plaque microenvironment, e.g., leading to excessive lipid accumulation, cytokine hyperactivation, hypoxia, apoptosis and necroptosis. The metabolic and functional transitions of plaque macrophages in response to plaque microenvironmental factors not only influence ongoing and imminent inflammatory responses within the lesions but also directly dictate atherosclerotic progression or regression. In this review, we discuss the origin of macrophages within plaques, their phenotypic diversity, metabolic shifts, and fate and the roles they play in the dynamic progression of atherosclerosis. It also describes how macrophages interact with other plaque cells, particularly T cells. Ultimately, targeting pathways involved in macrophage polarization may lead to innovative and promising approaches for precision medicine. Further insights into the landscape and biological features of macrophages within atherosclerotic plaques may offer valuable information for optimizing future clinical treatment for atherosclerosis by targeting macrophages.
Similar content being viewed by others

Facts
-
Macrophages metabolic characteristics and polarization play a crucial role in the initiation, growth, rupture, and healing stages of atheromatic plaque formation.
-
Hypoxia-related changes in macrophage metabolism and phenotype modulate atheromatic plaque growth by HIF-1α expression.
-
β-cyclodextrin treatment favor plaque regression promoting cholesterol efflux and anti-inflammatory properties by modulating macrophages metabolism.
-
The efferocytosis of apoptotic macrophages lead to immune suppression or inflammation resolution in atheromatic plaques.
Open Questions
-
What are the specific metabolic characteristics of macrophages that contribute to reduce plaque progression and stabilize vulnerable lesions in humans?
-
How do macrophage metabolic shifts in response to plaque microenvironmental factors affect the progression or regression of atherosclerosis?
-
Can targeting macrophage polarization pathways lead to effective and selective treatments for atherosclerosis?
-
How the identification of novel therapeutic targets to regulate the metabolism and polarization of macrophages at plaque sites may lead to innovative therapies for cardiovascular disease leading to effective precision medicine ?
Introduction
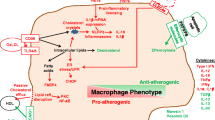
Atherosclerosis, a chronic inflammatory disease involving abnormal lipid metabolism, shares some similarities with autoimmune diseases due to the presence of immune responses directed against self-components within the arterial wall. Autoreactive effector T cells, which recognize self-antigens like oxidized low-density lipoproteins (oxLDL), play a pivotal role in triggering the inflammatory response associated with atherosclerotic lesions. Atherosclerosis involves the formation of lipid-rich plaques in the arterial intima (which also are characterized by a large population of immune, nonimmune, and apoptotic cells and apoptotic cell debris). As the pathological basis of most cardiovascular diseases, e.g., myocardial infarction, stroke and heart failure, with high morbidity and mortality rates, atherosclerosis has emerged as the leading cause of death worldwide [1, 2]. According to the ‘response to injury’ hypotheses, endothelium activation or damage is a crucial step in atherogenesis, involving endothelial dysfunction, increased permeability, formation of focal lesions, and a pro-inflammatory state. This damage can be induced by classical cardiovascular risk factors, including hyperlipemia, hypertension, hyperglycemia, obesity, and arterial wall shear stress [3,4,5,6]. Accordingly, the aggregation of low-density lipoproteins (LDLs) in the intima, pathological modifications (oxidation and acetylation), and increased expression of leukocyte adhesion molecules mediate the rolling and firm attachment of monocytes and lymphocytes to the damaged intima surface. After stimulation by chemokines and cytokines, monocytes enter the intima and differentiate into mature macrophages, which are able to ingest excessive lipoproteins and eventually form cholesterol-enriched foam cells, which are characterized by increased expression of lipid-processing genes [7, 8]. Interestingly, although the clearance of lipoproteins by macrophages seems to contribute to the prevention of deleterious lipid production, the excessive lipid metabolism in macrophages leads to alterations in their phenotypes and functions, ultimately causing their death. For example, foam cells cannot easily escape the intima and are thus trapped due to their diminished migration capacity [9, 10], leading to the accumulation and retention of foam cells in the arterial intima. During the progression of atherosclerosis, a large number of foam cells or macrophages undergo apoptosis induced by oxidized LDLs, cholesterol crystals, hypoxia, mitochondrial dysfunction or abundant death ligand protein expression. Apoptotic cells contribute to either immune suppression or inflammation resolution [11, 12]. However, in advanced plaques, efferocytosis is impaired; hence, abundant apoptotic cells are ineffectively eliminated, resulting in secondary necrosis together with the release of their lipid contents, inflammatory cytokines, cellular debris and damage-associated molecular patterns. These released substances not only trigger a stronger immune response to perpetuate plaque inflammation but also constitute the complex plaque microenvironment that contributes to necrotic core formation (Fig. 1).

A normal artery consists of three layers, the tunica adventitia, tunica media (containing abundant SMCs), and tunica intima (located in the subendothelial space). Under homeostatic conditions, almost no blood leukocytes accumulate in the endothelial layer. In contrast, when activated by proinflammatory cytokines or other cardiovascular risk factors, endothelial cells express leukocyte adhesion molecules (such as ICAM-1 and VCAM-1) and thereby cause the rolling and attachment of circulating monocytes, neutrophils and lymphocytes. In parallel, increased endothelium permeability promotes the entry of lipoproteins into the intima. Monocytes are recruited to the intima by chemokines (such as CCL2) and differentiate into mature macrophages mediated by M-CSF. Within the intima, macrophages take up excessive lipoproteins and form foam cells, which is a crucial step in initiating atherosclerosis. Recent evidence has demonstrated that foam cells can also be formed from macrophage-like cells that are produced via the transdifferentiation of SMCs. Furthermore, excessive lipid metabolism in foam cells results in inflammatory cytokine production and induces cell death (apoptosis). During the development of atherosclerosis, abundant apoptotic macrophages or neutrophil extracellular traps (NETs) within plaques cannot be removed by surrounding macrophages because of defective efferocytosis, leading to secondary necrosis accompanied by the release of cellular contents such as lipids, cell debris and DAMPs, which ultimately contribute to a lipid-rich necrotic core and unresolved inflammation. Notably, macrophages can be classified into five main subsets based on several scRNA-Seq studies on atherosclerotic plaques in mice; these subsets are namely res-like macrophages, TREM2hi macrophages, inflammatory macrophages, proliferating macrophages and IFNIC macrophages. Among these macrophages, res-like macrophages, with an M2-like phenotype, are distributed in the adventitia. TREM2hi macrophages, with powerful lipid catabolism capabilities, are regarded as foam cells and are likely derived from SMCs. Inflammatory macrophages, with upregulated inflammatory genes and pathway activation, are key contributors to plaque inflammation. Generated by BioRender.
Considerable attention is being directed to therapeutic strategies for atherosclerosis that are based on macrophage targeting. For example, a phenotypic shift in macrophages (from the M1-like phenotype to the M2-like phenotype) can be achieved by metabolic reprogramming because the metabolic states of macrophages determine their functionalattributes [13, 14]. Notably, M2-like macrophages rely extensively on the mitochondrial oxidative phosphorylation (OXPHOS) system [15], while M1-like macrophages are prone to exhibit anaerobic glycolysis [16].
Through advanced single-cell multiomics technologies (Fig. 2), scientists are able to distinguish discrete immune or nonimmune cells [17,18,19,20] in atherosclerotic plaques and analyse the functions of different plaque cell subsets and their interaction networks. In this review, considering recent advances in understanding atherosclerotic progression in humans and mice, we focus mainly on the origin, heterogeneity, metabolic pathways and death modalities of plaque macrophages in response to the complex plaque microenvironment.

Tissues at different stage of plaque development from atherosclerotic model mice or patients with atherosclerosis-related cardiovascular diseases were processed into tissue sections and single-cell suspensions for single-cell and multiomics analysis. a, b Using imaging mass cytometry and spactial transcriptomics to map in situ plaque microenvironment information such as cell type, metabolic characteristics and spatial distribution. c–f Single-cell transcriptome sequencing (scRNA-seq), cytometry by time of flight (CyTOF), cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) and single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) were performed to comprehensively map the metabolic, functional and epigenetic landscapes of plaque cells. Generated by BioRender.
The natural history of the human atherosclerotic plaques
The natural history of human atherosclerotic plaques in distinct vascular regions reveals a progressive clinical presentation; although some individuals remain asymptomatic throughout their lifespan, others suffer ischemic symptoms or potentially life-threatening events, e.g., acute myocardial infarction or stroke [21]. The current classification of human atherosclerotic plaques includes four different subgroups [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]: (i) stable plaques, (ii) vulnerable plaques, (iii) unstable thrombotic plaques and (iv) healed lesions. All stable plaques are characterized by a low level of inflammatory infiltrate [22,23,24,25].
• The typical fibrous cap of an atheroma comprises a large lipidic-necrotic core containing extracellular lipids, cholesterol crystals and necrotic cell debris, covered by a thick fibrous cap, whose thickness varies in different vascular regions: <65 µm in coronaries and <165 µm in carotids [26,27,28]. The amount of inflammatory infiltrate within the fibrous cap is modest.
• Vulnerable plaques, or thin fibrous cap atheromata (TCFAs), are “high risk” plaques, prone to rupture and thrombosis [29]. These plagues are characterized by abundant inflammatory cells, macrophages, T cells and a few smooth muscle cells. Nodules formed by eruptive calcification [30, 31] are large calcified structures protruding into the vascular lumen that form ectopic nodules. The thin fibrous cap frequently ruptures, and an overlying luminal thrombus is formed.
• Unstable plaques are associated with the presence of a luminal thrombus and correlated with the fast progression of atherosclerotic disease and the onset of acute symptoms [28,29,30,31,32]. Acute thrombi might undergo rupture, erosion, or calcification. A disrupted fibrous cap is infiltrated by numerous macrophages and lymphocytes and is associated with an overlying thrombus in continuity with the underlying necrotic core [33, 34]. Similar to other ectopic calcifications [35, 36], two different types of calcifications in carotid atheromatous plaques have been described [37]: hydroxyapatite and calcium oxalate nodules [38]. Among cells in plagues, macrophages are thought to be the main contributors to plaque calcification. Indeed, these cells exhibit strong phenotypic plasticity and differentiate into a variety of cell types depending on the surrounding environment [39]. M1 macrophages can directly release oncostatin M (OSM) to promote the differentiation of vascular smooth muscle cells (VSMCs) with osteoblastic phenotypes through activation of the JAK3-STAT3 pathway, thus promoting the formation of hydroxyapatite structures in plaques [40]. Hemorrhage within a plaque is commonly due to the rupture of small, newly formed and thin-walled vessels, which can easily break following an increase in pressure or the presence of bone spicules or cholesterol clefts. Sometimes, intraplaque bleeding is sudden and large, causing rapid occlusion of the vascular lumen, which leads to acute onset of clinical symptoms.
Inflammation factors and, in particular, different types of monocytes/macrophages play determinant roles in all phases of plaque evolution. M1-like macrophage infiltration has been described in early stages of atherosclerotic lesion development. Moreover, a topographic relationship among the inflammatory infiltrate, plaque rupture and thrombosis play pathogenetic roles in macrophagic cells at cap rupture sites in patients who die because of acute myocardial infarction. In contrast, M2-like macrophages seem to play a prominent role in the organization of thrombi, resulting in transformation into a healed lesion [41].
Morphological characterization of macrophages
Macrophages play a critical role in the elimination of pathogens, apoptotic cells, and cell debris [42, 43]. Additionally, they actively participate in tissue inflammation and regeneration [44,45,46]. The alteration of macrophage morphology essentially depends on their activation state or their responses microenvironment stimuli [47, 48]. Specifically, after stimulation with GM-CSF, macrophages differentiated from human monocytes are characterized by a small and round shape morphology. In the presence of M-CSF, macrophages exhibit an elongated shape and contain numerous vacuoles [49]. Understanding the relationship between the morphological characteristics and functional properties of macrophages during the development of diseases will help to effectively predict patient prognosis and further improve therapeutic outcomes [50].
The origin of macrophages in the plaque
Macrophages are readily recruited to plaque-prone sites and produced through haematopoiesis in the bone marrow [51, 52]. An additional source of plaque macrophages, namely, local proliferating macrophages, contributes to the formation of atherosclerotic plaques [53,54,55]. Moreover, in the context of atheromatic plaques, intimal vascular smooth muscle cells (VSMCs) have the ability to transdifferentiate into macrophage-like cells. These VSMCs not only take up lipoprotein and become VSMC-derived foam cells but also drive the formation of macrophage-like cells, mesenchymal stromal/stem cells and osteochondrogenic cells by phenotypic switching [56]. Additionally, aortic macrophages originating from yolk sac- and bone marrow-derived monocytes after birth are similar to resident macrophages in different tissues and organs and play crucial roles in maintaining physiological function and tissue homeostasis [57,58,59]. Thus, the diversity of macrophage sources [60] contributes to the heterogeneity of plaque macrophages.
The contribution of circulating monocytes to plaque macrophages development
Circulating monocytes in mice are generally classified into two major subsets, conventional Ly6Chigh monocytes (with high expression of CCR2 and CD62L and moderate expression of CX3CR1) and patrolling Ly6Clow monocytes (with high expression of CX3CR1 as well as low expression of CCR2 and CD62L) [57,58,59, 61]. The developmental trajectory from haematopoietic stem cells to monocytes or macrophages supports the view that Ly6Chigh monocytes give rise to Ly6Clow monocytes via preferential transition into Ly6Cint monocytes named intermediate monocytes [59, 62, 63]. In humans, according to the expression levels of CD14 and CD16, monocyte subsets are classified into CD14+ CD16− and CD14+ cells, which are thought to correspond to Ly6Chigh, Ly6Cint and Ly6Clow monocytes in mice, respectively [64, 65].
Interestingly, these monocyte subsets represent heterogeneity not only in the development trajectory but also in physiological function. For example, steady-state Ly6Clow monocytes are regarded as endothelial cell-protective monocytes that patrol the arterial vasculature, where they scavenge particles, including lipoproteins, cellular debris, and necrotic cells [66, 67]. An increase in the number of Ly6Clow monocytes under hyperlipidaemic and atherosclerotic conditions partially depended on CCR5, and depleting the number of patrolling Ly6Clow monocytes via knockout of transcription factor Nr4a1 (Nur77) determined the survival or death of Ly6Clow monocytes in ApoE/− mice, resulting in pronounced endothelial apoptosis and exacerbated atherosclerosis, clearly confirming that Ly6Clow monocytes are crucial for maintaining endothelial cell homeostasis [67]. Moreover, recent work based on an in-depth phenotype analysis of monocyte subsets demonstrated that LYN (Lck/yes novel tyrosine kinase) regulated the signaling pathways and lifespan in Ly6Clow monocytes, and Lyn deficiency led to the production of protective patrolling monocytes, impairing atherogenesis induced by a high-fat diet [68].
Conventional Ly6Chigh monocytes, regarded as the constituents of inflammatory monocyte populations, are able to rapidly respond to inflammatory signals, invade damaged sites, and ultimately differentiate into macrophages [65]. Specifically, in atherosclerosis, steady and sustained recruitment of blood-borne monocytes (Ly6Chigh monocytes) to lesions accelerates the progression of atherosclerosis, and Ly6Chigh monocytes are thus thought to be proinflammatory cells [8, 69]. A high level of Ly6Chigh monocytes in cases of hyperlipidaemia have been associated with monocytosis [69]; these monocytes are preferentially produced in bone marrow and from progenitor cells in the initial stage of atherosclerosis. During atherosclerotic development, the spleen, as an extramedullary site, gradually supplements the haematopoietic function of the bone marrow by generating Ly6Chigh monocytes that infiltrate plaque regions [51]. Moreover, inflammatory Ly6Chigh monocytes can be converted into M2-like macrophages in a STAT6-dependent manner, leading to atherosclerosis regression [70].
The contribution of local proliferation of macrophages to plaque macrophage formation
This is an open question [53, 71, 72]: local macrophage proliferation contributes to ~87% of the macrophages accumulated in advanced atherosclerotic lesions [53]. Recent evidence revealed that aortic intima resident macrophages (MacAIR) arise from bone marrow progenitors and are seeded into the aorta at birth and are sustained mainly through local proliferation in the a steady state, independent of the number of recruited monocytes [71]. Moreover, MacAIR cells can also become foam cells and promote the recruitment of monocytes, thereby contributing to the formation of early atherosclerotic lesions. Due to their limited proliferation during plaque progression, which is insufficient to drive the expansion of plaque macrophages, MacAIR cells rely heavily on monocytes for atherosclerotic plaque progression [71]. Interestingly, apoptotic cell degradation after macrophage efferocytosis induces macrophage proliferation, which promotes tissue repair during atherosclerosis regression [73]. Therefore, locally proliferating macrophages play multifaceted roles in different stages of atherosclerosis, and the plaque microenvironment, such as the level of ox-LDL [74], the number of apoptotic cells or their metabolites, may influence their proliferation [73].
The contribution of smooth muscle cell (SMC) transdifferentiation to plaque macrophages
SMCs have garnered attention for a long time as an important contributor to plaque growth [56]: at least a subset of foam cells in atherosclerosis originates from SMCs [75]. Furthermore, VSMCs coexpress CD68 (as a macrophage marker) and αSMA in human atherosclerotic plaques [76, 77], and SMCs can transdifferentiate into macrophage-like cells after cholesterol loading [78]. SMCs can undergo colony proliferation and be converted into macrophage-like cells that lose αSMA expression and that constitute a primary component of atherosclerotic lesions [55]. However, these data need to be evaluated with caution because macrophage markers (CD68 or MAC2) are also expressed in other myeloid cells, and the possibility of labeled cells differentiating into other cells (mesenchymal stem-like and osteochondrogenic cells) should be considered. Moreover, the transformation of SMCs into macrophage-like cells is governed by diverse factors: deficient Krüppel-like factor-4 (KLF4) in SMCs resulted in reduced SMC-derived macrophage-like cells and a profound decrease in lesion size [79]; BCLAF1 (BCL2 [B-cell lymphoma 2]-associated transcription factor-1) was a central transcription factor for the survival and transdifferentiation of SMCs into a macrophage-like phenotype under lipid stress conditions [80]. In contrast, activation of the NOTCH signaling pathway completely prevented the transformation of SMCs into macrophage-like cells in vitro [81]. Indeed, a portion of plaque macrophages are derived from SMCs. However, the true contribution of SMC transdifferentiation to the macrophage population in atherosclerosis needs to be determined, and the biological functions of these SMC-derived macrophages still need to be further substantiated.
The landscape and heterogeneity of aortic macrophages in atherosclerosis
Macrophages are a highly heterogeneous and plastic cell population that rapidly respond to microenvironmental signaling. Macrophage subsets were initially described as M1 and M2-like macrophages, which represent two extremes of the activated macrophage spectrum [82,83,84,85,86]. M1-like macrophages are the predominate macrophage cells in the rupture-prone shoulder regions of a plaque, while M2-like macrophages are the most abundant macrophage type in the plaque adventitia [84]. However, macrophages constitute a broad phenotype spectrum and extensive plasticity when exposed to local tissue signaling cues, including signaling by cytokines, pattern recognition receptor ligands, and other immunomodulatory molecules [87, 88].
In addition to M1/M2-like macrophage subsets, Mox, M4, M(Hb) and Mhem macrophages have been described in plaques [85, 89, 90]. Intraplaque Mox macrophages, with decreased phagocytic and chemotactic capacity, are activated by oxidized phospholipids and regulated by a key regulator nuclear erythroid-2 related factor (Nrf2), which modulates the expression of redox-regulating genes such as haem oxygenase-1 (HO-1), thioredoxin reductase 1 (TrxR1), and sulfiredoxin-1 (Srxn1) [91]. M4 macrophages, frequently found in the atheromas, are activated by the platelet chemokine CXCL4 [92] and characterized by the coexpression of matrix metalloproteinase-7 (MMP7) and Ca2+-binding protein S100A8 [93]. CXCL4-induced M4 macrophages seemingly play a harmful role due to a reduction in the atheroprotective hemoglobin receptor CD163 and a positive association with vulnerable plaques [94, 95]. In contrast, M(Hb) and Mhem macrophages activated by hemoglobin and haeme, respectively, play more atheroprotective roles attributed to their prevention of foam cell formation and lipid accumulation by promoting the expression of reverse cholesterol transport-associated genes, such as liver X receptor alpha (LXRα) and ATP-binding cassette transporters A1 (ABCA1) [96, 97]. Notably, CD163+/CD206+ (also known as mannose receptor, Mrc1) macrophages, M(Hb), often involved in autoimmune response, do not always confer protection against atherosclerosis because CD163+ macrophages can promote angiogenesis, vascular permeability and inflammatory cell recruitment via the CD163-HIF1α-VEGFA pathway [98, 99]. These studies emphasized the multifaceted roles of macrophage subsets in determining the plaque inflammatory state and influencing atherosclerosis progression.
Aortic macrophage subpopulations based on single-cell technologies
There are five main macrophage subsets in the atherosclerotic plaques of humans and mice; these cells include resident-like macrophages (Res-like Macs), inflammatory macrophages (inflammatory Macs), macrophages with high expression of triggered receptor expressed on myeloid cells 2 (TREM2hi Macs), proliferating macrophages (proliferating Macs) and interferon-inducible macrophages (IFNIC Macs) [100] (Figs. 1, 3).

Plaque macrophages can be classified into five main subsets based on several scRNA-seq studies in humans and mice, including Res-like macrophages, Foamy/TREM2hi macrophages, Inflammatory macrophages, Proliferating macrophages, and IFNIC macrophages (Type I IFN induced response signature). These macrophage subsets exhibit distinct gene characteristic, biological functions, and spatial distributions.
Res-like macs originate from embryonic CX3C motif chemokine receptor 1-positive (CX3CR1+) precursors and circulating monocytes after birth [101], and these cells are predominantly located in the adventitia of arteries under both healthy and disease conditions [100]. Moreover, single-cell studies revealed that Res-like Mac are involved in the process of endocytosis, and lysosomes and are enriched with proteins encoded by genes such as lymphatic vessel endothelial hyaluronan receptor 1 (Lyve1), Cx3cr1, Timd4, folate receptor β (Flor2), Cd206, Ccl8, factor XIIIa (F13a1), carbonyl reductase 2 (Cbr2), seleno-protein P (Sepp1), colony stimulating factor1 (Csf1) and platelet factor (Pf4), thereby presenting an M2-like phenotype [7, 101,102,103].
Inflammatory Macs, regarded as chemokinehigh macrophages [104] or nonfoamy macrophages [7], not only constitute plaque macrophage populations but are also considered to be the main drivers of inflammation [105,106,107] in the lesions because of the many inflammatory pathways involved in this subset of cells with high expression of inflammatory genes, including Ccl2-5, Cxcl1, Cxcl2, Il1β, Nlrp3, Nfkbia, Tnf and Ear1 [101], similar to the gene profile of M1-like macrophages. Moreover, higher expression of CCR2 in inflammatory macrophages suggests that these cells are likely derived from circulating monocytes.
TREM2hi Macs are foamy lipid-laden macrophages with high expression of Trem2, Gpnmb, Spp1 (secreted phosphoprotein 1), Mmp12, Mmp14, Cd9, and markers of lipid transport (Abca1, Lipa, Fabp4 and Fabp5) and lysosomal cathepsins (Ctsb, Ctsd and Ctsz), in atherosclerotic plaques but not in healthy aortas [7, 101, 108]. TREM2hi Macs, constituting a lipid-handling macrophage subset, seem to be endowed with powerful lipid catabolic and anabolic capabilities and present genetic features similar to those of osteoclasts, suggesting a role in plaque calcification. How TREM2hi Macs affect plaque calcification remains unclear. A possible reason is that plastic VSMCs may adapt to the complex plaque microenvironment and thus acquire osteoclastic and macrophagic phenotypes [101, 109]. Notably, although foamy TREM2hi Macs exhibit low expression of inflammatory genes, they do not establish a proinflammatory milieu [7].
Proliferating Macs and IFNIC Macs, two small clusters of macrophages, reside in healthy vessels and atherosclerotic plaques, exhibiting different transcriptional profiles depending on the conditions [7, 71, 103, 104]. These proliferating mac populations are enriched in Birc5 (baculoviral IAP repeat-containing -5, inhibiting apoptosis), Stmn1 (stathmin-1) and Mki67 (marker of proliferation Ki67), reflecting a proliferative state and likely functioning as pools of macrophages to maintain macrophage self-renewal and population numbers. The IFNIC Macs subset displays high expression of the interferon-stimulated genes, including Ifit3, lsg15 and Irf7. It has been reported that IFNIC Macs amplify post-MI (myocardial infarction) inflammation by initiating IRF3 [110]. However, IFNIC Macs are also present in the aorta in a steady state and are regarded as resembling as adventitial macrophages [71]. Therefore, the function of IFNIC Macs in aortic homeostasis or disease progression is still unclear and needs to be determined via further investigation in upcoming studies.
Sanin et al. [111] established a common framework of monocyte-derived macrophage activation, which is also applicable to the analysis of macrophage activation status during atherosclerotic progression and regression. Specifically, distinct macrophage activation stages were defined on the basis of four conserved activation pathways, including the “phagocytic pathway”, “oxidative stress pathway”, “inflammatory pathway”, and “remodeling pathway”, representing P1, P2, P3, and P4, respectively. Moreover, these authors found that phagocytic pathway macrophages are the predominant atherosclerotic plaque macrophages and the most abundant macrophages under a condition characterized by dietary and pharmacological intervention-induced atherosclerosis regression characterized by high expression of phagocytosis- associated genes, suggesting that these interventions accelerated the transition of late P2 stage cells into final P2 stage cells. The biological functions and metabolic changes of macrophages during the course of inflammatory responses and in tissue homeostasis are still under investigation.
Macrophage interactions with T cells in atherosclerotic plaques
Intercellular interactions within an atherosclerotic plaque orchestrate the pathogenesis of atherosclerosis and associated cardiovascular events [108,109,110,111,112]. Fernandez et al. [108] discovered that adaptive signals and ligands derived from T cells were involved in macrophage activation, polarization, migration, and foam cell formation through ligand‒receptor interactions (VCAN-TLR1/2 and HSP90B1-TLR2, which are involved in the activation of proinflammatory macrophages; APOE-LRP1 and LRPAP1-LDLR, which are involved in regulating lipid, including cholesterol, efflux). Similarly, ligands expressed by macrophages were also predicted to bind CD4+ and CD8+ T-cell receptors, which may have contributed to T-cell proliferation and the immune response. A ground-breaking study [112] characterized the clonal expansion of plaque effector CD4+ T cells that present with an activated cytotoxic phenotype (GZMA, GAMK) and increased expression of CD69, FOS, FOSB and JUN, suggesting antigen-specific activation of CD4+ T cells in plaques. Interestingly, the TREM2hi macrophage (foam cell) population was implicated in the clonal expansion of effector CD4+ T cells in atherosclerotic lesions mediated via multiple pathways associated with costimulation and immunological synapse formation; the factors involved included CD99, macrophage inhibitory factor (MIF), ANNEXIN, CD6 and CD40. These observations suggest an autoimmune component in atherosclerotic plaques or the circulatory system driven by autoreactive CD4+ T cells, apparently following their interaction with foam cells (Fig. 1). Therefore, targeting autoimmune components within plaques and blood and the costimulatory pathways in T cells and macrophages may be promising approaches to the treatment of atherosclerosis-related cardiovascular diseases.
Microenvironmental cues shape plaque macrophage metabolism and function
Here, we outline the microenvironmental factors driving alterations in macrophage functions alongside metabolic adaptation based on in vivo evidence and in vitro mechanistic research (Fig. 4).

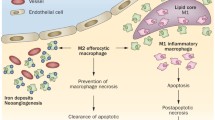
a In atherosclerotic plaques, macrophages are exposed to various lipids (including LDLs, ox-LDLs, oxPAPCs, and cholesterol crystals) that either promote (mostly) or attenuate a proatherogenic microenvironment. Production of intracellular cholesterol crystals and accumulation of free cholesterol following excessive lipid uptake by macrophages via receptor pathways and phagocytosis directly or indirectly lead to metabolic changes (i.e., increased glycolysis and OXPHOS rates), the production of proinflammatory cytokines and the activation of death pathways, ultimately resulting in atherosclerotic progression, which is blunted by effective cholesterol efflux. For example, β-cyclodextrin promotes atherosclerosis regression by activating LXR-targeting genes (such as ABCA1 and ABCG1) that mediate cholesterol metabolism and inflammatory responses in macrophages. b Moreover, hypoxic microenvironments jointly created by increased oxygen demand in inflammatory cells and vigorous metabolism (such as that in activated macrophages and lipid-loaded foam cells) inversely potentiate glycolysis, inflammatory gene expression, and necroptotic death in macrophages. c Cholesterol crystals can also trigger NETosis and the release of danger signals (MPO, MMPs, ROS, dsDNA) that prime macrophages for cytokine release (IL-1β). Recent works have shown that metabolites (including DNA, arginine, ornithine, and methionine) released from apoptotic cells during efferocytosis enhance continuous efferocytosis and injury resolution through different signaling pathways. d Additionally, macrophages in the plaque undergo distinct death modes (i.e., apoptosis, necroptosis, and pyroptosis), which influence atherosclerotic progression and plaque stability. The elimination of apoptotic cells via efficient efferocytosis induces immune suppression, while defective efferocytosis induces immune responses triggered by danger signals from necroptotic or pyroptotic cells. Generated by BioRender.
Lipid-mediated changes in macrophage metabolism and functions
Monocyte-derived and tissue-resident macrophages in the intima take up lipoproteins that are then retained and modified via diverse processes, including efferocytosis, micropinocytosis, and phagocytosis as well as through scavenger receptors (CD36, SR-A, SR-B1, LRP1, and LOX1) [8]; these modifications are beneficial changes that eliminate inflammatory lipids. However, excessive uptake of lipids by macrophages contributes to their transformation into foamy macrophages, leading to inhibited migration and entrapment in the intima, where free cholesterol-induced apoptosis of macrophages further results in the amplification of chronic inflammation [8].
An in vitro study mimicking macrophage foam cell formation via ox-LDLs showed that ox-LDLs increased the uptake of [16] F-fluorodeoxyglucose by macrophages and enhanced glycolysis in macrophages by upregulating GLUT1 and hexokinase expression. Mechanistically, this metabolic alteration was mediated by Nox2-dependent production of reactive oxygen species (ROS), which promote hypoxia-induced factor-1α activation and stabilization [113]. Similarly, Chen et al. [114] found that ox-LDL/CD36 signaling contributes to a metabolic shift from the mitochondrial oxidative phosphorylation (OXPHOS) to glycolysis, superoxide production, and proinflammatory cytokine expression in peritoneal macrophages ex vivo. In addition, increased rates of glycolysis and macrophage foam cell formation and inflammation induced by ox-LDLs were associated with pyruvate kinase M2 (PMK2), suggesting a close link between inflammation-related cellular dysfunction and metabolic changes in the context of atherosclerotic coronary artery disease [115]. Specifically, PKM2 induced by ox-LDLs interacts with sterol regulatory element-binding proteins (SREBP1) and increases lipid binding and uptake or inhibits cholesterol efflux, ultimately leading to the formation of foamy macrophages. Consistent with the aforementioned study, upregulated PKM2 expression was evident in atherosclerotic plaques in Ldlr−/− mice fed a high-fat Western diet compared with control mice fed a chow diet, and lack of PKM2 in myeloid cells reduced atherosclerotic lesion progression attributed to inhibited inflammation, impaired glycolytic activity, or enhanced efferocytosis that is typically mediated via the upregulation of LRP1 in macrophages [116].
Internalized lipoproteins and their associated lipids are digested in lysosomes, which subsequently release a large amount of fatty acid and free cholesterol. In addition, free cholesterol can be pumped out from cells to APOA1-deficient cells, with low lipid synthesis via ABCA1 and ABCG1, key lipid transporters regulated by the transcription factor heterodimeric-liver X receptor (LXR)–retinoid X receptor (RXR). Activation of this reverse cholesterol transport pathway leads to the accumulation of cytotoxic free cholesterol, which can traffic to the endoplasmic reticulum (ER) and activate the unfolded protein response (UPR), eventually resulting in ER stress-CHOP-Bcl2-associated X protein-mediated apoptosis in macrophages [117, 118]. Furthermore, enrichment of free cholesterol in foamy macrophage membranes can boost inflammatory signaling from lipid rafts, particularly via the activation of the TLR4 and NF-κB signaling pathways [119, 120]. Finally, cholesterol crystals formed by free cholesterol in macrophages can trigger NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome activation, which results in the secretion of IL-1β and IL-18. Accordingly, a Western diet or high-fat diet can induce “innate immunity reprogramming” in an NLRP3-dependent manner [121, 122]. In this scenario, ox-LDLs can induce trained immunity in human monocytes via epigenetic histone modifications; these cells are characterized by increased proinflammatory cytokine expression and enhanced formation into foam cells [123]. Interestingly, neutrophil extracellular trap formation triggered by cholesterol crystals licenses macrophages for the production of cytokines, which activate T helper 17 cells and lead to immune cell recruitment and amplification of immune responses in plaques [124, 125].
In addition to ox-LDLs, oxidized phospholipids composed of polyunsaturated fatty acids, which are also recognized and taken in by receptors (including CD36, SR-B1, Toll-like receptor (TLR)-2), TLR4, and E-type prostaglandin receptor (EP2)) and nonreceptors [126, 127], exhibited proinflammatory functions and atherogenicity in hypercholesterolaemic mice [128, 129]. In BMDMs primed with LPS, oxidized phospholipid 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholines (collectively known as oxPAPCs) potently drove a hypermetabolic state characterized by increased glycolysis and OXPHOS as well as glutaminolysis and oxaloacetate accumulation. These metabolic changes potentiate HIF-1α stabilization and further promote hyperinflammation by increasing IL-1β production [130]. Furthermore, oxPAPC-driven immunometabolic adaptation also occurs in atherosclerotic mice and hypercholesterolemic patients [130].
These findings support the notion that lipid-loaded foamy macrophages are proinflammatory cells that drive atherosclerotic lesion progression. However, lipid accumulation in macrophages does not always lead to activation or inhibition of inflammatory gene expression. For example, foamy macrophages in which desmosterol, a cholesterol biosynthetic intermediate, had accumulated, induced many homeostatic responses, including activation of LXRα/β gene expression, inhibition of SREBP1 and SERBP2 gene expression, alteration of fatty acid metabolism [131,132,133,134], and suppression of inflammatory response-related gene expression [135]. Subsequent research showed that depletion of desmosterol in macrophages promoted IFN-mediated responses and attenuated the expression of anti-inflammatory macrophage markers. Moreover, increased mitochondrial ROS production and NLRP3 inflammasome activation were observed in the desmosterol-depleted macrophages [136]. These clear discrepancies may be well explained by the results of recent single-cell studies on atherosclerosis, which revealed that nonfoamy but not foamy macrophages exhibited proinflammatory effects with distinct features that are also found in macrophages in atherosclerotic plaques [7]. Intimal foamy macrophages resemble TREM2hi macs by exhibiting high expression of genes involved in cholesterol metabolism, fatty acid transport, OXPHOS, proteasome and lysosome activity and PPAR signaling.
Hypoxia-driven changes in macrophage metabolism and functions
Hypoxia is evident in all stages of atherosclerotic lesion formation [137,138,139,140]. An imbalance between the supply and demand of oxygen [141,142,143] in the arterial wall leads to a hypoxic plaque microenvironment, mainly arising from the combination of increased cellular metabolic demand and reduced oxygen diffusion caused by thickening of the arterial wall during the development of atherosclerotic lesions [144]. Hypoxia is more likely to be caused by a combination of increased metabolic oxygen demand, especially in metabolically active inflammatory cells, not by impaired oxygen delivery because hypoxia is also evident in some subluminal (20–30 µm) foam cells or areas [138, 139], despite their location of these cells or areas being within the regions with oxygen diffusion (at distances of 100–250 µm [145]).
In humans, hypoxic regions colocalize with HIF, VEGF and macrophages and foam cells, and this colocalization is correlated with intraplaque angiogenesis [138]. In parallel, the expression of HIF-1α target genes (e.g., GLUT1, GLUT3 and HK1) is significantly increased during the progression from early to advanced atherosclerosis development, reflecting enhanced reliance on glycolysis in macrophages [138]. In mice, hypoxic areas contained abundant HIF-1α-expressing macrophage foam cells with high expression of GLUT1 [139]. In contrast, HIF-1α deficiency in myeloid cells not only led to reduced plaque and necrotic core sizes [146] but also inhibited GLUT1 expression in foam cell-rich plaque regions, which blunted hypoxia-mediated glucose uptake by macrophages in vitro. In addition, hypoxia induced the accumulation of sterol and decreased cholesterol efflux via ABCA1 in macrophages, which was substantially reversed by blocking HIF-1α expression [147]. Apparently, this metabolic switching [148] and proinflammatory activation in macrophages is attributable to the transcriptional activity of HIF-1α, which induces the expression of glycolytic enzymes and glucose transport receptors and simultaneously activates the production of inflammatory proteins. Interestingly, a shift in glucose metabolism towards glycolytic flux in response to hypoxia relies on appropriate mitochondrial ROS levels because ROS generated via mitochondrial complex III are essential for the stabilization of HIF-1α [149]. Similarly, ROS-mediated HIF-1α activation has been found in ox-LDL-stimulated macrophages [113], indicating that HIF-1α activation is jointly regulated by multiple factors such as lipids and hypoxia in plaques. Hence, hypoxia potentiates IL-1β and NLRP3 expression in LPS-stimulated human macrophages [150], and elevated IL-1β production mediated by HIF-1α is evident in macrophage-rich hypoxic plaque regions displaying increased HK2 and cleaved caspase-1 levels [150]. HIF-1α deficiency in macrophages results in the acquisition of certain phenotypes, including low inflammation-related gene [151,152,153] (e.g., Mcp1, osteopontin and iNOS) expression in vitro [139, 140, 144,145,146] and reduced macrophage necroptosis rates by regulating miR-210 activity (inhibiting oxidative phosphorylation and enhancing mitochondrial ROS production) and miR-383 activity (increasing ATP levels and inhibiting necroptosis) in vivo [146]. Collectively, these findings highlight that HIF-1α is a key metabolic regulator that accelerates atherosclerotic progression (Fig. 4b).
Macrophage death versus immune suppression in the plaques
The death of macrophages, cells thus considered “the walking dead” [154], is induced via distinct modalities, e.g., apoptosis, pyroptosis, necroptosis [154, 155], for details see [156]. In the atherosclerotic context, distinct factors contribute to apoptosis, e.g., TNFα, NO, hypoxia, cholesterol crystals, and ox-LDLs [157]. Depletion of p53, which regulates cell death [158,159,160], in macrophages resulted in deteriorative atherosclerosis in APOE3-Leiden transgenic mice [161]. Under homeostatic conditions, these apoptotic macrophages were efficiently cleared by surrounding macrophages through efferocytosis, leading to immune suppression or inflammation resolution [162]. Notably, apoptotic cell-derived metabolites and products (such as methionine [163], arginine [164], and DNA [73]) play vital roles in the efferocytosis-mediated resolution of inflammation [165] (Fig. 4c). For example, nucleotides produced by the hydrolysis of apoptotic cell DNA via phagolysosomal DNase2a activated the DNA-PK-mTORC2-Myc proliferation-related pathway, which expanded the pool of inflammation-resolving macrophages in vitro and in mice [73]. Moreover, apoptotic cell-derived arginine and ornithine are converted into putrescine, which promotes continual efferocytosis and inflammation resolution by enhancing Rac1 activation [164]. In advanced atherosclerotic contexts, the combination of accumulating apoptotic cells, lipid metabolism dysfunction, and defective efferocytosis in plaques results in secondary necrosis of uncleared apoptotic cells and the release of a large number of cellular components (i.e., DAMPs) into the plaque milieu, which contributes to lipid-rich necrotic core formation and vulnerable plaques. Cell death, a major outcome of advanced atherosclerotic plaques, is mediated by different cell death modalities [166,167,168,169,170,171], including necroptosis, pyroptosis, and ferroptosis (for an excellent recent review, see [155]) (Fig. 4d).
RIPK3 and MLKL have been demonstrated to mediate macrophage necroptosis during atherosclerosis development. In one study, macrophage-reduced primary necrosis and necrotic areas were discovered in advanced lesions in RIP3−/− LDLR−/− or Apoe−/− mice [170], as well as in MLKL-deficient Apoe−/− mice [171]. Cumulatively, the data suggest that RIPK1 is emerging as a master switch that controls inflammation and cell survival in atherosclerosis progression, and phosphorylation of RIPK1, RIPK3 and MLKL are definitive indicators of necroptosis activation. Pharmacological inhibition of the necroptosis pathway (e.g., necrostatin-1) may be an exciting therapeutic approach to atherosclerosis [172, 173].
Pyroptosis is also involved in atherosclerosis [174, 175]: ox-LDLs, cholesterol crystals, hypoxia-response factors, ROS, DAMPs, and PAMPs promote activation of the NLRP3 inflammasome, which likely drives the pyroptosis of macrophages and induce plaque instability [176,177,178]. Caspase-11 accelerates atherogenesis and ox-LDL-induced macrophage pyroptosis, which is reversed when the activation of GSDMD is blocked through caspase-11 deficiency [176]. In addition, the absence of melanoma 2 (AIM2) inflammasome-mediated pyroptosis and inflammation in macrophages have been demonstrated to exacerbate atherosclerosis in clonal hemtopoiesis [179]. Interestingly, this AIM2-dependent form of inflammation-aggravated atherosclerosis, with accelerated necrotic core formations was reversed by knockout of caspase-1/11 or GSDMD [179]. Therefore, targeting inflammasomes or the pyroptosis executioner GSDMD can inhibit atherosclerotic progression and facilitate plaque regression.
Conclusion
The metabolic characteristics of macrophages and their polarization not only influence the inflammatory responses within a lesion but also play a pivotal role in determining atherosclerosis progression or regression, thus providing valuable insights into the dynamic nature of atherosclerosis [180, 181]. In principle, the modulation of macrophage function can be exploited for the intervention of disease by reducing plaque progression and stabilizing vulnerable lesions. Accordingly, both efferocytosis and β-cyclodextrin have been investigated [182, 183]. Although at very high pharmaceutical doses, β-cyclodextrin treatment reduced plaque size and facilitated atherosclerosis regression through macrophage reprograming in ApoE−/− mice [184]. β-cyclodextrin promoted the consumption of free cholesterol to generate 27-hydroxycholesterol, which in turn activated LXR-targeted gene expression, contributing to enhanced cholesterol efflux and acquisition of anti-inflammatory properties in macrophages (Fig. 4a). The identification of novel therapeutic targets to regulate the metabolism and polarization of macrophages at plaque sites (Fig. 2) may lead to innovative therapies for cardiovascular disease in addition to the traditional management of risk factors and the use of standard-of-care lipid-lowering drugs, leading to effective precision medicine [185].
Availability of data and materials
Not applicable.
References
Organization WH the top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death, 2019.
Rana JS, Khan SS, Lloyd-Jones DM, Sidney S. Changes in mortality in Top 10 causes of death from 2011 to 2018. J Gen Intern Med. 2021;36:2517–8.
Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–33.
Shantsila A, Dwivedi G, Shantsila E, Butt M, Beevers DG, Lip GY. Persistent macrovascular and microvascular dysfunction in patients with malignant hypertension. Hypertension. 2011;57:490–6.
Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–85.
Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, et al. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol. 2020;17:52–63.
Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 2018;123:1127–42.
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21.
Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119:136–45.
van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–43.
Tajbakhsh A, Rezaee M, Kovanen PT, Sahebkar A. Efferocytosis in atherosclerotic lesions: malfunctioning regulatory pathways and control mechanisms. Pharm Ther. 2018;188:12–25.
Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J. 2016;80:2259–68.
Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20:589–610.
Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406.
Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24.
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470.e413.
Gupta S, Amatullah H, Tsoporis JN, Wei K, Monteiro APT, Ektesabi AM, et al. DJ-1 binds to rubicon to impair LC-3 associated phagocytosis. Cell Death Differ. 2022;29:2024–33.
Li Q, Wang Y, Sun Q, Knopf J, Herrmann M, Lin L, et al. Immune response in COVID-19: what is next? Cell Death Differ. 2022;29:1107–22.
Shen JL, Doherty J, Allen E, Fortier TM, Baehrecke EH. Atg6 promotes organismal health by suppression of cell stress and inflammation. Cell Death Differ. 2022;29:2275–87.
Hsu CG, Chávez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022;29:1790–803.
Liao B, Dong S, Xu Z, Gao F, Zhang S, Liang R. MiR-19b-3p regulated by BC002059/ABHD10 axis promotes cell apoptosis in myocardial infarction. Biol Direct. 2022;17:20.
Servadei F, Anemona L, Cardellini M, Scimeca M, Montanaro M, Rovella V, et al. The risk of carotid plaque instability in patients with metabolic syndrome is higher in women with hypertriglyceridemia. Cardiovasc Diabetol. 2021;20:98.
Rovella V, Anemona L, Cardellini M, Scimeca M, Saggini A, Santeusanio G, et al. The role of obesity in carotid plaque instability: interaction with age, gender, and cardiovascular risk factors. Cardiovasc Diabetol. 2018;17:46.
Mauriello A, Sangiorgi GM, Virmani R, Trimarchi S, Holmes DR, Kolodgie FD, et al. A pathobiologic link between risk factors profile and morphological markers of carotid instability. Atherosclerosis. 2010;208:572–80.
Montanaro M, Scimeca M, Toschi N, Bonanno E, Giacobbi E, Servadei F, et al. Effects of risk factors on in situ expression of proinflammatory markers correlated to carotid plaque instability. Appl Immunohistochem Mol Morphol. 2021;29:741–9.
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–75.
Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. Role of inflammation in atherosclerosis. J Nucl Med: Off Publ Soc Nucl Med. 2007;48:1800–15.
Spagnoli LG, Mauriello A, Sangiorgi G, Fratoni S, Bonanno E, Schwartz RS, et al. Extracranial thrombotically active carotid plaque as a risk factor for ischemic stroke. JAMA. 2004;292:1845–52.
Tomaniak M, Katagiri Y, Modolo R, de Silva R, Khamis RY, Bourantas CV, et al. Vulnerable plaques and patients: state-of-the-art. Eur Heart J. 2020;41:2997–3004.
Cardellini M, Rovella V, Scimeca M, Anemona L, Bischetti S, Casella S, et al. Chronic kidney disease is linked to carotid nodular calcification, an unstable plaque not correlated to inflammation. Aging Dis. 2019;10:71–81.
Torii S, Sato Y, Otsuka F, Kolodgie FD, Jinnouchi H, Sakamoto A, et al. Eruptive calcified nodules as a potential mechanism of acute coronary thrombosis and sudden death. J Am Coll Cardiol. 2021;77:1599–611.
Mauriello A, Servadei F, Sangiorgi G, Anemona L, Giacobbi E, Liotti D, et al. Asymptomatic carotid plaque rupture with unexpected thrombosis over a non-canonical vulnerable lesion. Atherosclerosis. 2011;218:356–62.
Tesauro M, Mauriello A, Rovella V, Annicchiarico-Petruzzelli M, Cardillo C, Melino G, et al. Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med. 2017;281:471–82.
Mauriello A, Servadei F, Zoccai GB, Giacobbi E, Anemona L, Bonanno E, et al. Coronary calcification identifies the vulnerable patient rather than the vulnerable Plaque. Atherosclerosis. 2013;229:124–9.
Scimeca M, Bonfiglio R, Menichini E, Albonici L, Urbano N, De Caro MT, et al. Microcalcifications drive breast cancer occurrence and development by macrophage-mediated epithelial to mesenchymal transition. Int J Mol Sci. 2019;20.
Scimeca M, Giocondo R, Montanaro M, Granaglia A, Bonfiglio R, Tancredi V, et al. BMP-2 Variants in breast epithelial to mesenchymal transition and microcalcifications origin. Cells 2020;9.
Scimeca M, Anemona L, Granaglia A, Bonfiglio R, Urbano N, Toschi N, et al. Plaque calcification is driven by different mechanisms of mineralization associated with specific cardiovascular risk factors. Nutr Metab Cardiovascular Dis: NMCD. 2019;29:1330–6.
Montanaro M, Scimeca M, Anemona L, Servadei F, Giacobbi E, Bonfiglio R,et al. The paradox effect of calcification in carotid atherosclerosis: microcalcification is correlated with plaque instability. Int J Mol Sci. 2021;22.
Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arteriosclerosis Thrombosis Vasc Biol. 2014;34:724–36.
Zhang X, Li J, Qin JJ, Cheng WL, Zhu X, Gong FH, et al. Oncostatin M receptor beta deficiency attenuates atherogenesis by inhibiting JAK2/STAT3 signaling in macrophages. J Lipid Res. 2017;58:895–906.
Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell Mol Life Sci. 2020;77:1919–32.
Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398–414.
Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264:182–203.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62.
Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol. 2021;18:579–87.
Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. 2022;19:384–408.
Heinrich F, Lehmbecker A, Raddatz BB, Kegler K, Tipold A, Stein VM, et al. Morphologic, phenotypic, and transcriptomic characterization of classically and alternatively activated canine blood-derived macrophages in vitro. PLoS ONE. 2017;12:e0183572.
Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–26.
Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. 2008;180:5707–19.
Donadon M, Torzilli G, Cortese N, Soldani C, Di Tommaso L, Franceschini B, et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med. 2020;217.
Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–74.
Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci USA. 2006;103:10340–5.
Farahi L, Sinha SK, Lusis AJ. Roles of macrophages in atherogenesis. Front Pharmacol 2021;12:785220. https://doi.org/10.3389/fphar.2021.785220.
von Ehr A, Bode C, Hilgendorf I. Macrophages in atheromatous plaque developmental stages. Front Cardiovasc Med. 2022;9:865367.
Yurdagul A Jr. Crosstalk between macrophages and vascular smooth muscle cells in atherosclerotic plaque stability. Arterioscler Thromb Vasc Biol. 2022;42:372–80.
Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16:727–44.
Frodermann V, Nahrendorf M. Macrophages and Cardiovascular Health. Physiol Rev. 2018;98:2523–69.
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804.
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91.
Fang J, Feng C, Chen W, Hou P, Liu Z, Zuo M, et al. Redressing the interactions between stem cells and immune system in tissue regeneration. Biol Direct. 2021;16:18.
Nagenborg J, Goossens P, Biessen EAL, Donners M. Heterogeneity of atherosclerotic plaque macrophage origin, phenotype and functions: Implications for treatment. Eur J Pharm. 2017;816:14–24.
Mildner A, Schonheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, et al. Genomic characterization of murine monocytes reveals C/EBPbeta transcription factor dependence of Ly6C(-) cells. Immunity. 2017;46:849–862.e847.
Thomas GD, Hanna RN, Vasudevan NT, Hamers AA, Romanoski CE, McArdle S, et al. Deleting an Nr4a1 super-enhancer subdomain ablates Ly6C(low) monocytes while preserving macrophage gene function. Immunity. 2016;45:975–87.
Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–19.
Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. 2018;49:595–613.
Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153:362–75.
Quintar A, McArdle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ Res. 2017;120:1789–99.
Roberts ME, Barvalia M, Silva J, Cederberg RA, Chu W, Wong A, et al. Deep phenotyping by mass cytometry and single-cell RNA-sequencing reveals LYN-regulated signaling profiles underlying monocyte subset heterogeneity and lifespan. Circ Res. 2020;126:e61–e79.
Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205.
Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Investig. 2017;127:2904–15.
Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. 2020;21:1194–204.
Lhotak S, Gyulay G, Cutz JC, Al-Hashimi A, Trigatti BL, Richards CD, et al. Characterization of proliferating lesion-resident cells during all stages of atherosclerotic growth. J Am Heart Assoc. 2016;5.
Gerlach BD, Ampomah PB, Yurdagul A Jr, Liu C, Lauring MC, Wang X, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. 2021;33:2445–2463.e2448.
Hamilton JA, Myers D, Jessup W, Cochrane F, Byrne R, Whitty G, et al. Oxidized LDL can induce macrophage survival, DNA synthesis, and enhanced proliferative response to CSF-1 and GM-CSF. Arteriosclerosis Thrombosis Vasc Biol. 1999;19:98–105.
Wissler RW. The arterial medial cell, smooth muscle, or multifunctional mesenchyme? Circulation. 1967;36:1–4.
Andreeva ER, Pugach IM, Orekhov AN. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis. 1997;135:19–27.
Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–9.
Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci USA. 2003;100:13531–6.
Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37.
Rykaczewska U, Zhao Q, Saliba-Gustafsson P, Lengquist M, Kronqvist M, Bergman O, et al. Plaque evaluation by ultrasound and transcriptomics reveals BCLAF1 as a regulator of smooth muscle cell lipid transdifferentiation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2022;42:659–76.
Zhang Z, Huang J, Wang Y, Shen W. Transcriptome analysis revealed a two-step transformation of vascular smooth muscle cells to macrophage-like cells. Atherosclerosis. 2022;346:26–35.
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604.
Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–8.
Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262:153–66.
Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43.
Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88.
Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res. 2016;119:414–7.
Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17.
Li H, Cao Z, Wang L, Liu C, Lin H, Tang Y, et al. Macrophage subsets and death are responsible for atherosclerotic plaque formation. Front Immunol. 2022;13:843712.
Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–46.
Zhuo C, Ruan Q, Zhao X, Shen Y, Lin R. CXCL1 promotes colon cancer progression through activation of NF-κB/P300 signaling pathway. Biol Direct. 2022;17:34.
Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+in vitro and in vivo. 2015;21:255-65.
Domschke G, Gleissner CA. CXCL4-induced macrophages in human atherosclerosis. Cytokine. 2019;122:154141.
Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. 2010;106:203–11.
Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166–77.
Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res. 2012;110:20–33.
Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest. 2018;128:1106–24.
Pourcet B, Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest. 2018;128:910–2.
Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. 2020;250:705–14.
Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17:159–68.
Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127:402–26.
Park I, Goddard ME, Cole JE, Zanin N, Lyytikainen LP, Lehtimaki T, et al. C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat Commun. 2022;13:215.
Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019;4.
Gangoda L, Schenk RL, Best SA, Nedeva C, Louis C, D’Silva DB, et al. Absence of pro-survival A1 has no impact on inflammatory cell survival in vivo during acute lung inflammation and peritonitis. Cell Death Differ. 2022;29:96–104.
Tang P, Virtue S, Goie JYG, Png CW, Guo J, Li Y, et al. Regulation of adipogenic differentiation and adipose tissue inflammation by interferon regulatory factor 3. Cell Death Differ. 2021;28:3022–35.
Marques RM, Gonzalez-Nunez M, Walker ME, Gomez EA, Colas RA, Montero-Melendez T, et al. Loss of 15-lipoxygenase disrupts Treg differentiation altering their pro-resolving functions. Cell Death Differ. 2021;28:3140–60.
Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–88.
Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600.
King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481–7.
Sanin DE, Ge Y, Marinkovic E, Kabat AM, Castoldi A, Caputa G, et al. A common framework of monocyte-derived macrophage activation. Sci Immunol. 2022;7:eabl7482.
Depuydt MAC, Schaftenaar FH, Prange KHM, Boltjes A, Hemme E, Delfos L, et al. Single-cell T cell receptor sequencing of paired human atherosclerotic plaques and blood reveals autoimmune-like features of expanded effector T cells. Nat Cardiovas Res. 2023.
Lee SJ, Thien Quach CH, Jung KH, Paik JY, Lee JH, Park J, et al. Oxidized low-density lipoprotein stimulates macrophage 18F-FDG uptake via hypoxia-inducible factor-1alpha activation through Nox2-dependent reactive oxygen species generation. J Nucl Med. 2014;55:1699–705.
Chen Y, Yang M, Huang W, Chen W, Zhao Y, Schulte ML, et al. Mitochondrial metabolic reprogramming by CD36 signaling drives macrophage inflammatory responses. Circ Res. 2019;125:1087–102.
Kumar A, Gupta P, Rana M, Chandra T, Dikshit M, Barthwal MK. Role of pyruvate kinase M2 in oxidized LDL-induced macrophage foam cell formation and inflammation. J Lipid Res. 2020;61:351–64.
Doddapattar P, Dev R, Ghatge M, Patel RB, Jain M, Dhanesha N, et al. Myeloid cell PKM2 deletion enhances efferocytosis and reduces atherosclerosis. Circ Res. 2022;130:1289–305.
Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–32.
Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–92.
Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–206.
Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–47.
Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098.
Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172:162–175.e114.
Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014;34:1731–8.
Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20.
Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight 2020;5.
Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA. Role of phospholipid oxidation products in atherosclerosis. Circ Res. 2012;111:778–99.
Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S, et al. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. 2011;51:1903–9.
Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. 2018;558:301–6.
Kamstrup PR, Hung MY, Witztum JL, Tsimikas S, Nordestgaard BG. Oxidized phospholipids and risk of calcific aortic valve disease: the copenhagen general population study. Arterioscler Thromb Vasc Biol. 2017;37:1570–8.
Di Gioia M, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, et al. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol. 2020;21:42–53.
Favaloro B, Tamburro A, Angelucci S, Luca AD, Melino S, di Ilio C, et al. Molecular cloning, expression and site-directed mutagenesis of glutathione S-transferase from Ochrobactrum anthropi. Biochem J. 1998;335:573–9.
Fazi B, Melino S, De Rubeis S, Bagni C, Paci M, Piacentini M, et al. Acetylation of RTN-1C regulates the induction of ER stress by the inhibition of HDAC activity in neuroectodermal tumors. Oncogene 2009;28:3814–24.
Melino S, Nepravishta R, Bellomaria A, Di Marco S, Paci M. Nucleic acid binding of the RTN1-C C-terminal region: toward the functional role of a reticulon protein. Biochemistry. 2009;48:242–53.
Angelucci S, Sacchetta P, Moio P, Melino S, Petruzzelli R, Gervasi P, et al. Purification and characterization of glutathione transferases from the sea bass (Dicentrarchus labrax) liver. Arch Biochem Biophys. 2000;373:435–41.
Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–52.
Zhang X, McDonald JG, Aryal B, Canfran-Duque A, Goldberg EL, Araldi E, et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci USA 2021;118.
Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999;19:870–6.
Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–65.
Aarup A, Pedersen TX, Junker N, Christoffersen C, Bartels ED, Madsen M, et al. Hypoxia-inducible factor-1alpha expression in macrophages promotes development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:1782–90.
Karshovska E, Wei Y, Subramanian P, Mohibullah R, Geissler C, Baatsch I, et al. HIF-1alpha (Hypoxia-Inducible Factor-1alpha) promotes macrophage necroptosis by regulating miR-210 and miR-383. Arterioscler Thromb Vasc Biol. 2020;40:583–96.
Yuan J, Zhu Q, Zhang X, Wen Z, Zhang G, Li N, et al. Ezh2 competes with p53 to license lncRNA Neat1 transcription for inflammasome activation. Cell Death Differ. 2022;29:2009–23.
Vallelian F, Buzzi RM, Pfefferlé M, Yalamanoglu A, Dubach IL, Wassmer A, et al. Heme-stress activated NRF2 skews fate trajectories of bone marrow cells from dendritic cells towards red pulp-like macrophages in hemolytic anemia. Cell Death Differ. 2022;29:1450–65.
Liang Y, Sun X, Wang M, Lu Q, Gu M, Zhou L, et al. PP2Acα promotes macrophage accumulation and activation to exacerbate tubular cell death and kidney fibrosis through activating Rap1 and TNFα production. Cell Death Differ. 2021;28:2728–44.
Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19:526–37.
Torres Filho IP, Leunig M, Yuan F, Intaglietta M, Jain RK. Noninvasive measurement of microvascular and interstitial oxygen profiles in a human tumor in SCID mice. Proc Natl Acad Sci USA. 1994;91:2081–5.
Karshovska E, Wei Y, Subramanian P, Mohibullah R, Geißler C, Baatsch I, et al. HIF-1α (Hypoxia-Inducible Factor-1α) promotes macrophage necroptosis by regulating miR-210 and miR-383. Arteriosclerosis Thrombosis Vasc Biol. 2020;40:583–96.
Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, et al. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circulation Res. 2011;109:1141–52.
Gallo M, Paludi D, Cicero DO, Chiovitti K, Millo E, Salis A, et al. Identification of a conserved N-capping box important for the structural autonomy of the prion alpha 3-helix: the disease associated D202N mutation destabilizes the helical conformation. Int J Immunopathol Pharm. 2005;18:95–112.
Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–6.
Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ Res. 2014;115:875–83.
Hwang SJ, Ahn BJ, Shin MW, Song YS, Choi Y, Oh GT, et al. miR-125a-5p attenuates macrophage-mediated vascular dysfunction by targeting Ninjurin1. Cell Death Differ. 2022;29:1199–210.
Feng Z, Zhou J, Liu Y, Xia R, Li Q, Yan L, et al. Epithelium- and endothelium-derived exosomes regulate the alveolar macrophages by targeting RGS1 mediated calcium signaling-dependent immune response. Cell Death Differ. 2021;28:2238–56.
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29:133–46.
Kavurma MM, Rayner KJ, Karunakaran D. The walking dead: macrophage inflammation and death in atherosclerosis. Curr Opin Lipido. 2017;28:91–98.
Puylaert P, Zurek M, Rayner KJ, De Meyer GRY, Martinet W. Regulated necrosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2022;42:1283–306.
Vitale I, Pietrocola F, Guilbaud E, Aaronson SA, Abrams JM, Adam D, et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023;30:1097–154.
Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–7.
Panatta E, Butera A, Celardo I, Leist M, Melino G, Amelio I. p53 regulates expression of nuclear envelope components in cancer cells. Biol Direct. 2022;17:38.
Butera A, Roy M, Zampieri C, Mammarella E, Panatta E, Melino G, et al. p53-driven lipidome influences non-cell-autonomous lysophospholipids in pancreatic cancer. Biol Direct. 2022;17:6.
Panatta E, Zampieri C, Melino G, Amelio I. Understanding p53 tumour suppressor network. Biol Direct. 2021;16:14.
Van Vlijmen BJ, Gerritsen G, Franken AL, Boesten LS, Kockx MM, Gijbels MJ, et al. Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice. Circ Res. 2001;88:780–6.
Sun EW, Shi YF. Apoptosis: the quiet death silences the immune system. Pharmacol Therapeutics. 2001;92:135–45.
Ampomah PB, Cai B, Sukka SR, Gerlach BD, Yurdagul A Jr, Wang X, et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat Metab. 2022;4:444–57.
Yurdagul A Jr, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 2020;31:518–533.e510.
Mao Y. Apoptotic cell-derived metabolites in efferocytosis-mediated resolution of inflammation. Cytokine Growth Factor Rev. 2021;62:42–53.
Tummers B, Green DR. The evolution of regulated cell death pathways in animals and their evasion.
Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–36.
Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–97.
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65.
Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–10.
Rasheed A, Robichaud S, Nguyen M-A, Geoffrion M, Wyatt H, Cottee ML, et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis. Arteriosclerosis Thrombosis Vasc Biol. 2020;40:1155–67.
Karunakaran D, Nguyen MA, Geoffrion M, Vreeken D, Lister Z, Cheng HS, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce NF-kappaB activation and atherogenesis in mice. Circulation. 2021;143:163–77.
Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224.
He X, Fan X, Bai B, Lu N, Zhang S, Zhang L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol Res. 2021;165:105447.
Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X, Liu NF. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022;13:467.
Jiang M, Sun X, Liu S, Tang Y, Shi Y, Bai Y, et al. Caspase-11-gasdermin D-mediated pyroptosis is involved in the pathogenesis of atherosclerosis. Front Pharmacol. 2021;12:657486.
Diaz-Garcia E, Sanz-Rubio D, Garcia-Tovar S, Alfaro E, Cubero P, Gil AV, et al. Inflammasome activation mediated by oxidized LDL in patients with sleep apnea and early subclinical atherosclerosis. Eur Respir J. 2022.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61.
Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301.
Artyomov MN, Van den Bossche J. Immunometabolism in the Single-Cell Era. Cell Metab. 2020;32:710–25.
Fernandez DM, Giannarelli C. Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat Rev Cardiol. 2022;19:43–58.
Mahjoubin-Tehran M, Kovanen PT, Xu S, Jamialahmadi T, Sahebkar A. Cyclodextrins: potential therapeutics against atherosclerosis. Pharm Ther. 2020;214:107620.
Gao C, Liu C, Chen Q, Wang Y, Kwong CHT, Wang Q, et al. Cyclodextrin-mediated conjugation of macrophage and liposomes for treatment of atherosclerosis. J Control Release. 2022;349:2–15.
Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra350.
Ganini C, Amelio I, Bertolo R, Bove P, Buonomo OC, Candi E, et al. Global mapping of cancers: the Cancer Genome Atlas and beyond. Mol Oncol. 2021;15:2823–40.
Acknowledgements
We thank MA and GM for their helpful and constructive criticisms.
Funding
This work has been supported by the MUR-PNRR M4C2I1.3 PE6 project PE00000019 Heal Italia (CUP: E83C22004670001) to GM, EC, MA, MF; and by Associazione Italiana per la Ricerca contro il Cancro (AIRC) to GM (IG 2022 ID 27366; 2023-2027), and to EC (IG#22206; 2019-2023). Work has been also partially supported by Regione Lazio through LazioInnova Progetto Gruppo di Ricerca n 85-2017-14986; n 33 & 55-2021-T0002E0001.
Author information
Authors and Affiliations
Contributions
PH, MA, GM, and MF conceived the project; PH, JF, and ZL prepared the first draft; PH, GM, MA, and MF wrote the manuscript; PH prepared figures. All of the authors have approved this submitted version.
Corresponding authors
Ethics declarations
Competing interests
GM and MA are members of the Editorial Board of Cell Death Differentiation/Disease. The authors declare no other conflict of interest.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hou, P., Fang, J., Liu, Z. et al. Macrophage polarization and metabolism in atherosclerosis. Cell Death Dis 14, 691 (2023). https://doi.org/10.1038/s41419-023-06206-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06206-z
- Springer Nature Limited
This article is cited by
-
Association between triglyceride glucose-waist to height ratio and coronary heart disease: a population-based study
Lipids in Health and Disease (2024)
-
Dysregulated cellular metabolism in atherosclerosis: mediators and therapeutic opportunities
Nature Metabolism (2024)
-
Prediction model for spinal cord injury in spinal tuberculosis patients using multiple machine learning algorithms: a multicentric study
Scientific Reports (2024)
-
Saponins from Allii Macrostemonis Bulbus attenuate atherosclerosis by inhibiting macrophage foam cell formation and inflammation
Scientific Reports (2024)
-
Molecular profiling of a bladder cancer with very high tumour mutational burden
Cell Death Discovery (2024)