
Abstract
Mitochondria are essential organelles that play critical roles in energy metabolism, apoptosis and various cellular processes. Accumulating evidence suggests that mitochondria are also involved in cancer development and progression. The mitochondrial unfolded protein response (UPRmt) is a complex cellular process that is activated when the protein-folding capacity of the mitochondria is overwhelmed. The core machinery of UPRmt includes upstream regulatory factors, mitochondrial chaperones and proteases. These components work together to eliminate misfolded proteins, increase protein-folding capacity, and restore mitochondrial function. Recent studies have shown that UPRmt is dysregulated in various cancers and contributes to tumor initiation, growth, metastasis, and therapeutic resistance. Considering the pivotal role of the UPRmt in oncogenesis, numerous compounds and synthetic drugs targeting UPRmt-related components induce cancer cell death and suppress tumor growth. In this review, we comprehensively summarize recent studies on the molecular mechanisms of UPRmt activation in C. elegans and mammals and elucidate the conceptual framework, functional aspects, and implications of the UPRmt for cancer therapy. In summary, we paint a developmental landscape of the UPRmt in different types of cancer and offer valuable insights for the development of novel cancer treatment strategies by targeting the UPRmt.
Similar content being viewed by others

Facts
-
Various molecules and multiple mechanisms participate in the UPRmt activation in C. elegans and mammals.
-
Activation of UPRmt promotes the invasion and metastasis of cancer cells.
-
The fluctuation of UPRmt is associated with a variety of physiological processes and diseases.
-
UPRmt manipulation is a potential therapeutic target for treating human cancers.
Open Questions
-
How do seemingly independent key components interact with each other in the molecular mechanism of UPRmt activation?
-
Do different signaling pathways of UPRmt jointly regulate the occurrence and development of tumors?
-
What is the clinical efficacy of potential anti-cancer drugs targeting UPRmt?
-
By what mechanism does overactivation of UPRmt negatively affect cancer cells, and can we use this process to treat tumors?
Introduction
Mitochondria, referred to as the “powerhouses of the cell”, have a multitude of functions that are essential to cell survival and organismal health [1, 2]. Mitochondria not only produce the majority of the cell’s ATP through oxidative phosphorylation (OXPHOS) but also regulate calcium levels and apoptosis within the cell [3, 4]. Mitochondria are also the hub of various metabolic pathways, including the TCA cycle, fatty acid oxidation and amino acid metabolism [5, 6]. Considering the critical roles of mitochondria, the number and function of mitochondria are strictly regulated to adapt to various environmental stimuli.
As a complex and finely regulated organelle, the proper functioning of mitochondria often faces internal or external pressures. Fortunately, cells have the ability to cope with the stress conditions they encounter. Due to the presence of multiple compartments in cells, different and intricate pathways have evolved in the cytoplasm, endoplasmic reticulum (ER) and mitochondria to ensure dynamic balance of proteins, known as heat shock response (HSR) and ER stress (UPRER) and mitochondrial unfolded protein reactions (UPRmt), respectively. They are precisely coordinated, not only requiring close communication with the nucleus, but also influencing each other [7]. For example, classical ER stress inducers can modulate the expression of three core transcriptional regulators, including activating transcription factor 4 (ATF4), ATF5 and CCAAT/enhancer-binding protein homology protein (CHOP), which are required for UPRmt induction [8, 9] (for crosstalk between UPRER and UPRmt, see Supplementary file S1). Due to the fact that mitochondria are a major source of reactive oxygen species (ROS), ROS have also become an important medium for communication between mitochondrial stress and other cellular stresses (for ROS and UPRmt, see Box 1). More importantly, the crosstalk between mitochondria and other organelles allows for the integration of UPRmt with broader cellular stress responses, ensuring the maintenance of cellular homeostasis and the resolution of stress.
The association between the UPRmt and cancer development has been gradually explored. Mitochondria play important roles in all stages of cancer cell development [10]. During tumor growth, mitochondria exposed to stress also facilitate cancer development because mitochondria have unique mechanisms to maintain their stability, including the UPRmt. Activation of the UPRmt improves the invasion and metastasis of cancer cells [11]. Cancer cells slyly utilize this self-repair mechanism of mitochondria to accelerate their proliferation. Thus, in cancer cells, the UPRmt is hijacked and exploited for the repair of mitochondria and the promotion of oncogenesis. Disrupting the proteostasis in cancer cells by targeting UPRmt is considered a novel anticancer therapeutic strategy.
Overview of UPRmt
The UPRmt is a relatively independent life process that was first discovered in mammalian cells. After arduous efforts, the UPRmt is analyzed in detail in nematodes. The UPRmt is relatively conserved between nematodes and mammals, but more complex regulatory mechanisms in mammals have been gradually discovered [12, 13]. The UPRmt of mammals is regulated by different signaling pathways and has multiple executors. The existence of multiple pathways provides a level of redundancy to the system. If one pathway or regulatory factor becomes dysfunctional, other pathways can compensate to maintain the cell’s ability to respond to mitochondrial stress and ensure cell survival.
The molecular mechanism of the UPRmt in C. elegans
ATFS-1 and UPRmt
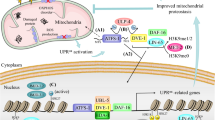
Accumulating evidence suggests that the bZIP transcription factor ATFS-1 (activating transcription factor associated with stress-1) plays a pivotal role in the UPRmt [13, 14]. It is activated in response to mitochondrial stress and functions as a central transcriptional regulator of the UPRmt in C. elegans. ATFS-1 contains a nuclear localization sequence (NLS) and a mitochondrial targeting sequence (MTS) [12], making ATFS-1 a medium for mitochondria to transmit signals to the nucleus [12, 14]. Under normal conditions, ATFS-1 enters mitochondria to facilitate its degradation. Following mitochondrial stress, ATFS-1 moves more to the nucleus, possibly due to a reduction in mitochondrial membrane potential or a change in the efficiency of transporting proteins into mitochondria [14,15,16]. Then ATFS-1 promotes the expression of UPRmt-related genes by directly binding to the promoters, thereby supporting cells to overcome mitochondrial dysfunction (Fig. 1). A recent interesting study proposes that changes in tRNA processing in C. elegans could activate the UPRmt [17]. The upregulation of nuclear HOE-1 increases 3’-tRNA processing, resulting in the accumulation of nuclear ATFS-1 and DVE-1.

In C. elegans, the misfolded proteins in mitochondria are digested by ClpP and transported to the cytoplasm by HAF-1. The reduced efficiency of protein input is an indicator of mitochondrial dysfunction and triggers UPRmt. During UPRmt, ATFS-1 moves to the nucleus to promote the expression of many UPRmt-related genes, resulting in increased synthesis of mitochondrial proteins, such as HSP10 and LONP1, to help cells survive. The chromatin remodeling factors UBL-5 and DVE-1 form complexes that interact with promoters of UPRmt-related genes, which may help ATFS-1 bind to nuclear DNA. Additionally, several chromatin remodeling factors function dependently of ATFS-1. In mammals, ATF5 plays a similar role to ATFS-1. Phosphorylated eIF2α upregulates the expression of ATF4 and CHOP, and then CHOP dimerizes with C/EBPβ. The expression of ATF5 is affected by ATF4 and CHOP. Independently, AKT phosphorylates and activates ERα to modulate NRF1 and HTRA2 expression. HSF1 binds with SSBP1 and modifies chromosomes to increase the transcription of HSP10, HSP60 and HSPA9 to stimulate UPRmt. Moreover, PHF8 and JMJD3 affect the expression of UPRmt-related genes through a conserved epigenetic mechanism. Figure was generated utilizing Biorender.com.
In the process of ATFS-1-activated UPRmt, mitochondrial misfolded proteins are digested into small peptides by the mitochondrial matrix-localized caseinolytic protease P (ClpP), and transported to the cytoplasm by inner mitochondrial membrane-located HAF-1 (Fig. 1) [18]. Both ClpP and HAF-1 participate in UPRmt activation. The contribution of HAF-1 in peptide efflux prevents ATFS-1 from entering the mitochondria [14]. A recent study demonstrates that mitochondria activate the target of rapamycin complex 1 (TORC1) through v-ATPase- and Rheb-dependent mechanisms following stress, resulting in increased translation of ATFS-1 to ensure that enough ATFS-1 translocate to the nucleus to stimulate the UPRmt (Fig. 1) [19]. In fact, the regulatory effect of ATFS-1 on mitochondrial function may far exceed our current understanding. For instance, ATFS-1 promotes mtDNA replication by promoting the binding of mitochondrial DNA replication polymerase (POLG) to mtDNA [20]. Additionally, ATFS-1 interferes with the assembly of mitochondrial preinitiation transcription complexes to inhibit transcription and aid mtDNA repair to ensure mitochondrial function [21]. With the deepening of research, more and more functions of ATFS-1 have been described.
Chromatin remodeling factors and UPRmt
During the movement of ATFS-1 to the nucleus, ubiquitin-like protein 5 (UBL-5) is upregulated with the assistance of ATFS-1 and defective proventriculus homolog protein (DVE-1) [22]. Subsequently, UBL-5 and DVE-1 form a complex that interacts with the promoters of UPRmt-related genes for chromatin remodeling, which may facilitate the binding of ATFS-1 to nuclear DNA (Fig. 1) [12]. The discovery of the homologs of DVE-1 and UBL-5 in mammals (SATB2 and Ubl5) again confirms the conservation of UPRmt. Notably, there are many chromatin remodeling factors that play a role in UPRmt activation. For example, the cytosolic protein LIN-65 is an indispensable component for full activation of UPRmt and plays a specific role in mitochondrial stress but not in cytoplasmic or ER stress [23]. LIN-65 moves toward the nucleus under the action of histone methyltransferase MET-2 during mitochondrial stress to dimethylate histone H3K9 to silence most of the chromatin (Fig. 1). Moreover, LIN-65 and MET-2 also affect the expression and distribution of DVE-1 in the nucleus, while DVE-1 influences the expression and distribution of LIN-65, suggesting that there is crosstalk among these factors [23]. Genes are almost turned off by LIN-65 and MET-2, but if transcription of all genes is suppressed, this clearly contradicts with the effect of UPRmt. Therefore, the expression of some substances produced in large quantities during the UPRmt, such as chaperone HSP-6 and protease YMEL-1, requires special mechanisms for targeted activation. JMJD-1.2 and JMJD-3.1, conserved histone demethylases of the JumonjiC (JmjC)-domain-containing protein family, are involved in longevity and are necessary for UPRmt excitation [24]. They act upstream of ATFS-1 and perform similar but not identical modifications to histones. JMJD-1.2 and JMJD-3.1 regulate the status of UPRmt-associated genes through their demethylase activity, allowing these essential genes to be transcribed to relieve stress on mitochondria (Fig. 1).
CREB-binding protein-1 (CBP-1) is the worm ortholog of the mammalian acetyltransferase CBP/p300. CBP-1 enhances the protective response to polystyrene nanoparticles in certain tissues and regulates UPRmt activation in C. elegans [25]. Specifically, CBP-1 can convert histone methylation to acetylation to promote the transcription of UPRmt-related genes (Fig. 1) [26]. This step occurs downstream of JMLD-1.2 and JMLD-3.1 and upstream of ATFS-1, transmitting mitochondrial emergency signals to the nucleus. In mammals, PHF8 and JMJD3 also play similar roles as homologs of JMJD-1.2 and JMJD-3.1, affecting the expression of UPRmt-related genes [27]. CBP-1 also directly acetylates ATFS-1, indicating that CBP-1 may play multiple and reversible roles in the UPRmt.
In C. elegans, histone deacetylase HDA-1, the homolog of mammalian histone deacetylase (HDAC), cooperates with DVE-1 to activate UPRmt [28]. Consistently, in mammals, HDAC1/2, together with SATB2, maintains mitochondrial homeostasis and assists other chromatin-remodeling enzymes in their function. Overall, when UPRmt is triggered, many epigenetic modification-related factors are systematically regulated to activate UPRmt [29].
In addition to the UPRmt triggered by internal or external stresses on the cell itself, stressed mitochondria also communicate with other mitochondria in distant cells, and signals from the UPRmt can be transmitted between different tissues. The regulation of stress response by this cell non-autonomous mechanism is considered an important way to affect body health [29]. (for further discussion about cell non-autonomous UPRmt, see Supplementary file S2).
The molecular mechanism of the UPRmt in mammals
ATF5 and UPRmt
Accumulating evidence proves that various stresses lead to ATF5-mediated UPRmt activation, and loss of ATF5 reduces the expression of UPRmt-related genes. ATF5 is the homolog of ATFS-1 in mammals. ATF5 plays similar roles as ATFS-1, and ATF5 can compensate for the loss of ATFS-1 under stress [12]. ATF5 also has MTS and NLS; thus, it can translocate into mitochondria and sense their state, thereby transmitting signals to the nucleus. Under normal conditions, ATF5 localizes to mitochondria and maintains the survival state of mitochondria. Knockdown of ATF5 significantly reduces basal, overall and maximal respiration [30]. During mitochondrial stress, ATF5 translocates into the nucleus and forms dimers with other transcription factors to regulate downstream target genes, including mitochondrial chaperones and proteases (Fig. 1) [10, 31]. Thus, ATF5 acts specifically in mitochondria, and ATF5 activity correlates with mitochondrial protein input efficiency [32]. A recent study reveals that tubular interstitial injury is related to the activation of the UPRmt, and ATF5 expression in the kidney is positively correlated with the expression of UPRmt-related molecules. Knockout of ATF5 alleviates tubular oxidative stress and apoptosis by coordinating the UPRmt pathway [33]. Moreover, inhibition of ATF5 activity leads to inhibition of the UPRmt, resulting in defects in monocyte development [34]. Knockdown of ATF5 reduces the production of proinflammatory cytokines and tumor necrosis factor-α (TNF-α) by inhibiting the UPRmt; thus, ATF5 may become a therapeutic target for alleviating neuroinflammatory processes [35].
ATF4, CHOP and UPRmt
ATF4 and CHOP are also important bZIP transcription factors but do not contain MTS. ATF4 can induce the expression of CHOP, which dimerizes with another transcription factor C/EBPβ [10]. Notably, the promoters of UPRmt-associated genes, such as HSP60, ClpP and mtDNAJ, contain a binding element of CHOP, demonstrating that CHOP participates in the UPRmt [22]. Moreover, ATF4 and CHOP assist ATF5 in activating the UPRmt. Under the influence of CHOP and ATF4, ATF5 accumulates in the nucleus during mitochondrial stress and responds to stress through transcriptional programs. The results of the protein-protein interactive network suggest that the interactions among ATF4, ATF5 and CHOP may determine the specificity of UPRmt (Fig. 2A). Since these transcription factors are involved in both the UPRmt and UPRER, revealing the selective role of these mediators will help us to better understand the activation process of the UPRmt.

A The protein-protein interaction (PPI) network is analyzed using the GeneMANIA database (http://genemania.org/). B Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of 10 core UPRmt-related genes were performed using the R package clusterProfiler and visualized using the R package enrichplot. The gene-disease associations in DisGeNET database were explored using the R package disgenet2r. C The correlations between gene expression and the sensitivity of drugs (top 30) in pan-cancer were analyzed using The Genomics of Drug Sensitivity in Cancer (GDSC) database. Positive correlation suggests that increased gene expression may contribute to drug resistance, whereas negative correlation implies that elevated gene expression may enhance drug sensitivity. D Heatmap showing the association between expression of core UPRmt-related genes and OS in pan-cancer through univariate Cox regression analysis. The result is visualized using the R package ggplot2. The red circle represents a correlation between high expression of gene and poor prognosis in cancer patients, while the blue circle represents a correlation between low expression of gene and poor prognosis.
A recent study demonstrates that the v-ATPase/mTORC1 complex located on the surface of lysosomes facilitates cells to process stress signals from mitochondria more efficiently [36]. Specifically, during mitochondrial stress, mTORC1 is activated by v-ATPase on the lysosome surface and then phosphorylates ATF4, ensuring the function of ATF4 in the UPRmt. These findings again suggest that UPRmt activation involves interactions between mitochondria and other organelles.
Estrogen receptor alpha (ERα) and UPRmt
In the mitochondrial intermembrane space (MIS), protein kinase B (PKB)/AKT phosphorylates and activates ERα to regulate the expression of nuclear respiratory factor 1 (NRF1) and mitochondrial protease high-temperature requirement serine protease A2 (HTRA2, also known as OMI) (Fig. 1) [27]. NRF1 binds to SIRT7 and transports SIRT7 to the nucleus to modify chromatin, and HTRA2 degrades misfolded proteins to facilitate cells to cope with crises. In the G93A-SOD1 mouse model of amyotrophic lateral sclerosis (ALS), there is a significant sex difference in the activation of the UPRmt pathway in MIS [37]. Increased expression and proteasome activity of HTRA2 are observed in female mice but not in male mice, which is consistent with ERα being more active in female mice. This coincided with a longer lifespan in female mice and a higher incidence of ALS in men. However, CHOP and HSP60 are not involved in this ERα-dependent process; thus, ERα-involved UPRmt pathways may be independent and complementary to typical UPRmt transcriptional responses.
HSF1 and UPRmt
HSF1, an important orchestrator of HSR, is indispensable for upregulating the mitochondrial chaperones during UPRmt [38]. Under normal conditions, HSP70 binds to HSF1 and inhibits its activity. Mitochondria stress induces ROS generation and causes the oxidation of DNAJA1 to enhance its binding to HSP70 and other mitochondrial protein precursors, leading to HSF1 release and activation [39]. Therefore, DNAJA1 indirectly activates HSF1 to regulate the expression of ATF5 and mitochondrial chaperones, indicating that HSF1 acts as a signal transfer of oxidative stress to the UPRmt effector. Interestingly, the PP2A-regulated dephosphorylation of HSF1 facilitates the generation of mitochondrial stress-specific variant of HSF1, thereby selectively inducing small HSPs to maintain proteostasis in response to mitochondrial stress in C. elegans [40]. The transcription activity of HSF1 is influenced by some interacting proteins to regulate the expression of UPRmt downstream effectors. For example, mitochondrial single-stranded DNA binding protein 1 (SSBP1) binds to HSF1 and modifies chromosomes to increase the transcription of HSP10, HSP60 and HSPA9 to stimulate the UPRmt (Fig. 1) [41]. Interestingly, HSF1 also accumulated in the mitochondria to exacerbate the pathogenesis of Huntington’s disease (HD). Mechanistically, mitochondrial HSF1 induced mitochondrial fragmentation by phosphorylating and activating dynamin-related protein 1 (Drp1), and also impeded mtDNA stabilization and SSBP1 oligomer formation [42]. Moreover, the peptide inhibitor DH1 markedly inhibits the interaction between HSF1 and Drp1 to suppress the mitochondrial localization of HSF1, thus alleviating mitochondrial dysfunction and HD symptoms [43]. In conclusion, HSF1 is involved in UPRmt process through the mediation of expression of mitochondrial function-associated genes. HSF1 activity is regulated by many co-factors, which enables us to better understand the functional network of HSF1 in UPRmt.
SIRT3 and UPRmt
Sirtuins (SIRTs) are NAD+-dependent deacetylases and play important roles in the regulation of energy metabolism and stress response, among which SIRT3 is shown to affect mitochondrial metabolism by regulating the UPRmt (for further discussion about SIRT3 and UPRmt, see Supplementary file S3).
UPRmt and cancer
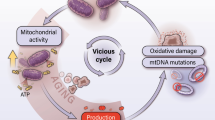
Cancer cells often experience increased mitochondrial stress. Activation of UPRmt guarantees the survival and apoptosis escape of cancer cells. Mitochondrial stress can indeed be both a consequence and a cause of cancer. Various tumorigenesis- and UPRmt-related factors (oxidative stress, DNA damage and mutagenesis, increased energy demand and metabolic rewiring) influence and promote each other, forming a positive cycle. A growing body of evidence shows that numerous components of UPRmt widely participate in tumorigenesis.
A recent study reveals that mitochondrial DNA promotes the selective activation of the UPRmt [44]. A significant increase in tumor incidence is observed in mice with UPRmt, accompanied by tumor invasion, demonstrating that UPRmt activation exacerbates the occurrence and invasion of tumors. Mechanistically, the pregnancy-associated plasma protein A (PAPP-A)/discoidin receptor 2 (DDR2) pathway is associated with the UPRmt to initiate liver cancer. PAPP-A is a protease involved in the degradation of insulin-like-growth-factor-binding proteins 4/5 (IGFBP4/5) and also acts as an oncogene. PAPP-A could activate DDR2 to promote the invasion and metastasis of cancer cells through the ERK/SNAIL signaling [45].
Additionally, the pan-cancer analysis results demonstrate that the 10 pivotal UPRmt-related genes, which have been thoroughly studied in previous literature and selected to be detailed in this review, are associated with overall survival (OS) in certain cancers. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses suggest that the core UPRmt-related genes are significantly enriched in the UPR-associated signaling pathways, such as cellular response to incorrect protein and cellular response to chemical stress (Fig. 2B). Furthermore, these genes are also interlinked with many human diseases, including mitochondrial diseases and cancers (Fig. 2B). More importantly, these key UPRmt-related genes are negatively correlated with the sensitivity of chemotherapy drugs according to the GDSC database (Fig. 2C). The above findings all demonstrate that the UPRmt signaling pathway participates in regulating the occurrence and development of tumors through different molecular mechanisms.
Relationships between UPRmt-related proteins and oncogenesis
The roles of ATF5 in oncogenesis
ATF5 is a key regulator of the UPRmt in mammals and is involved in the tumorigenesis of a variety of cancers [46,47,48]. ATF5 is dramatically overexpressed and correlated with poorer prognosis in many cancer patients (Fig. 2D) [49]. As an upstream regulator, E74-like ETS transcription factor 1 (ELF1), playing diverse roles in lymphocyte development, angiogenesis and cancer, stimulates ATF5 gene transcription by directly binding to its promoter (Fig. 3) [50]. Moreover, ATF5 overexpression is strongly linked with both clinicopathological characteristics and relapse-free survival rates in bladder urothelial carcinoma patients. Mechanistically, ATF5 promotes the transcription of disheveled-1 (DVL1), a potent activator of Wnt/β-catenin, and increases tumor sphere formation ability through the ATF5/DVL1/Wnt/β-catenin axis (Fig. 3) [51]. ATF5 knockdown suppresses the proliferation, migration and invasion abilities of esophageal cancer cells. ATF5 can bind to hypoxia-inducible factor 1α (HIF-1α), an oxygen-sensing transcriptional regulator orchestrating a complex of adaptive cellular responses to hypoxia and oncogenesis, to act as a coactivator and form a transcription complex, thereby regulating the expression of HIF-1α target genes (Fig. 3) [52]. Decreased ATF5 expression inhibits angiogenesis and tumor growth in vivo [53]. Additionally, activation of Maf1, a master repressor of Pol III-dependent transcription and mechanistic target of rapamycin (mTOR) downstream effector, alleviates ionizing radiation-induced UPRmt in lung cancer cells. ATF5 is needed for this process, as evidenced by the fact that silencing ATF5 greatly reduced the Maf1 inhibition-induced upregulation of HSP60 and HSPA9, suggesting that Maf1 modulated the UPRmt in an ATF5-dependent manner [54]. In summary, ATF5 not only elevates the expression of antiapoptotic genes but also increases the levels of factors that regulate growth and metabolism, thereby contributing to radiotherapy resistance and promoting tumor cell invasion [8, 55, 56].

A ATF5 regulates the expression of tumorigenic genes, such as HSP60. ATF5 binds to HIF-1α to regulate the transcription of target genes. These downstream genes enhance the proliferation, migration and invasion of cancer cells. B Phosphorylated HSF1 plays a role in tumor immune escape by modulating the expression of HSPs and PD-L1. HSF1 also interacts with p53 and ERα to regulate gene expression to facilitate oncogenesis. C HSPA9 interacts with ANT3, HIF-1α or TGR5 to promote tumor development and metastasis. HSPA9 also interacts with AR-V7 to enhance ubiquitination and degradation of AR-V7 to overcome drug resistance. D LONP1 is deacetylated by SIRT3 and degraded by ubiquitination, thus preventing tumor development. LONP1 inhibits apoptosis by stabilizing p53. LONP1 attenuates the generation of ROS or facilitates cancer cells to adapt to anoxic environment under the influence of AKT1 or HIF-1α. Figure was generated utilizing Biorender.com.
The roles of ATF4 in oncogenesis
Dysregulation of ATF4 expression has been implicated in various types of cancer [57,58,59]. Higher ATF4 expression is significantly correlated with worse overall survival in gastric cancer patients, indicating that ATF4 is an independent prognostic factor [60]. Silencing ATF4 strongly blocks cell proliferation, invasion, migration and cell cycle progression and increases drug sensitivity, possibly by modulating autophagy and asparagine metabolism [61]. Inhibition of glutaminolysis upregulates and activates ATF4 by reducing FTO-mediated m6A modification and YTHDF2-mediated mRNA degradation, ultimately positively regulating CHOP transcription to trigger autophagy [62, 63]. Targeting ATF4-regulated protective autophagy enhances the anticancer effect of glutaminolysis inhibition [64]. In pancreatic ductal adenocarcinoma (PDAC), TGF-β1 secreted from cancer-associated fibroblasts (CAFs) drives the elevation of ATF4 expression through the SMAD2/3 pathway to modulate cancer cell proliferation, migration and stemness. ATF4 then directly promotes the expression of multidrug resistance protein 1 (MRP1, also known as ABCC1) by binding its promoter, leading to gemcitabine resistance in PDAC [65]. Depletion of ATF4 also decreases metastasis and tumor growth in breast cancer by mediating the TGF-β/SMAD and mTOR/RAC1-RHOA pathways [66]. The EWS-FLI1 fusion protein directly activates ATF4 transcription by binding to its promoter. ATF4 then controls the transcription of target genes involved in the core serine synthesis pathway in Ewing sarcoma [67]. Kristen rat sarcoma (KRAS), the most common mutated oncogene in human cancers, cooperates with NRF2 to upregulate ATF4 expression during nutrient stress, thereby modulating the amino acid uptake and asparagine biosynthesis in lung cancer [68]. Asparagine synthetase (ASNS) is an important target gene of ATF4 that modulates protein synthesis, promotes tumor growth and suppresses cell death [69]. Inhibition of ASNS sensitizes lung cancer cells to L-asparaginase, a cornerstone drug in the treatment of acute lymphoblastic leukemia (ALL). Ribosomal protein L41 (RPL41) is a tumor suppressor and its downregulation and deletions are frequently detected in human cancers [70]. RPL41 promotes ATF4 translocation to the cytoplasm, increases the ATF4 phosphorylation, thus accelerating ATF4 degradation and sensitizing lung cancer and retinoblastoma cells to chemotherapeutic drugs [71]. Moreover, the eIF2α/ATF4 axis is associated with radioresistance through the modulation of glutathione biosynthesis and ROS generation in breast cancer [57]. Elevated ATF4 expression also correlates with resistance to multiple DNA-interacting drugs. For instance, cisplatin upregulates ATF4 expression, and ATF4-deficient cancer cells exhibit elevated sensitivity to cisplatin-induced cell death [72]. Mechanistically, ATF4 promotes multidrug resistance by directly regulating the transcription of SIRT1 and STAT3 [73]. The circadian transcription factor Clock promotes the transcription of ATF4 following cisplatin treatment, thereby mediating drug resistance by regulating the genes for glutathione metabolism [74]. YAP and TAZ are transcriptional coactivators in Hippo signaling cascade and promote cell proliferation, tissue regeneration and tumorigenesis. Notably, YAP/TAZ inhibits ferroptosis in liver cancer and is associated with sorafenib resistance. Activated YAP/TAZ promotes ATF4 nuclear localization, increases its transcriptional activity and cooperates with ATF4 to induce SLC7A11 transcription to overcome sorafenib-triggered ferroptosis [75]. Together, ATF4 coordinates and integrates various cellular processes by regulating different genes to facilitate tumorigenesis and chemoresistance.
The roles of HSF1 in oncogenesis
HSF1 not only maintains proteostasis by inducing HSP expression in response to stresses but also regulates tumorigenesis through multiple network pathways and target genes [76,77,78]. An increasing number of studies has summarized that HSF1 achieves its cancer-promoting effects by inhibiting cell apoptosis, accelerating cell proliferation and migration, reprogramming metabolic programs, and supporting the tumor microenvironment [79, 80]. Consistent with its oncogenic role, HSF1 is overexpressed or overactivated in a broad spectrum of cancers and negatively correlated with the prognosis of cancer patients (Fig. 2D) [81]. The pan-cancer analysis of HSF1 reveals that HSF1 is involved in diverse cancer-associated signaling pathways, and is closely associated with immune cell infiltration and efficacy of immunotherapy [78, 82]. PIM2-regulated phosphorylation of HSF1 at Thr120 regulates PD-L1 expression by binding to its promoter and facilitates oncogenesis (Fig. 3) [82]. HSF1 is also activated in stromal fibroblasts, a component of the tumor microenvironment (TME), leading to remodeling of the extracellular matrix that promotes colon cancer development [83]. Moreover, thrombospondin 4 (TSP-4) secreted from CAFs binds to integrin α2 to promote HSF1 phosphorylation at Ser326 to support the malignant phenotypes of gallbladder cancer cells, including cell proliferation, epithelial-mesenchymal transition (EMT) and cancer stemness [84]. Activated HSF1 further controls the expression of TGF-β1 to help the transdifferentiation of fibroblasts into CAFs, thereby forming positive feedback [84]. HSF1 interacts with ERα and cooperates to regulate some common genes in breast cancer (Fig. 3) [85]. Moreover, HSF1 is a target of FTO-regulated and YTHDF2-dependent m6A modification to accelerate the development and progression of multiple myeloma [86]. Additionally, the ABL2/HSF1/E2F axis is needed for the brain metastasis of lung cancer [87]. The ERK/FBXW7/HSF1/MDR1 pathway is closely linked with chemoresistance [88]. Inhibition of HSF1 significantly overcomes resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), which is the standard treatment for NSCLC patients with EGFR mutations, by downregulating the expression of HSPs [89].
Numerous high-throughput screening studies have identified many molecules that act as HSF1 inhibitors [81, 90, 91]. However, the search for drugs targeting HSF1 is challenging due to the broad roles of transcription factors and the lack of specificity of small molecules. However, targeting the upstream regulatory factors or signaling pathways of HSF1 for drug screening provides an indirect pharmacological strategy to more easily inhibit the tumor-promoting effect of HSF1. Notably, combining HSF1-targeted cancer therapy with other chemotherapeutic agents and treatment strategies has become an effective oncology strategy and has been initially validated in several studies [92].
The roles of HSPA9 in oncogenesis
HSPA9, also called mitochondrial HSP70/GRP75/mortalin, is a survival-promoting protein and plays an important role in mitochondrial function. Recent literature has demonstrated that HSPA9 expression is upregulated and associated with poor prognosis in many types of cancer patients (Fig. 2D) [93,94,95]. Additionally, circulating HSPA9 levels are elevated in colorectal cancer patients and correlated with worse prognosis, indicating that HSPA9 is a useful prognostic biomarker [96,97,98,99]. HSPA9 not only modulates the Raf/MEK/ERK signaling pathway but also promotes the PP1α-MER1/2 interaction to increase MEK1/2 dephosphorylation in KRAS and BRAF mutant cancer cells [100, 101]. Moreover, HSPA9 blocks the degradation of high mobility group A1 (HMGA1) and promotes the activation of the HMGA1/JNK/c-Jun axis in lung cancer to stimulate cancer cell growth and metastasis [102]. In breast cancer, HSPA9 facilitates oncogenesis through the Wnt/GSK3β/β-catenin pathway [103]. Thus, knockdown of HSPA9 markedly suppresses cell proliferation, inhibits EMT, induces cell cycle arrest and initiates cell death in various cancer cells [104]. In vemurafenib-resistant BRAF-mutant melanoma cells, HSPA9 depletion triggers cell death in a MEK/ERK- and ANT/CypD-dependent manner, indicating that HSPA9 is an effective target in drug-resistant tumors [105]. With the gradual identification of HSPA9-interacting proteins, the molecular mechanism of HSPA9-regulated oncogenesis has gradually emerged. Proteomic analysis identifies adenine nucleotide translocase 3 (ANT3) as an interacting protein of HSPA9 and indicates that HSPA9 can disrupt the interaction between ANT3 and cyclophilin D (CypD) in BRAF mutant cancer cells (Fig. 3) [106]. In prostate cancer, HSPA9 maintains the protein stability of sine oculis homeobox 1 (SIX1), a developmental transcriptional regulator frequently overexpressed in human cancers, by inhibiting its polyubiquitination and degradation through recruiting the deubiquitinating enzyme ubiquitin-specific protease 1 (USP1). Either genetic or pharmacological inhibition of the HSPA9-SIX6-USP1 complex effectively impedes progression of prostate cancer (PC) and castration resistance [107]. Androgen receptor splice variant 7 (AR-V7) is a form of ligand-independent and constitutively activating variant of androgen receptor (AR), and plays an important role in castration-resistant PC. The HSPA9-AR-V7 complex can recruit the E3 ligase SIAH2 to enhance the ubiquitination and degradation of AR-V7 to overcome drug resistance in PC (Fig. 3) [108]. Moreover, HSPA9 also interacts with HIF-1α to promote the association of HIF-1α with voltage-dependent anion channel 1 (VDAC1) and hexokinase II (HK-II) in the mitochondrial outer membrane, thereby enhancing resistance to apoptosis under hypoxic conditions [109]. HSPA9 is also associated with cytoplasmic sequestration and retention of p53 [110, 111]. Furthermore, Takeda-G-protein receptor-5 (TGR5) accelerates cancer cell proliferation and migration and promotes cell death resistance partially by interacting with HSPA9. Knockdown of HSPA9 reversed various oncogenic phenotypes mediated by TGR5 [112].
The above literature suggests that the development of small-molecule compounds or drugs targeting HSPA9 is a promising strategy for cancer therapy. MKT-077, a specific inhibitor of HSPA9, exhibits anticancer effects by inducing apoptosis and necrosis and reducing resistance to oxidative stress in bladder cancer (Table 1 and Fig. 4) [113]. Additionally, several analogs of MKT-077, such as JG-98, JG-194 and JG-231, have been synthesized to bind to HSPA9 and suppress cancer cell growth, thereby enhancing the clinical translational value of MKT-077 (Table 1) [114, 115].

With the progression of tumor, cancer cells face problems such as increased ROS levels and hypoxia, which puts mitochondria in a state of stress, manifested as mitochondrial dysfunction and protein misfolding. Meanwhile, UPRmt is stimulated. ATF5 and other UPRmt regulators transmit signals between mitochondria and nucleus, and promote the transcription of UPRmt-related genes. Transcribed mitochondrial chaperonins and proteases enter mitochondria to facilitate proteins to fold correctly or eliminate misfolded proteins, allowing the mitochondria to restore proteostasis and normal function. Inhibitors of UPRmt-related proteins suppress the activity of upstream UPRmt regulators or downstream functional proteins, thus making it difficult for mitochondria to recover healthy state and ultimately accelerating cancer cell death. Therefore, these inhibitors of UPRmt are considered as potential drugs for cancer treatment. Figure was generated utilizing Biorender.com.
The roles of HSP60 in oncogenesis
HSP60 (HSPD1), another important mitochondrial chaperone, performs essential tasks for maintaining mitochondrial proteostasis. In breast cancer, accumulated misfolded mitochondrial proteins activate the UPRmt and upregulate HSP60 and HSP10 expression. Altered HSP60 expression is associated with a variety of cancers, serving as a tool for diagnosis and prognosis of cancer patients (Fig. 2D) [116, 117]. Elevated HSP60 expression activates the ERK1/2 pathway and promotes the growth of adenocarcinoma cells [118]. Moreover, HSP60 upregulates the proto-oncogene MYC and impedes the function of the tumor suppressor p53, thereby allowing cancer cells to grow and invade surrounding tissues [119]. HSP60 downregulation increases the intracellular ROS levels and activates the AMPK-mTOR pathway, thereby inhibiting the proliferation of glioblastoma cells [120].
A number of HSP60 inhibitors have been identified which can interact with HSP60 or alter its post-translational modifications (Table 1) [121]. For example, the synthetic small-molecule KHS101 disrupts energy metabolism in glioblastoma and NSCLC by inhibiting the folding activity of HSP60 [122]. Doxorubicin, a medication used to treat various cancers, binds to HSP60 to promote its acetylation, ubiquitination and degradation (Table 1) [123]. HSP60 inhibitors represent a promising avenue in cancer research by targeting the protein folding machinery of cancer cells, potentially leading to cell death and reduced tumor growth (Table 1) [124,125,126,127].
The roles of LONP1 in oncogenesis
LONP1 and ClpP are the two major ATP-dependent proteases in mammals, involved in regulating protein assembly, folding and degradation, as well as controlling mitochondrial function under stress conditions [128]. Mitochondrial proteotoxic stress caused by loss of LONP1 activates the UPRmt via ATFS-1 in C. elegans [129]. LONP1 is a multifaceted enzyme with different functions, including proteolysis, chaperone activity and binding of mtDNA [130, 131]. LONP1 is upregulated by several stress stimuli. LONP1 degrades not only misfolded or damaged proteins but also apoptosis- and metabolism-related proteins, thus helping to protect the mitochondria from oxidative damage [132, 133]. LONP1 is activated in various human cancer tissues, and its overexpression is associated with prognosis in multiple types of cancer (Fig. 2D) [134]. As one of the molecular mechanisms, LONP1 is transcriptionally upregulated by CHOP and CEB/P, two UPRmt signature proteins [132]. Moreover, LONP1 is preferentially mediated by the p53 and β-catenin. Knockdown of LONP1 inhibits cervical cancer cell proliferation, migration and invasion by influencing mitophagy and autophagy [121]. LONP1 is involved in metabolic reprogramming in cancer cells by remodeling the OXPHOS complex. When cells lack LONP1, severe mitochondrial dysfunction occurs. However, high level of LONP1 can induce a shift from oxidative respiration to glycolytic metabolism [128, 135]. LONP1 promotes the transformation of metabolic mode to glycolysis during carcinogenesis to provide a large amount of energy for rapid cell proliferation [130]. LONP1 is deacetylated by SIRT3 at the K145, making it more susceptible to ubiquitination and degradation (Fig. 3) [136]. With increasing age, the loss of SIRT3 increases the degree of acetylation of LONP1, which promotes the occurrence and progression of tumors. Oxidative stress also induces LONP1 expression, which in turn promotes the proliferation and migration of cancer cells [130]. Under hypoxic conditions, AKT1 phosphorylates LONP1 at Ser173 and Ser181 to enhance its protease activity, which attenuates ROS effects and promotes cancer cell migration (Fig. 3) [137]. Thus, interfering with this phosphorylation process inhibits tumor growth and metastasis. During anoxia, LONP1 is upregulated by HIF-1α, and its enzyme activity is enhanced by the cytochrome c oxidase (COX), thereby facilitating cancer cell adaptation to anoxic environments (Fig. 3) [132]. Additionally, LONP1 inhibits apoptosis by stabilizing p53 in extreme environments, and cells with p53 deletion have stronger proliferation ability under the influence of LONP1 [130]. LONP1 ablation also blocks tumor metastasis by inhibiting EMT and extracellular matrix remodeling through the c-Jun N-terminal kinase (JNK) pathway in human pancreatic cancer [138]. More importantly, LONP1 participates in ferroptosis by regulating the peroxidase GPX4 and NRF2/Keap1 signaling pathways, providing a cancer treatment strategy via ferroptosis [139]. LONP1 also assists cancer cell migration and invasion by promoting CAFs formation in the TME and leads to PD-L1-mediated immune escape [134].
Under the conditions of artificial induction of colon carcinoma, Lonp1+/– mice show lower tumor incidence and milder symptoms than wild-type mice. LONP1 is also essential for the metastasis of cancer cells in vivo. Silencing LONP1 results in a significant reduction in the melanoma cell metastases [128].
Several chemical inhibitors have the potential to inhibit LONP1 activity to prevent cancer progression and sensitize cancer cells to chemotherapy (Table 1 and Fig. 4). The synthetic oleanane 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), obtusilactone A (OA) and (-)-Sesamin directly bound to LONP1 to inhibit its activity (Table 1) [140, 141]. Furthermore, overexpression of LONP1 can salvage the inhibition of proteasome activity by proteasome inhibitors; thus, therapeutic strategies targeting LONP1 may affect the effectiveness of existing treatment modalities (Table 1 and Fig. 4) [142].
The roles of ClpP in oncogenesis
ClpP is an ATP-dependent protease in the mitochondrial matrix that degrades misfolded or denatured proteins [143]. ClpP is overexpressed in many human cancers and associated with poor prognosis in breast and lung cancer [144]. ClpP expression in metastatic lesions is higher and affects the proliferation and metastasis of tumor cells [145]. Inhibition of ClpP suppresses the proliferation, migration and invasion of cancer cells, which is manifested by a blocked cell cycle and a lower degree of metastasis [146]. Moreover, ClpP inhibition also triggers oxidative stress, which subsequently decreases the expression of the cell motility effector caveolin-1, resulting in impaired energy metabolism and reduced metastatic potential [147]. In breast cancer, silencing ClpP decreases the activity of SRC and AKT and phosphorylation of PI3K, thereby interfering with the proliferation, migration and apoptosis of breast cancer cells [148]. However, overexpression of ClpP in ovarian cancer cells reduces cell motility and represses cell migration and invasion by inducing mitochondrial respiratory chain disorder [149]. Activation of ClpP also prevents cancer cell growth by disrupting mitochondrial structure and function, and causes cell death in a p53-independent manner [146, 150]. Therefore, too low or too high levels of ClpP lead to cancer cell death through different mechanisms in different cancers.
ClpP expression is markedly associated with drug resistance, and high expression of ClpP reduces apoptosis during chemotherapy, while silencing ClpP improves chemotherapeutic response [146]. ClpP enhances resistance to cisplatin in ovarian cancer cells by suppressing mitophagy and exacerbating cellular stress [151]. Thus, targeting ClpP to increase the drug sensitivity is beneficial for cancer treatment [152].
Drugs developed based on ClpP can achieve the ultimate goal of treating cancer by modulating ClpP activity (Table 1 and Fig. 4). For example, TG42 and TG53, modified analogs of the first ClpP inhibitor trans-β-lactones, inhibit ClpP proteolytic activity, reduce cell migration and induce apoptosis in liver cancer cells (Table 1) [145, 153]. Interestingly, some ClpP agonists were also identified. The small molecule ONC201 and its analogs activate ClpP to disrupt respiratory chain integrity, thus affecting mitochondrial function and exhibiting anticancer properties (Table 1) [150, 154,155,156,157]. Since the effect of ClpP on cancer cells is closely associated with its expression level, whether increasing or inhibiting ClpP activity is more effective in treating cancer needs to be further explored (Fig. 4).
Conclusions and perspective
The UPRmt, triggered by multiple mitochondrial stresses, is a vital process by which mitochondria ensure the correct folding and homeostasis of proteins. With the continuous understanding of the UPRmt, the molecular mechanism model of the UPRmt is gradually being constructed and improved, providing a solid foundation for its further application in disease treatment. However, the undisclosed parts of its triggering mechanism have also drawn the attention of researchers. Additionally, it is not clear whether there is an interaction between several seemingly independent pathways that stimulate the UPRmt and whether one pathway is dominant in certain cancer cells. Furthermore, many examples of cellular non-autonomous UPRmt have been found, but the differences in mechanism, excitation and consequences between cell-induced and non-autonomous UPRmt are still unclear. We wonder whether a centain type of cancer cell is more inclined to rely on the UPRmt for survival. However, the possible side effects of UPRmt on healthy cells should be noted.
The UPRmt, as an emergency measure taken by mitochondria, is of great significance in maintaining the growth and proliferation of cancer cells (for UPRmt in cancer treatment, see Box 2) (Fig. 4). However, the UPRmt is a complex process with dual roles. Faced with a certain amount of protein misfolding or ROS, cells activate the UPRmt to maintain intracellular proteostasis and help cells survive. However, when exposed to excessive stress, mitochondria initiate mitophagy and even apoptosis [158]. Moderate UPRmt activation exhibits the potential for treating neurodegenerative diseases and heart disease, but excessive activation of the UPRmt promotes cardiomyocyte apoptosis, the death of dopaminergic neurons, and mitochondrial dysfunction in the ALS mouse model [159]. These findings suggest that there may be crossover signaling pathways between the UPRmt and cell death pathways. These phenomena have led us to realize that the benefits of the UPRmt appear to be variable and fragile (Fig. 5). For a period of time after mitochondrial stress, cells continue to enhance UPRmt to save themselves. However, when the stress intensity reaches the maximum tolerance limit, cells may clear the damaged mitochondria through mitophagy to ensure the survival of the entire cell (Fig. 5). If the cells cannot maintain the high activation of UPRmt to save themselves, a large number of cells will suffer cell death (Fig. 5). This model we predict has been described in the UPRER [160, 161]. Although more detailed studies are lacking, overactivation of UPRmt has more complex and multi-dimensional effects, which may be an effective therapeutic strategy for cancer (Box 2). To this end, more sensitive detection methods and biomarkers of the UPRmt may be helpful.

When the stress faced by cells reaches a certain level, UPRmt is triggered. Within a certain range, cells repair themselves, which helps maintain proteostasis and survive. A certain degree of UPRmt can prolong lifespan, showing beneficial effects on organisms. The activation of UPRmt intensifies as the cells are subjected to increased stress. Up to a certain point, the effects of UPRmt on cells are reversed, and high level of UPRmt have some negative effects on cells, such as protein dyshomeostasis, mitophagy and apoptosis. However, the mechanisms by which cells perceive and judge stress intensity are still unclear, and the adverse effects of excessively high level of UPRmt on cells need to be explored. More importantly, under what circumstances do cells choose to abandon self-rescue and initiate apoptosis, as well as the mechanisms that play a role in this critical turning point require further research. Figure was generated utilizing Biorender.com.
In conclusion, UPRmt, as a mitochondrial stress mechanism, is highly associated with tumor progression and has great potential for clinical application (Box 2). The UPRmt also affects aging and many other human diseases; thus, targeting the UPRmt is a relatively new and promising therapeutic strategy [162].
References
Suomalainen A, Nunnari J. Mitochondria at the crossroads of health and disease. Cell. 2024;187:2601–27.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54.
Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61:667–76.
Suliman HB, Piantadosi CA. Mitochondrial quality control as a therapeutic target. Pharm Rev. 2016;68:20–48.
Eisner V, Picard M, Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20:755–65.
Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:124.
Bilen M, Benhammouda S, Slack RS, Germain M. The integrated stress response as a key pathway downstream of mitochondrial dysfunction. Curr Opin Physiol. 2022;27:100555.
Keerthiga R, Pei DS, Fu A. Mitochondrial dysfunction, UPR(mt) signaling, and targeted therapy in metastasis tumor. Cell Biosci. 2021;11:186.
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6.
Inigo JR, Chandra D. The mitochondrial unfolded protein response (UPR(mt)): shielding against toxicity to mitochondria in cancer. J Hematol Oncol. 2022;15:98.
Kenny TC, Germain D. mtDNA, metastasis, and the mitochondrial unfolded protein response (UPR(mt)). Front Cell Dev Biol. 2017;5:37.
Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol. 2019;11:a033944.
Melber A, Haynes CM. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018;28:281–95.
Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–90.
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58:123–33.
Kumar M, Sharma S, Mazumder S. Role of UPR(mt) and mitochondrial dynamics in host immunity: it takes two to tango. Front Cell Infect Microbiol. 2023;13:1135203.
Held JP, Feng G, Saunders BR, Pereira CV, Burkewitz K, Patel MR. A tRNA processing enzyme is a key regulator of the mitochondrial unfolded protein response. Elife. 2022;11:e71634.
Zhou Z, Fan Y, Zong R, Tan K. The mitochondrial unfolded protein response: a multitasking giant in the fight against human diseases. Ageing Res Rev. 2022;81:101702.
Li TY, Gao AW, Li X, Li H, Liu YJ, Lalou A, et al. V-ATPase/TORC1-mediated ATFS-1 translation directs mitochondrial UPR activation in C. elegans. J Cell Biol. 2023;222:e202205045.
Yang Q, Liu P, Anderson NS, Shpilka T, Du Y, Naresh NU, et al. LONP-1 and ATFS-1 sustain deleterious heteroplasmy by promoting mtDNA replication in dysfunctional mitochondria. Nat Cell Biol. 2022;24:181–93.
Dai CY, Ng CC, Hung GCC, Kirmes I, Hughes LA, Du Y, et al. ATFS-1 counteracts mitochondrial DNA damage by promoting repair over transcription. Nat Cell Biol. 2023;25:1111–20.
Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–6.
Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell. 2016;165:1197–208.
Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–23.
Liu H, Tian L, Qu M, Wang D. Acetylation regulation associated with the induction of protective response to polystyrene nanoparticles in Caenorhabditis elegans. J Hazard Mater. 2021;411:125035.
Li TY, Sleiman MB, Li H, Gao AW, Mottis A, Bachmann AM, et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat Aging. 2021;1:165–78.
Wang G, Fan Y, Cao P, Tan K. Insight into the mitochondrial unfolded protein response and cancer: opportunities and challenges. Cell Biosci. 2022;12:18.
Shao LW, Peng Q, Dong M, Gao K, Li Y, Li Y, et al. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat Commun. 2020;11:4639.
Thompson MA, De-Souza EA. A year at the forefront of proteostasis and aging. Biol Open. 2023;12:bio059750.
Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26:2037–43.
Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2018;19:109–20.
Smyrnias I. The mitochondrial unfolded protein response and its diverse roles in cellular stress. Int J Biochem Cell Biol. 2021;133:105934.
Liu Y, Zhang L, Zhang S, Liu J, Li X, Yang K, et al. ATF5 regulates tubulointerstitial injury in diabetic kidney disease via mitochondrial unfolded protein response. Mol Med. 2023;29:57.
Zhao Y, Zhao C, Guo H, Zhang Z, Xu H, Shi M, et al. mTORC2 orchestrates monocytic and granulocytic lineage commitment by an ATF5-mediated pathway. iScience. 2023;26:107540.
Zhu J, Lee MJ, An JH, Oh E, Chung W, Heo JY. ATF5 attenuates the secretion of pro-inflammatory cytokines in activated microglia. Int J Mol Sci. 2023;24:3322.
Li TY, Wang Q, Gao AW, Li X, Sun Y, Mottis A, et al. Lysosomes mediate the mitochondrial UPR via mTORC1-dependent ATF4 phosphorylation. Cell Discov. 2023;9:92.
Riar AK, Burstein SR, Palomo GM, Arreguin A, Manfredi G, Germain D. Sex specific activation of the ERα axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum Mol Genet. 2017;26:1318–27.
Katiyar A, Fujimoto M, Tan K, Kurashima A, Srivastava P, Okada M, et al. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio. 2020;10:1135–48.
Sutandy FXR, Gößner I, Tascher G, Münch C. A cytosolic surveillance mechanism activates the mitochondrial UPR. Nature. 2023;618:849–54.
Williams R, Laskovs M, Williams RI, Mahadevan A, Labbadia J. A mitochondrial stress-specific form of HSF1 protects against age-related proteostasis collapse. Dev Cell. 2020;54:758–72.e5.
Tan K, Fujimoto M, Takii R, Takaki E, Hayashida N, Nakai A. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat Commun. 2015;6:6580.
Liu C, Fu Z, Wu S, Wang X, Zhang S, Chu C, et al. Mitochondrial HSF1 triggers mitochondrial dysfunction and neurodegeneration in Huntington’s disease. EMBO Mol Med. 2022;14:e15851.
Rao Y, Li C, Hu YT, Xu YH, Song BB, Guo SY, et al. A novel HSF1 activator ameliorates non-alcoholic steatohepatitis by stimulating mitochondrial adaptive oxidation. Br J Pharm. 2022;179:1411–32.
Chattopadhyay M, Jenkins EC, Lechuga-Vieco AV, Nie K, Fiel MI, Rialdi A, et al. The portrait of liver cancer is shaped by mitochondrial genetics. Cell Rep. 2022;38:110254.
Jenkins EC, Brown SO, Germain D. The multi-faced role of PAPP-A in post-partum breast cancer: IGF-signaling is only the beginning. J Mammary Gland Biol Neoplasia. 2020;25:181–9.
Angelastro JM. Targeting ATF5 in cancer. Trends Cancer. 2017;3:471–4.
Deng P, Haynes CM. Mitochondrial dysfunction in cancer: Potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol. 2017;47:43–9.
Sears TK, Angelastro JM. The transcription factor ATF5: role in cellular differentiation, stress responses, and cancer. Oncotarget. 2017;8:84595–609.
Chen A, Qian D, Wang B, Hu M, Lu J, Qi Y, et al. ATF5 is overexpressed in epithelial ovarian carcinomas and interference with its function increases apoptosis through the downregulation of Bcl-2 in SKOV-3 cells. Int J Gynecol Pathol. 2012;31:532–7.
Hu M, Li H, Xie H, Fan M, Wang J, Zhang N, et al. ELF1 transcription factor enhances the progression of glioma via ATF5 promoter. ACS Chem Neurosci. 2021;12:1252–61.
Zhou J, Tian H, Zhi X, Xiao Z, Chen T, Yuan H, et al. Activating transcription factor 5 (ATF5) promotes tumorigenic capability and activates the Wnt/b-catenin pathway in bladder cancer. Cancer Cell Int. 2021;21:660.
He F, Xiao H, Cai Y, Zhang N. ATF5 and HIF1α cooperatively activate HIF1 signaling pathway in esophageal cancer. Cell Commun Signal. 2021;19:53.
Arias A, Lamé MW, Santarelli L, Hen R, Greene LA, Angelastro JM. Regulated ATF5 loss-of-function in adult mice blocks formation and causes regression/eradication of gliomas. Oncogene. 2012;31:739–51.
Lai C, Zhang J, Tan Z, Shen LF, Zhou RR, Zhang YY. Maf1 suppression of ATF5-dependent mitochondrial unfolded protein response contributes to rapamycin-induced radio-sensitivity in lung cancer cell line A549. Aging (Albany NY). 2021;13:7300–13.
Liu DX, Qian D, Wang B, Yang JM, Lu Z. p300-Dependent ATF5 acetylation is essential for Egr-1 gene activation and cell proliferation and survival. Mol Cell Biol. 2011;31:3906–16.
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118:2840–8.
Bai X, Ni J, Beretov J, Wasinger VC, Wang S, Zhu Y, et al. Activation of the eIF2α/ATF4 axis drives triple-negative breast cancer radioresistance by promoting glutathione biosynthesis. Redox Biol. 2021;43:101993.
Du J, Liu H, Mao X, Qin Y, Fan C. ATF4 promotes lung cancer cell proliferation and invasion partially through regulating Wnt/β-catenin signaling. Int J Med Sci. 2021;18:1442–8.
Liu J, Qi YB. Activation of LXRβ inhibits proliferation, promotes apoptosis, and increases chemosensitivity of gastric cancer cells by upregulating the expression of ATF4. J Cell Biochem. 2019;120:14336–47.
Wang M, Lu Y, Wang H, Wu Y, Xu X, Li Y. High ATF4 expression is associated with poor prognosis, amino acid metabolism, and autophagy in gastric cancer. Front Oncol. 2021;11:740120.
Wang SF, Chang YL, Liu TY, Huang KH, Fang WL, Li AF, et al. Mitochondrial dysfunction decreases cisplatin sensitivity in gastric cancer cells through upregulation of integrated stress response and mitokine GDF15. Febs j. 2024;291:1131–50.
Mu N, Lei Y, Wang Y, Wang Y, Duan Q, Ma G, et al. Inhibition of SIRT1/2 upregulates HSPA5 acetylation and induces pro-survival autophagy via ATF4-DDIT4-mTORC1 axis in human lung cancer cells. Apoptosis. 2019;24:798–811.
Shen Z, Liu P, Sun Q, Li Y, Acharya R, Li X, et al. FTO inhibits UPR(mt)-induced apoptosis by activating JAK2/STAT3 pathway and reducing m6A level in adipocytes. Apoptosis. 2021;26:474–87.
B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–99.
Wei L, Lin Q, Lu Y, Li G, Huang L, Fu Z, et al. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-β1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021;12:334.
González-González A, Muñoz-Muela E, Marchal JA, Cara FE, Molina MP, Cruz-Lozano M, et al. Activating transcription factor 4 modulates TGFβ-induced aggressiveness in triple-negative breast cancer via SMAD2/3/4 and mTORC2 signaling. Clin Cancer Res. 2018;24:5697–709.
Jiménez JA, Apfelbaum AA, Hawkins AG, Svoboda LK, Kumar A, Ruiz RO, et al. EWS-FLI1 and Menin converge to regulate ATF4 Activity in ewing sarcoma. Mol Cancer Res. 2021;19:1182–95.
Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. 2018;33:91–107.e6.
Fujita Y, Ito M, Nozawa Y, Yoneda M, Oshida Y, Tanaka M. CHOP (C/EBP homologous protein) and ASNS (asparagine synthetase) induction in cybrid cells harboring MELAS and NARP mitochondrial DNA mutations. Mitochondrion. 2007;7:80–8.
Wang A, Xu S, Zhang X, He J, Yan D, Yang Z, et al. Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J Pathol. 2011;225:285–92.
Geng W, Ren J, Shi H, Qin F, Xu X, Xiao S, et al. RPL41 sensitizes retinoblastoma cells to chemotherapeutic drugs via ATF4 degradation. J Cell Physiol. 2021;236:2214–25.
Li J, Ma X, Xu F, Yan Y, Chen W. Babaodan overcomes cisplatin resistance in cholangiocarcinoma via inhibiting YAP1. Pharm Biol. 2024;62:314–25.
Zhu H, Chen X, Chen B, Chen B, Fan J, Song W, et al. Activating transcription factor 4 mediates a multidrug resistance phenotype of esophageal squamous cell carcinoma cells through transactivation of STAT3 expression. Cancer Lett. 2014;354:142–52.
Igarashi T, Izumi H, Uchiumi T, Nishio K, Arao T, Tanabe M, et al. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene. 2007;26:4749–60.
Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med. 2021;13:e14351.
Cantó C. The heat shock factor HSF1 juggles protein quality control and metabolic regulation. J Cell Biol. 2017;216:551–3.
Jiang S, Tu K, Fu Q, Schmitt DC, Zhou L, Lu N, et al. Multifaceted roles of HSF1 in cancer. Tumour Biol. 2015;36:4923–31.
Li J, Labbadia J, Morimoto RI. Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 2017;27:895–905.
Dai C. The heat-shock, or HSF1-mediated proteotoxic stress, response in cancer: from proteomic stability to oncogenesis. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160525.
Dai C, Sampson SB. HSF1: Guardian of proteostasis in cancer. Trends Cell Biol. 2016;26:17–28.
Wang G, Cao P, Fan Y, Tan K. Emerging roles of HSF1 in cancer: cellular and molecular episodes. Biochim Biophys Acta Rev Cancer. 2020;1874:188390.
Yang T, Ren C, Lu C, Qiao P, Han X, Wang L, et al. Phosphorylation of HSF1 by PIM2 induces PD-L1 expression and promotes tumor growth in breast cancer. Cancer Res. 2019;79:5233–44.
Levi-Galibov O, Lavon H, Wassermann-Dozorets R, Pevsner-Fischer M, Mayer S, Wershof E, et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat Commun. 2020;11:6245.
Shi Y, Sun L, Zhang R, Hu Y, Wu Y, Dong X, et al. Thrombospondin 4/integrin α2/HSF1 axis promotes proliferation and cancer stem-like traits of gallbladder cancer by enhancing reciprocal crosstalk between cancer-associated fibroblasts and tumor cells. J Exp Clin Cancer Res. 2021;40:14.
Vydra N, Janus P, Kus P, Stokowy T, Mrowiec K, Toma-Jonik A, et al. Heat shock factor 1 (HSF1) cooperates with estrogen receptor α (ERα) in the regulation of estrogen action in breast cancer cells. Elife. 2021;10:e69843.
Xu A, Zhang J, Zuo L, Yan H, Chen L, Zhao F, et al. FTO promotes multiple myeloma progression by posttranscriptional activation of HSF1 in an m(6)A-YTHDF2-dependent manner. Mol Ther. 2022;30:1104–18.
Hoj JP, Mayro B, Pendergast AM. The ABL2 kinase regulates an HSF1-dependent transcriptional program required for lung adenocarcinoma brain metastasis. Proc Natl Acad Sci USA. 2020;117:33486–95.
Mun GI, Choi E, Lee Y, Lee YS. Decreased expression of FBXW7 by ERK1/2 activation in drug-resistant cancer cells confers transcriptional activation of MDR1 by suppression of ubiquitin degradation of HSF1. Cell Death Dis. 2020;11:395.
Lee S, Jung J, Lee YJ, Kim SK, Kim JA, Kim BK, et al. Targeting HSF1 as a therapeutic strategy for multiple mechanisms of EGFR inhibitor resistance in EGFR mutant non-small-cell lung cancer. Cancers (Basel). 2021;13:2987.
Dong B, Jaeger AM, Thiele DJ. Inhibiting heat shock factor 1 in cancer: A unique therapeutic opportunity. Trends Pharm Sci. 2019;40:986–1005.
Chin Y, Gumilar KE, Li XG, Tjokroprawiro BA, Lu CH, Lu J, et al. Targeting HSF1 for cancer treatment: mechanisms and inhibitor development. Theranostics. 2023;13:2281–300.
Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets. 2009;13:469–78.
Cheng W, Zhang B, Zikeliyar M, Wang J, Jian H, Wu K, et al. Elevated Mortalin correlates with poor outcome in hepatocellular carcinoma. Ann Diagn Pathol. 2019;42:59–63.
Dundas SR, Lawrie LC, Rooney PH, Murray GI. Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J Pathol. 2005;205:74–81.
Jin H, Ji M, Chen L, Liu Q, Che S, Xu M, et al. The clinicopathological significance of Mortalin overexpression in invasive ductal carcinoma of breast. J Exp Clin Cancer Res. 2016;35:42.
Cui X, Li Z, Piao J, Li J, Li L, Lin Z, et al. Mortalin expression in pancreatic cancer and its clinical and prognostic significance. Hum Pathol. 2017;64:171–8.
Jubran R, Kocsis J, Garam N, Maláti É, Gombos T, Barabás L, et al. Circulating mitochondrial stress 70 protein/mortalin and cytosolic Hsp70 in blood: Risk indicators in colorectal cancer. Int J Cancer. 2017;141:2329–35.
Xu M, Jin T, Chen L, Zhang X, Zhu G, Wang Q, et al. Mortalin is a distinct bio-marker and prognostic factor in serous ovarian carcinoma. Gene. 2019;696:63–71.
Xu M, Zhang Y, Cui M, Wang X, Lin Z. Mortalin contributes to colorectal cancer by promoting proliferation and epithelial-mesenchymal transition. IUBMB Life. 2020;72:771–81.
Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, et al. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013;33:4051–67.
Wu PK, Hong SK, Park JI. Steady-state levels of phosphorylated mitogen-activated protein kinase kinase 1/2 determined by Mortalin/HSPA9 and protein phosphatase 1 Alpha in KRAS and BRAF tumor cells. Mol Cell Biol. 2017;37:e00061–17.
Qiao GB, Wang RT, Wang SN, Tao SL, Tan QY, Jin H. GRP75-mediated upregulation of HMGA1 stimulates stage I lung adenocarcinoma progression by activating JNK/c-JUN signaling. Thorac Cancer. 2021;12:1558–69.
Wei B, Cao J, Tian JH, Yu CY, Huang Q, Yu JJ, et al. Mortalin maintains breast cancer stem cells stemness via activation of Wnt/GSK3β/β-catenin signaling pathway. Am J Cancer Res. 2021;11:2696–716.
Starenki D, Sosonkina N, Hong SK, Lloyd RV, Park JI. Mortalin (GRP75/HSPA9) promotes survival and proliferation of thyroid carcinoma Cells. Int J Mol Sci. 2019;20:2069.
Wu PK, Hong SK, Park JI. Mortalin depletion induces MEK/ERK-dependent and ANT/CypD-mediated death in vemurafenib-resistant B-Raf(V600E) melanoma cells. Cancer Lett. 2021;502:25–33.
Wu PK, Hong SK, Chen W, Becker AE, Gundry RL, Lin CW, et al. Mortalin (HSPA9) facilitates BRAF-mutant tumor cell survival by suppressing ANT3-mediated mitochondrial membrane permeability. Sci Signal. 2020;13:eaay1478.
Liao Y, Liu Y, Shao Z, Xia X, Deng Y, Cai J, et al. A new role of GRP75-USP1-SIX1 protein complex in driving prostate cancer progression and castration resistance. Oncogene. 2021;40:4291–306.
Liao Y, Liu Y, Xia X, Shao Z, Huang C, He J, et al. Targeting GRP78-dependent AR-V7 protein degradation overcomes castration-resistance in prostate cancer therapy. Theranostics. 2020;10:3366–81.
Mylonis I, Kourti M, Samiotaki M, Panayotou G, Simos G. Mortalin-mediated and ERK-controlled targeting of HIF-1α to mitochondria confers resistance to apoptosis under hypoxia. J Cell Sci. 2017;130:466–79.
Gestl EE, Anne Böttger S. Cytoplasmic sequestration of the tumor suppressor p53 by a heat shock protein 70 family member, mortalin, in human colorectal adenocarcinoma cell lines. Biochem Biophys Res Commun. 2012;423:411–6.
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y, Reddel RR, Kaul SC. Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res. 2002;274:246–53.
Li AD, Xie XL, Qi W, Wang WB, Ma JJ, Zhao DQ, et al. TGR5 promotes cholangiocarcinoma by interacting with mortalin. Exp Cell Res. 2020;389:111855.
Pagliarone AC, Castañeda ED, Santana JPP, de Oliveira CAB, Robeldo TA, Teixeira FR, et al. Mitochondrial heat shock protein mortalin as potential target for therapies based on oxidative stress. Photodiagnosis Photodyn Ther. 2021;34:102256.
Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, et al. Analogs of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med Chem Lett. 2013;4:1042–7.
Shao H, Li X, Moses MA, Gilbert LA, Kalyanaraman C, Young ZT, et al. Exploration of Benzothiazole rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (Hsp70). J Med Chem. 2018;61:6163–77.
Mittal S, Rajala MS. Heat shock proteins as biomarkers of lung cancer. Cancer Biol Ther. 2020;21:477–85.
Buttacavoli M, Di Cara G, D’Amico C, Geraci F, Pucci-Minafra I, Feo S, et al. Prognostic and functional significant of heat shock proteins (HSPs) in breast cancer unveiled by multi-omics approaches. Biology (Basel). 2021;10:247.
Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018;9:161.
Tsai YP, Teng SC, Wu KJ. Direct regulation of HSP60 expression by c-MYC induces transformation. FEBS Lett. 2008;582:4083–8.
Guo J, Zhu S, Deng H, Xu R. HSP60-knockdown suppresses proliferation in colorectal cancer cells via activating the adenine/AMPK/mTOR signaling pathway. Oncol Lett. 2021;22:630.
Tang Y, Zhou Y, Fan S, Wen Q. The multiple roles and therapeutic potential of HSP60 in cancer. Biochem Pharm. 2022;201:115096.
Polson ES, Kuchler VB, Abbosh C, Ross EM, Mathew RK, Beard HA, et al. KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci Transl Med. 2018;10:eaar2718.
Marino Gammazza A, Campanella C, Barone R, Caruso Bavisotto C, Gorska M, Wozniak M, et al. Doxorubicin anti-tumor mechanisms include Hsp60 post-translational modifications leading to the Hsp60/p53 complex dissociation and instauration of replicative senescence. Cancer Lett. 2017;385:75–86.
Chitre S, Ray AM, Stevens M, Doud EH, Liechty H, Washburn A, et al. Bis-aryl-α,β-unsaturated ketone (ABK) chaperonin inhibitors exhibit selective cytotoxicity to colorectal cancer cells that correlates with levels of aberrant HSP60 in the cytosol. Bioorg Med Chem. 2022;75:117072.
Caruso Bavisotto C, Marino Gammazza A, Lo Cascio F, Mocciaro E, Vitale AM, Vergilio G, et al. Curcumin affects HSP60 folding activity and levels in neuroblastoma cells. Int J Mol Sci. 2020;21:661.
Indira M, Surendranath Reddy EC, Kamala Prasad V, Satyanarayana Swamy V, Kakarla RR, Venkata Krishna Reddy M, et al. Environmentally friendly and efficient TBHP-mediated catalytic reaction for the synthesis of substituted benzimidazole-2-ones: In-silico approach to pharmaceutical applications. Environ Res. 2024;252:118760.
Bi F, Wang J, Zheng X, Xiao J, Zhi C, Gu J, et al. HSP60 participates in the anti-glioma effects of curcumin. Exp Ther Med. 2021;21:204.
Quirós PM, Español Y, Acín-Pérez R, Rodríguez F, Bárcena C, Watanabe K, et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8:542–56.
Taouktsi E, Kyriakou E, Smyrniotis S, Borbolis F, Bondi L, Avgeris S, et al. Organismal and cellular stress responses upon disruption of mitochondrial Lonp1 protease. Cells. 2022;11.
Gibellini L, De Gaetano A, Mandrioli M, Van Tongeren E, Bortolotti CA, Cossarizza A, et al. The biology of Lonp1: More than a mitochondrial protease. Int Rev Cell Mol Biol. 2020;354:1–61.
Shetty R, Noland R, Nandi G, Suzuki CK. Powering down the mitochondrial LonP1 protease: a novel strategy for anticancer therapeutics. Expert Opin Ther Targets. 2024;28:9–15.
Pinti M, Gibellini L, Liu Y, Xu S, Lu B, Cossarizza A. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell Mol Life Sci. 2015;72:4807–24.
Zanini G, Selleri V, Malerba M, Solodka K, Sinigaglia G, Nasi M, et al. The role of Lonp1 on mitochondrial functions during cardiovascular and muscular diseases. Antioxidants (Basel). 2023;12:598.
Ma H, Chen W, Fan W, He H, Huang F. Roles of LonP1 in oral-maxillofacial developmental defects and tumors: A novel insight. Int J Mol Sci. 2022;23:13370.
Quirós PM, Bárcena C, López-Otín C. Lon protease: A key enzyme controlling mitochondrial bioenergetics in cancer. Mol Cell Oncol. 2014;1:e968505.
Wu L, Yan X, Sun R, Ma Y, Yao W, Gao B, et al. Sirt3 restricts tumor initiation via promoting LONP1 deacetylation and K63 ubiquitination. J Transl Med. 2023;21:81.
Ghosh JC, Seo JH, Agarwal E, Wang Y, Kossenkov AV, Tang HY, et al. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene. 2019;38:6926–39.
Liu C, Wang H, Li H, Chen X, Wu X, Lu B, et al. Inhibition of LONP1 suppresses pancreatic cancer progression via c-Jun N-terminal kinase pathway-meditated epithelial-mesenchymal transition. Pancreas. 2019;48:629–35.
Wang H, Liu C, Zhao Y, Zhang W, Xu K, Li D, et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun. 2020;522:1063–8.
Lee J, Pandey AK, Venkatesh S, Thilagavathi J, Honda T, Singh K, et al. Inhibition of mitochondrial LonP1 protease by allosteric blockade of ATP binding and hydrolysis via CDDO and its derivatives. J Biol Chem. 2022;298:101719.
Wang HM, Cheng KC, Lin CJ, Hsu SW, Fang WC, Hsu TF, et al. Obtusilactone A and (-)-sesamin induce apoptosis in human lung cancer cells by inhibiting mitochondrial Lon protease and activating DNA damage checkpoints. Cancer Sci. 2010;101:2612–20.
Maneix L, Sweeney MA, Lee S, Iakova P, Moree SE, Sahin E, et al. The mitochondrial protease LonP1 promotes proteasome inhibitor resistance in multiple myeloma. Cancers (Basel). 2021;13:843.
Mabanglo MF, Bhandari V, Houry WA. Substrates and interactors of the ClpP protease in the mitochondria. Curr Opin Chem Biol. 2022;66:102078.
Li X, Zou Y, Li T, Wong TKF, Bushey RT, Campa MJ, et al. Genetic variants of CLPP and M1AP are associated with risk of non-small cell lung cancer. Front Oncol. 2021;11:709829.
Cormio A, Sanguedolce F, Pesce V, Musicco C. Mitochondrial caseinolytic protease P: A possible novel prognostic marker and therapeutic target in cancer. Int J Mol Sci. 2021;22:6228.
Luo B, Ma Y, Zhou Y, Zhang N, Luo Y. Human ClpP protease, a promising therapy target for diseases of mitochondrial dysfunction. Drug Discov Today. 2021;26:968–81.
Seo JH, Rivadeneira DB, Caino MC, Chae YC, Speicher DW, Tang HY, et al. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 2016;14:e1002507.
Luo J, Zeng B, Tao C, Lu M, Ren G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ. 2020;8:e8754.
Kou X, Ding H, Li L, Chao H. Caseinolytic protease P (CLPP) activated by ONC201 inhibits proliferation and promotes apoptosis in human epithelial ovarian cancer cells by inducing mitochondrial dysfunction. Ann Transl Med. 2021;9:1463.
Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell. 2019;35:721–37.e9.
Kou X, Yang X, Zhao Z, Li L. HSPA8-mediated stability of the CLPP protein regulates mitochondrial autophagy in cisplatin-resistant ovarian cancer cells. Acta Biochim Biophys Sin (Shanghai). 2024;56:356–65.
Zhang Y, Maurizi MR. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim Biophys Acta. 2016;1862:252–64.
Gronauer TF, Mandl MM, Lakemeyer M, Hackl MW, Meßner M, Korotkov VS, et al. Design and synthesis of tailored human caseinolytic protease P inhibitors. Chem Commun (Camb). 2018;54:9833–6.
Wang P, Zhang T, Wang X, Xiao H, Li H, Zhou LL, et al. Aberrant human ClpP activation disturbs mitochondrial proteome homeostasis to suppress pancreatic ductal adenocarcinoma. Cell Chem Biol. 2022;29:1396–408.e8.
Fan Y, Wang J, Fang Z, Pierce SR, West L, Staley A, et al. Anti-tumor and anti-invasive effects of ONC201 on ovarian cancer cells and a transgenic mouse model of serous ovarian cancer. Front Oncol. 2022;12:789450.
Zhang J, Luo B, Sui J, Qiu Z, Huang J, Yang T, et al. IMP075 targeting ClpP for colon cancer therapy in vivo and in vitro. Biochem Pharm. 2022;204:115232.
Graves PR, Aponte-Collazo LJ, Fennell EMJ, Graves AC, Hale AE, Dicheva N, et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem Biol. 2019;14:1020–9.
Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M, et al. Activation of the mitochondrial unfolded protein response: a new therapeutic target? Biomedicines. 2022;10:1611.
Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, Reche-López D, Álvarez-Córdoba M, Munuera-Cabeza M, et al. mtUPR modulation as a therapeutic target for primary and secondary mitochondrial diseases. Int J Mol Sci. 2023;24:1482.
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–38.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102.
Zhu L, Zhou Q, He L, Chen L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic Biol Med. 2021;163:125–34.
Acknowledgements
We want to thank Mr. Hanyao Guo for the GO and KEGG analyses of core UPRmt-related genes.
Funding
This work was supported by the Guiding Funds of Central Government for Supporting the Development of the Local Science and Technology (236Z3003G), One Hundred Person Project of Hebei Province (E2016100019), Key Development Fund of Hebei Normal University (L2024ZD16), and Interdisciplinary Research Fund of Hebei Normal University (L2024J04).
Author information
Authors and Affiliations
Contributions
XZ, YF and KT wrote this manuscript; XZ and KT drew the figures in the manuscript; Ke Tan proofread and supervised the manuscript writing process; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Francesca Pentimalli
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Fan, Y. & Tan, K. A bird’s eye view of mitochondrial unfolded protein response in cancer: mechanisms, progression and further applications. Cell Death Dis 15, 667 (2024). https://doi.org/10.1038/s41419-024-07049-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-07049-y
- Springer Nature Limited