
Abstract
Acute myeloid leukemia (AML) is a hematological malignancy characterized by cytogenetic and genomic alterations. Up to now, combination chemotherapy remains the standard treatment for leukemia. However, many individuals diagnosed with AML develop chemotherapeutic resistance and relapse. Recently, it has been pointed out that leukemic stem cells (LSCs) are the fundamental cause of drug resistance and AML relapse. LSCs only account for a small subpopulation of all leukemic cells, but possess stem cell properties, including a self-renewal capacity and a multi-directional differentiation potential. LSCs reside in a mostly quiescent state and are insensitive to chemotherapeutic agents. When LSCs reside in a bone marrow microenvironment (BMM) favorable to their survival, they engage into a steady, continuous clonal evolution to better adapt to the action of chemotherapy. Most chemotherapeutic drugs can only eliminate LSC-derived clones, reducing the number of leukemic cells in the BM to a normal range in order to achieve complete remission (CR). LSCs hidden in the BM niche can hardly be targeted or eradicated, leading to drug resistance and AML relapse. Understanding the relationship between LSCs, the BMM, and the generation and evolution laws of LSCs can facilitate the development of effective therapeutic targets and increase the efficiency of LSCs elimination in AML.
Similar content being viewed by others


Facts
-
Abnormal BM niche may be involved in the pathogenesis of AML.
-
The BMM exerts a selective pressure for the clonal evolution in leukemia, and oxidative stress may represent another one.
-
The protection from BMM and the clonal evolution of LSC may responsible for drug resistance in leukemia.
-
Target BMM may be a potential therapeutic strategy for leukemia treatment.
Open questions
-
What is the exact role and mechanism of BMM in leukemogenesis?
-
How to avoid damage to normal hematopoietic and hematopoietic supporting cells while eradicating leukemia cells?
-
How to effectively repair the abnormal BMM?
-
How to effectively unblock the protection of BMM on AML cells?
Introduction
Acute myeloid leukemia (AML) is a group of highly heterogeneous clonal diseases. The constant accumulation of acquired somatic mutations in hematopoietic stem cells (HSCs) is the primary pathogenic mechanism of AML. Recently, the emergence of novel chemotherapeutic drugs and the progress of HSC transplantation have considerably improved the remission rate and disease-free survival of AML patients. However, the high relapse rate of AML, which is largely attributed to the residual leukemic stem cells (LSCs), remains an unsettled thorny issue. LSCs represent a subpopulation of malignant cells with unlimited self-renewal capacity and multi-directional differentiation potential. Despite representing a small proportion of all leukemic cells (approximately 0.1–1%), LSCs are considered the origin of leukemia and the primary cause of relapse after treatment, as well as of drug resistance [1]. LSCs are closely related to the bone marrow microenvironment (BMM). The BMM not only provides a habitat for LSCs but also offers the support for the survival and development of LSCs while alleviating the pressure imposed by chemotherapy on LSCs. This feature of the BMM can lead to the generation of dominant clones, resulting in leukemia relapse and drug resistance. Therefore, an in-depth investigation of the relationship between LSCs and the BMM, as well as the generation and evolution laws of LSCs, will facilitate the understanding of the pathogenesis and drug resistance mechanism of AML.
Abnormal BM niche involvement in the pathogenesis of AML
The BM is the principal hematopoietic organ consisting of HSCs and the hematopoietic microenvironment. The latter is also known as the stem cell niche, which consists of bone marrow stromal cells (BMSCs) and extracellular matrix (ECM). Normal hematopoiesis is the process whereby HSCs self-renew and the progenitor cells proliferate and differentiate in the BMM [2].
BMSCs are integral components of the hematopoietic microenvironment and play an indispensable role in maintaining the stability, homing, proliferation, and differentiation of HSCs [3]. Accumulated evidence demonstrates that the dysfunction of BMSCs impairs the BMM, thereby affecting normal hematopoiesis. Besides, the dysfunction of BMSCs will trigger and promote the development of malignant hematological diseases. Many studies have reported that AML is linked with a reduction in BMSCs. In most AML cases, cytogenetic abnormalities of BMSCs are usually found [4,5,6,7]. Besides, BMSCs derived from AML patients may exhibit molecular and functional alterations, e.g., higher senescence, decreased proliferation, reduced clonogenic potential, impaired in vitro osteogenic and adipogenic differentiation, imbalanced regulation of endogenous hematopoiesis, and increased support of leukemia growth [8,9,10,11].
Recently, several research groups have used animal models to explore the role of BMM in leukemia. According to Raaijmakers et al., selectively knocking out Dicer1 (a gene required for RNA and microRNA processing) in mesenchymal osteoprogenitors impaired osteoblast differentiation both in vitro and in vivo. In mouse models, the loss of Dicer1 in osteoprogenitors induced an evident hematopoietic disorder, increased apoptosis, and proliferation of hematopoietic stem and progenitor cell (HSPC). These knockout animals exhibited key features of human myelodysplastic syndromes (MDS), including the propensity to develop AML [12]. A study by Kode et al. demonstrated that activating mutations of β-catenin in mouse osteoblasts stimulated the expression of Notch ligand jagged 1, which subsequently altered the differentiation potential of myeloid and lymphoid progenitors through the activation of Notch signaling, and ultimately led to the development of AML [13]. These findings suggested that functional abnormalities caused by genetic mutations in BMSCs may play a crucial role in leukemogenesis. Yilmaz et al. implanted HSCs with tumor suppresser gene phosphatase and tensin homolog (PTEN) knocked out into irradiated recipient mice. These PTEN-deficient HSCs gradually depleted over time after a brief multilineage differentiation, while no evidence of myeloproliferation or development of leukemia was discovered in any recipient mice. In contrast, the implantation of PTEN-deficient whole bone marrow cells led to myeloproliferative progression to leukemia/lymphoma. These results suggested that BMSCs may play a more critical role in the malignant transformation of normal hematopoietic cells into leukemia cells [14]. The occurrence of donor cell leukemia in clinical AML cases further indicates that the hematopoietic microenvironment may be involved in the pathology of AML. However, the concrete mechanism underlie the hematopoietic microenvironment deterioration is still poorly understood. Recent evidence suggested that oxidative stress may play a key role: Huang et al. investigated the effect of iron overload (IO) on BMSCs in MDS and AML patients. The results showed that IO could promote the ROS generation and apoptosis in BMSCs, decrease expression of hematopoietic regulation-related genes such as VEGFA, CXCL12, and TGF-β1, and activate the Wnt/β-catenin signaling pathway, and it was supposed that these alterations may be associated with the MDS progression [15]. Li et al. showed that daunorubicin (DNR) treatment led to senescence of mouse BMSCs, increased ROS production, as well as mitochondrial dysfunction. When oxidatively damaged BMSCs were co-cultured with HSCs in vitro, these stromal cells generated more ROS, leading to increased genomic instability in HSCs [16]. In addition, BMSCs injury might also lead to impaired immune surveillance and dysregulated cytokine secretion, disrupted BMM homeostasis, and ultimately led to abnormal proliferation of hematopoietic cells, all of which may be potential causes of malignant transformation of hematopoietic cells [16]. However, the exact role and mechanism of BMM in leukemogenesis still need to be elucidated by more extensive and in-depth studies.
Origins and generation of LSC
It is generally believed that LSC result from mutation accumulation in HSC. Long-term HSCs are capable of self-renewal. Long-term exposure to carcinogens will cause the accumulation of genetic mutations in these HSCs, increasing the risk of malignant transformation of these cells. Hope et al. performed continuous tracking of the transplanted LSCs and highlighted that LSCs are not functionally homogeneous but, similarly to the normal HSC compartment, comprised distinct hierarchically arranged LSC classes. This finding supported the hypothesis that LSCs originate from normal HSCs [17]. In another study, Cozzio et al. adopted transgenic technology to analyze three types of cells with different differentiation grades: HSCs, common myeloid progenitors, and the lineal descendent granulocytic/monocytic-restricted progenitors. These three types of cells could survive and proliferate in vitro after mixed lineage leukemia (MLL) fusion genes were introduced. The mice could be successfully transfected with these cells, which induced leukemia at the same speed [18]. Thus, it was speculated that LSCs have a wide variety of sources. LSCs may be derived from HSCs and progenitor cells that are earlier in the development stage and also from leukemia progenitor cells that are more mature.
According to the AML two-hit model, two major types of genetic mutations play a pivotal role in the pathogenesis of AML. Type I genetic mutation involves the tyrosine kinases in the signal transduction pathway, including the signaling molecules FMS-like tyrosine kinase-3 (FLT3), stem cell factor receptor (c-Kit), and break point cluster region-abelson (BCR-ABL). Persistently activated tyrosine kinases confer special benefits for the proliferation and survival of hematopoietic precursor cells [19]. Type II mutation involves disrupting transcription factors or transcriptional coactivators, such as acute myeloid leukemia-1 transcription factor/eight-twenty-one corepressor (AML1/ETO), mixed-lineage leukemia/ALL1-fused gene from chromosome 9 protein (MLL/AF9), and promyelocytic leukemia/retinoic acid receptor α (PML/RARα) fusion gene. These abnormal changes can lead to disorders of differentiation, maturation, and apoptosis of hematopoietic progenitor cells (HPC) [20]. In mouse models, when genetic abnormalities of type I occurred alone, chronic myeloid leukemia (CML)-like changes were noted; when genetic abnormalities of type II occurred alone, MDS-like changes were noted. Under normal situations, leukemia occurs when these two types of genetic abnormalities occur simultaneously [21]. So far, it remains uncertain how the two types of genetic abnormalities work synergistically.
Recently, a large number of studies have shown that AML patients carry repeated mutations of many important epigenetic mediators. Among them, mutations of genes related to DNA methylation are most common, including DNA methyltransferase 3A (DNMT3A) and isocitrate dehydrogenase 1 and 2 (IDH1/2) mutations. Genetic modeling of individual epigenetic factors has shown that most of these mutations will affect the HSC compartment and alter hematopoietic differentiation, in some cases leading to myelodysplasia, stem cell expansion, or other preleukemic conditions [22,23,24]. Besides, mutations of other epigenetic modifiers, such as additional sex combs-like (ASXL1) and enhancer of zeste homolog 2 (EZH2), also play an important role in the occurrence of AML (Table 1). However, epigenetic mutations alone are not sufficient to transform HSCs, indicating that sequential acquisition of mutations is required [25]. In a recent study, Uckelmann et al. reported that in Npm1c/Dnmt3a mutant knock-in mice, a model of AML development, the use of a small molecule (VTP-50469) can reverse the self-renewal of myeloid progenitor cells before leukemia transformation. This finding indicates that individuals at high risk of developing AML might benefit from targeted epigenetic therapy in a preventative setting [26].
Evolution of LSC
Recent studies have shown that AML cell populations at relapse may have evolved from either the dominant clonal or minor subclonal cell populations present at diagnosis, accompanied by the potential acquisition of additional mutations. AML cells adapt to the environment by steady, continuous clonal evolution and better survive under chemotherapy pressure [27,28,29,30].
The clonal evolution in leukemia is a multi-step, dynamical evolutionary process. Corces-Zimmerman et al. analyzed the persistence of preleukemic mutations in patients in remission. It was discovered that mutations in preleukemia were present in CD34+ progenitor cells and various mature cells, indicating that preleukemic HSCs can survive the induction chemotherapy, thereby confirming these cells as a reservoir for the reevolution of relapsed disease [31]. Moreover, they tentatively proposed several patterns of clonal evolution leading to AML relapse based on the existing whole genome sequencing results: (1) treatment refractory primary disease, (2) further evolution of a dominant clone present at diagnosis, (3) outgrowth of a subclone present at diagnosis; (4) further evolution of disease from a preleukemic HSC [31]. Miles et al. employed single-cell DNA sequencing to analyze the samples from 123 patients with myeloid malignancies to determine the clonal features of myeloid malignancies. The results showed that AML is dominated by a small number of clones. Among different types of genetic mutations, epigenetic mutations usually occur at the early stage of leukemogenesis, and these mutations tend to occur simultaneously. In contrast, mutations of genes related to the cell signaling pathways occur much later, which are mainly found in distinct subclones, consistent with increased clonal diversity. Besides, it was found that increased clonal diversity in AML did not coincide with the difference in the number of mutations within the largest clone, suggesting that increased mutational burden within a clone is not the primary driver of clonal dominance [32].
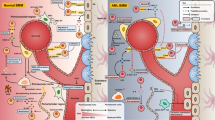
A major distinctive feature of cancer cells is their genetic instability. Cancer cells may evolve along paths under different selective pressures to gain survival and development advantages, a primary reason for continuous clonal evolution in leukemia. The BMM exerts a selective pressure for the clonal evolution in leukemia. It is not only a shelter for leukemic cells but also supports the survival and development of leukemia cells by interacting with them, thereby relieving the chemotherapeutic pressure and promoting the generation of dominant clones (see section 4). Elevated reactive oxygen species (ROS) levels are hallmark features of leukemia cells [33, 34], and oxidative stress induced by high level of ROS may represent another selective pressure for the clonal evolution in leukemia. The sources of ROS levels in AML cells are complex and diverse. Among them, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and mitochondrial electron transport chain (mtETC) are considered to be the main sources of ROS in leukemia cells [35]. However, Hole et al. found that AML cells treated with NOX inhibitors could more effectively inhibit the production of superoxide compared with mtETC inhibitors and mitochondrial ROS scavengers, therefore suggesting that ROS in AML cells is primarily derived from NOX [36]. It was reported that, oncogenic kinases like BCR-ABL1, FLT3-internal tandem duplication (ITD), or c-KIT can promote the generation of endogenous ROS by increasing the activities of RAC GTPases or membrane-bound NOX [37,38,39,40]. In addition, chemotherapy and chronic inflammation can also increase ROS generation, thereby elevating oxidative stress [37, 41, 42]. Although quiescent AML LSCs have a strong ability to maintain the redox balance, they still suffer from oxidative damage to DNA caused by high ROS levels. Such damage increases genomic instability and drives the genetic evolution of LSCs under the synergistic action of the leukemic microenvironment, from which LSCs gain a competitive advantage [35, 43] (Fig. 1). Further investigations are needed to reveal the specific mechanism by which LSCs evolve genetically.

Different from HSC, which mainly obtains energy through glycolysis, LSC mainly relies on oxidative phosphorylation (OXPHOS) to support cell metabolism and survival, thus producing a relatively high ROS level [94]. Chemotherapeutic drugs and chronic inflammation also promote ROS production [37, 41, 42]. In addition, oncogenes such as FLT3(ITD) and BCR-ABL1 can also facilitate intracellular ROS production through NOX or RAC2-MRC cIII pathway [95, 96]. High levels of ROS not only lead to mutagenic reactions in the DNA, but also inhibit DNA repair enzymes, resulting in genomic instability, which may be an important driver of LSC evolution [97]. Rac Rac GTPase; TCA tricarboxylic acid; MRC-cIII mitochondrial respiratory chain complex III.
Interactions between AML cells and the BMM
The interactions between LSCs and the microenvironment are fundamental to survival, therapeutic resistance, and relapse of leukemia. In the BMM, hypoxia induces the synthesis of hypoxia-inducible factor-1α (HIF-1α), which plays a key role in cell migration and survival. In human AMLcells, HIF-1α knowdown leads to increased apoptosis and impaired cell engraftment [44]. Furthermore, hypoxia can also upregulate C-X-C chemokine receptor type 4 (CXCR4) expression in AML cells, the cognate receptor of chemokine ligand 12 (CXCL12), and promote CXCL12 expression in BMSCs via HIF-1α [45, 46]. Relying upon the interaction between CXCR4 and CXCL12, as well as the adhesion effect mediated by other surface adhesion molecules, AML cells dwell in the BM niches to maintain their survival [44,45,46,47]. Besides, the interaction between BMM and AML cell can also lead to the activation of phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), wingless-related integration site (Wnt), NOTCH, and other signal pathways, which play key roles in the regulation of AML cells’ maintenance, proliferation, and differentiation (Fig. 2). A series of in vitro and in vivo experiments confirmed that AML cells can remodel the BMM by inducing the abnormal proliferation and differentiation of BM mesenchymal stem and progenitor cells (MSPCs), thereby favoring disease development and progression [48, 49]. Evidence suggests that AML cell-derived exosomes can stimulate the expression of Dickkopf-1 (DKK1), thereby inducing downregulation of HSC-supporting factors in BMSCs and reducing their ability to support normal hematopoiesis [50]. Furthermore, AML cell-derived exosomes can also downregulate critical retention factors, such as stem cell factor (SCF) and CXCL12, leading to the mobilization of HSPC out of the BM [51]. In a recent study, Scoville et al. found that human AML blasts can activate the transcription factor aryl hydrocarbon receptor (AHR) pathway and induce miR-29b expression in natural killer (NK) cells, thereby impairing NK cells maturation and function, which may be one of the important mechanisms underlying AML cells’ immune escape [52].

LSC interact and adhere to various niche cells (such as MSCs, osteoblasts, adipocytes, and endothelial cells) and various ECM molecules secreted by them. The interaction between LSC and BMM can activate many important signaling pathways, thereby regulating the biological function of LSC and remodeling BMM accordingly. (1) The CXCL12/CXCR4 axis plays a key role in LSC maintenance and can activate multiple signaling pathways, such as PI3K/AKT/mTOR, to regulate the survival and proliferation of LSC [98]; (2) The interaction between VLA4 from LSC cells and fibronectin (FN) from MSCs activates the PI3K/AKT/BCL2 pathway, allowing LSCs to be resistant to cytotoxic drugs [99]; (3) The binding of E-selectin to CD44 activates the Wnt [100] and PI3K/AKT/NF-κB signaling pathway [101], and promotes LSC survival; (4) The binding of Jagged to Notch activates the Notch signaling pathway, and the intracellular domain NICD of Notch is then released and translocated into the nucleus to activate the transcription of related genes [102]; (5) Hypoxia can promote the HIF-1α-VEGF signaling pathway and angiogenesis. In addition, NF-κB can promote the production of MMPs and VEGF, which in turn accelerates angiogenesis [103]; (6) LSC-secreted exosomes can induce the expression of DKK1 in MSCs, a suppressor of normal hematopoiesis and osteogenesis, thereby leading to the loss of osteoblasts [50]. BCL-2 B-cell lymphoma 2; Fn fibronectin; MMPs matrix metalloproteinases; mTOR mammalian target of rapamycin; NF-κB nuclear transcription factor-κB; OPN osteopontin; VLA4 very late antigen 4; Wnt wingless-type protein.
In severely hypoxic BM tissues, leukemic cells exhibit the strong adaptive ability and promote angiogenesis via multiple mechanisms. Such a capacity is needed to meet the demand for oxygen and nutrients in rapidly growing leukemic cells. Hypoxia has been reported to promote AML cells’ upregulation of transcription and expression of several angiogenic factors, such as vascular endothelial growth factor (VEGF), thus increasing the angiogenic activity of endothelial cells [53, 54]. Angiopoietin/Tyrosine Kinase With Immunoglobin-like And Epidermal Growth Factor-like Domain-2 (Ang/Tie-2) axis is another signaling pathway that is significantly associated with angiogenesis [55]. Tie-2 receptor is always overexpressed in AML cells, which promotes angiogenesis via interaction with Ang-1 [56]. The Ang-1/Tie-2 interactions also facilitates the adhesion of AML cells to BM niche, and helps to maintain the quiescent and anti-apoptotic state of LSC in BM niche [57, 58] (Fig. 2). In vitro experiments showed that Tie-2-blocking antibodies had a growth inhibitory effect on human AML cells co-cultured with microvascular endothelial cells [58]. However, the application of antiangiogenic drugs is often found ineffective in clinical studies, indicating that the deterioration of BMM is not simply due to angiogenesis. Recently, Passaro and colleagues reported that in human AML patient-derived xenografts, significant abnormalities in the bone marrow vasculature were observed. The engrafted AML not only promoted angiogenesis but also increased the permeability of blood vessels. Further analysis showed that the increased vascular permeability was associated with hypoxia-induced increases in ROS and NO. Cytarabine (AraC) treatment achieved only transient remission, and it was supposed that this may be relevant to the increase of vascular permeability, since increased vascular permeability not only leads to poor drug delivery, but also favors the tumor growth and enhances metastatic potential. The combination of nitric oxide synthase inhibitors and chemotherapy successfully restored normal vasculature and delayed leukemia, leading the way to combine leukemia-niche therapies in clinical trials [59].
Fatty acid metabolism is the main metabolic pathway for AML cell survival in the adipocyte-rich BMM [60]. The study by Shafat et al. found that AML cells can alter adipocyte metabolic processes, inducing lipolysis of triglyceride to fatty acid, and the fatty acid was then transferred to AML cells to fuel FAO, thereby promoting the survival and proliferation of AML cells [61]. Tunneling nanotubes (TNTs), or membrane nanotubes, are an important means of communication between eukaryotes [62]. Recent studies have shown that AML cells propel mitochondrial transfer from BMSCs to AML cells via TNTs under the stimulation of ROS, which generated from NOX. As a result, more energy is generated for survival of AML cells by mitochondrial OXPHOS. However, such a phenomenon was not observed in HSCs that derive energy primarily from anaerobic glycolysis [63]. These studies demonstrated that AML cells acquire conspicuous survival benefits by interacting with BMSCs.
Targeted therapy for AML
LSCs usually reside in a quiescent state, that is, the G0 phase, when the LSCs replicate slowly but have an unlimited self-renewal capacity. This biological characteristic of LSC make it insensitive to the cytotoxicity of the conventional cell cycle-targeting chemotherapy. Besides, the expression of multi-drug resistance proteins in LSCs results in poor clinical outcomes. In recent years, the advancement of both biomedical technology as well as the understanding of AML drug resistance mechanisms strongly promote the development of AML-targeted drugs. Drugs targeting AML cell surface antigens, mutant genes, and intracellular signaling pathways have become the current research trend and focus (Table 2), while the exploration of new targets is also ongoing. NOX is a major source of pro-survival ROS in AML cells and is therefore an ideal target to inhibit pro-survival signaling in AML cells. Among NOX family members, NOX1, NOX2, and NOX4 are expressed in human CD34+ hematopoietic progenitor cells [64, 65]. NOX2 inhibitors have been reported to induce reduction of intracellular ROS in FLT3-ITD AML cells, inhibition of downstream growth and survival pathways of FLT3, and increased apoptosis associated with induction of mitochondrial ROS and restoration of p38MAPK [66]. Setanaxib, an inhibitor with anti-NOX1 and NOX4 functions, has anti-proliferative effects on both FLT3-ITD-positive and negative AML cells, and has strong synergistic effects with cytotoxic drugs anthracyclines such as daunorubicin. This anti-proliferative effect may be mediated by amplification of oxidative stress signaling by promoting ROS production from sources other than NOX [67]. It was reported that a variety of chemotherapeutic drugs such as daunorubicin, cytarabine, and decitabine can also induce apoptosis in leukemia cells via ROS generation [68,69,70]. However, high levels of ROS inevitably cause oxidative damage to hematopoietic and hematopoietic supporting cells while eliminating leukemia cells.
Targeting the weaknesses in the metabolism of leukemia cells has attracted much attention in recent years. Fatty acid metabolism is the main metabolic pathway for AML cells to maintain survival, and drug-resistant LSCs have a higher FAO rate, suggesting that FAO is related to the drug resistance of LSCs [71]. However, intervention using the FAO inhibitor-avocatin-B was ineffective. Further investigation showed that glucose uptake and glycolysis were increased while inhibiting the FAO in AML cells, which promoted AML cell survival [60]. This finding suggested that targeting a single metabolic pathway has limitations as leukemic cells may escape through metabolic adaptation. Similarly, although the combination of Bcl-2 inhibitor venetoclax and azacitidine (ven/aza) inhibited OXPHOS in vivo and resulted in an effective eradication of LSCs [72, 73], ven/aza failed to eradicate LSCs in relapsed/refractory (R/R) patients, suggesting that metabolic properties had altered. Metabolomic analysis revealed that nicotinamide metabolism was elevated in relapsed LSCs, which activated amino acid metabolism and fatty acid oxidation to drive OXPHOS, and therefore avoided the cytotoxicity of ven/aza treatment [74, 75]. Recently, several research teams started to use nicotinamide phosphoribosyltransferase (NAMPT) as a new intervention. NAMPT is the rate-limiting enzyme in nicotinamide metabolism, and its application can selectively eradicate R/R LSC [74, 76]. These findings suggested that targeting metabolic weaknesses in AML cells may be an effective strategy.
Hypoxia is an important feature of the leukemia microenvironment and also an ideal target in AML therapy. In the hypoxic environment, the maintenance of neutral pH represents a key survival mechanism for leukemic cells. In the hypoxic environment, Carbonic Anhydrases IX and XII (CA IX/XII) function as transmembrane proteins that mediate intracellular pH value regulation, which is critical for leukemic cell survival, because maintaining a neutral pH represents a key survival mechanism for tumor cells in hypoxia [77]. The study by Chen et al. recently showed that dual CA IX/XII inhibitor FC531 can significantly reduce the pH level and induce apoptosis in AML cells in vitro with a synergistic effect with cytarabine. Furthermore, FC531 exhibited anti-leukemia effect in single-agent mode or in combination with cytarabine in vivo, which significantly improved the survival rate of AML mice [77]. TH-302 is a 2-nitroimidazole-linked prodrug that can be activated under hypoxia to generate potent cytotoxin bromo-isophosphoramide mustard (Br-iPM). In vitro experiments showed that TH-302 could inhibit the proliferation of AML cells and promote cell apoptosis by reducing the expression of HIF-1α. Additionally, in vivo experiments demonstrated that TH-302 could effectively prevent disease progression in AML mice and prolong mouse life expectancy [78, 79]. Moreover, TH-302 showed synergistic anti-leukemia effects in the FLT3-ITD AML model when combined with the tyrosine kinase inhibitor - Sorafenib [79]. It should be emphasized that the protection and repair of BMM are as crucial as eliminating leukemia cells. In addition to the aforementioned repair of the BM vasculature, the restoration of redox balance is also a hot topic of current concern. Theoretically, scavenging excessively high levels of ROS with antioxidants should foster the repair of BMM, thereby boosting the restoration of normal hematopoiesis. However, the use of antioxidants is not recommended clinically because malignant cells may also be protected. Several recent studies have shown that a variety of natural compounds, such as curcumin, quercetin, and resveratrol could protect normal cells from oxidative damage, exhibited excellent anti-leukemia effects, and synergized well with chemotherapeutic drugs, suggesting that natural antioxidants may have broad application prospects in clinical adjuvant therapy [80,81,82,83,84]. Notably, malignant niches are frequently coupled with inflammatory responses, however, the impact of inflammation on hematopoietic microenvironment homeostasis is poorly understood. Recently, an in vivo and in vitro study by Habbel et al. confirmed that AML cells could secrete inflammatory cytokines and activate the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathways in both AML blasts and BMSCs, thereby increasing the ROS generation and AML proliferation. Blockade of this inflammatory response signaling pathway resulted in the suppression of AML cells, suggesting that improving the hematopoietic microenvironment by inhibiting the inflammatory response is essential to amplify the anti-leukemia effect [85]. Blocking the connection between AML cells and BMM and unblocking the protection of BMM on AML cells are both critical for eliminating leukemia cells. It was demonstrated that the application of anti-CD44, anti-CD82, and anti-CD98 monoclonal antibodies could block the adhesion between the microenvironment and AML cells, thereby inhibiting the survival of AML cells [86,87,88]. The CXCL12/CXCR4 axis is an important mechanism regulating the interaction between the microenvironment and AML cells. CXCR4 inhibitors, such as plerixafor, BL-8040, and LY2510924, could disrupt the chemotaxis mediated by the CXCL12/CXCR4 axis and dislodge the leukemic cells from their protective BM niches to increase the sensitivity of leukemia cells to chemotherapy [89,90,91]. However, one major defect of inhibiting CXCL12-CXCR4 interaction is the release of a large number of leukemic cells into the peripheral blood. In that case, the potential of leukemic cells infiltrating extramedullary organs will be enhanced. At present, nearly all open-label clinical trials using CXCR4 inhibitors as chemosensitizers involve the combination of the CXCR4 inhibitors and different chemotherapeutic drugs to increase the possibility of the activated leukemic cells being killed [92, 93].
Conclusion
BMM is not only the place where LSCs are generated, but also serves as a sanctuary for these malignant cells and provides support for their clonal evolution, reinforcing its involvement in the occurrence of drug resistance and recurrence of leukemia. Drug resistance mechanisms of leukemia mediated by BMM are highly complex. Various factors, including the adhesion between the leukemic cells and the microenvironment, signal transduction, and gene expression regulation, are involved, which form a complex regulatory network. The key components of the network may be new targets for leukemia treatment. Therapies targeting the interactions between BMM and leukemic cells are new strategies in the management AML, which are also the hotspots for research on the drug resistance mechanism of leukemia. However, the existing studies have some limitations, i.e., most are in vitro studies and cannot fully mimic the BMM under pathological conditions. Neither can such studies realistically reproduce the clonal evolution of leukemia in the BMM. Therefore, it is hard to precisely depict the real inner relationship between clonal evolution, heterogeneity, and drug resistance of AML. Another urgent problem is that the selectivity of targeted drugs needs to be further improved. BMM harbors leukemic cells and normal HSCs as well. Targeted drugs with poor selectivity may adversely affect the protective effect of the BMM on HSCs or even cause direct harm to HSCs, resulting in adverse reactions such as BM suppression. Therefore, constant optimization and more efforts are necessary in these fields. It is believed that in the near future, with the emergence of more perfect leukemia models and the development of new generation of analysis technology, the law of leukemia occurrence and development will be more deeply recognized, thus facilitating the precise treatment of leukemia.
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during this study.
References
Jiang Y, Xu P, Yao D, Chen X, Dai H. CD33, CD96 and death associated protein kinase (DAPK) expression are associated with the survival rate and/or response to the chemotherapy in the patients with acute myeloid leukemia (AML). Med Sci Monit. 2017;23:1725–32.
Chen Y, Liang Y, Luo X, Hu Q. Oxidative resistance of leukemic stem cells and oxidative damage to hematopoietic stem cells under pro-oxidative therapy. Cell Death Dis. 2020;11:291.
Granéli C, Thorfve A, Ruetschi U, Brisby H, Thomsen P, Lindahl A, et al. Novel markers of osteogenic and adipogenic differentiation of human bone marrow stromal cells identified using a quantitative proteomics approach. Stem Cell Res. 2014;12:153–65.
Forte D, García-Fernández M, Sánchez-Aguilera A, Stavropoulou V, Fielding C, Martín-Pérez D, et al. Bone marrow mesenchymal stem cells support acute myeloid leukemia bioenergetics and enhance antioxidant defense and escape from chemotherapy. Cell Metab. 2020;32:829–43.e9.
Huang JC, Basu SK, Zhao X, Chien S, Fang M, Oehler VG, et al. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015;5:e302.
Kouvidi E, Stratigi A, Batsali A, Mavroudi I, Mastrodemou S, Ximeri M, et al. Cytogenetic evaluation of mesenchymal stem/stromal cells from patients with myelodysplastic syndromes at different time-points during ex vivo expansion. Leuk Res. 2016;43:24–32.
Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schümann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol. 2007;35:221–9.
Pievani A, Donsante S, Tomasoni C, Corsi A, Dazzi F, Biondi A, et al. Acute myeloid leukemia shapes the bone marrow stromal niche in vivo. Haematologica 2021;106:865–70.
Blau O, Baldus CD, Hofmann WK, Thiel G, Nolte F, Burmeister T, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011;118:5583–92.
Kim JA, Shim JS, Lee GY, Yim HW, Kim TM, Kim M, et al. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015;75:2222–31.
Weickert MT, Hecker JS, Buck MC, Schreck C, Rivière J, Schiemann M, et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci Rep. 2021;11:5944.
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010;464:852–7.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014;506:240–4.
Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006;441:475–82.
Huang L, Liu Z, Liu H, Ding K, Mi F, Xiang C, et al. Iron overload impairs bone marrow mesenchymal stromal cells from higher-risk MDS patients by regulating the ROS-related Wnt/β-catenin pathway. Stem Cells Int. 2020;2020:8855038.
Li Y, Xue Z, Dong X, Liu Q, Liu Z, Li H, et al. Mitochondrial dysfunction and oxidative stress in bone marrow stromal cells induced by daunorubicin leads to DNA damage in hematopoietic cells. Free Radic Biol Med. 2020;146:211–21.
Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43.
Cozzio A, Passegué E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35.
Zhao X, Liu HQ, Wang LN, Yang L, Liu XL. Current and emerging molecular and epigenetic disease entities in acute myeloid leukemia and a critical assessment of their therapeutic modalities. Semin Cancer Biol. 2020;S1044-579X:30246–47.
Lim HP. Integrative genomic and molecular characterisation of newly identified cooperating events in acute myeloid leukaemia with NPM1 mutation. Universität Ulm. 2021. https://doi.org/10.18725/OPARU-39194.
Kitamura T, Inoue D, Okochi-Watanabe N, Kato N, Komeno Y, Lu Y, et al. The molecular basis of myeloid malignancies. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90:389–404.
Park DJ, Kwon A, Cho BS, Kim HJ, Hwang KA, Kim M, et al. Characteristics of DNMT3A mutations in acute my eloid leukemia. Blood Res. 2020;55:17–26.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33.
Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell. 2016;30:337–48.
Xie X, Feng M, Wang Q, Wang J, Yin R, Li Y, et al. Cellular and molecular state of myeloid leukemia stem cells. Adv Exp Med Biol. 2019;1143:41–57.
Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020;367:586–90.
Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer. 2019;58:839–49.
Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica 2012;97:966–74.
Yilmaz M, Wang F, Loghavi S, Bueso-Ramos C, Gumbs C, Little L, et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019;9:7.
Zhan D, Zhang Y, Xiao P, Zheng X, Ruan M, Zhang J, et al. Whole exome sequencing identifies novel mutations of epigenetic regulators in chemorefractory pediatric acute myeloid leukemia. Leuk Res. 2018;65:20–4.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111:2548–53.
Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020;587:477–82.
Chen Y, Li J, Zhao Z. Redox control in acute lymphoblastic leukemia: From physiology to pathology and therapeutic opportunities. Cells 2021;10:1218.
Robinson AJ, Davies S, Darley RL, Tonks A. Reactive oxygen species rewires metabolic activity in acute myeloid leukemia. Front Oncol. 2021;11:632623.
Kurosawa S, Doki N, Hino Y, Sakaguchi M, Fukushima K, Shingai N, et al. Occurrence of donor cell-derived lymphoid blast crisis 24 years following related bone marrow transplantation for chronic myeloid leukemia. Intern Med. 2016;55:395–7.
Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013;122:3322–30.
Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20:158–73.
Moloney JN, Stanicka J, Cotter TG. Subcellular localization of the FLT3-ITD oncogene plays a significant role in the production of NOX- and p22phox-derived reactive oxygen species in acute myeloid leukemia. Leuk Res. 2017;52:34–42.
Maraldi T, Prata C, Vieceli Dalla Sega F, Caliceti C, Zambonin L, Fiorentini D, et al. NAD(P)H oxidase isoform Nox2 plays a prosurvival role in human leukaemia cells. Free Radic Res. 2009;43:1111–21.
Chen YF, Liu H, Luo XJ, Zhao Z, Zou ZY, Li J, et al. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit Rev Oncol Hematol. 2017;112:21–30.
Habbel J, Arnold L, Chen Y, Möllmann M, Bruderek K, Brandau S, et al. Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro. Blood Adv. 2020;4:3000–10.
Demircan MB, Mgbecheta PC, Kresinsky A, Schnoeder TM, Schröder K, Heidel FH, et al. Combined activity of the redox-modulating compound setanaxib (GKT137831) with cytotoxic agents in the killing of acute myeloid leukemia cells. Antioxidants 2022;11:513.
Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–35.
Vukovic M, Guitart AV, Sepulveda C, Villacreces A, O’Duibhir E, Panagopoulou TI, et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J Exp Med. 2015;212:2223–34.
Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 2009;113:1504–12.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64.
Li L, Zhao L, Man J, Liu B. CXCL2 benefits acute myeloid leukemia cells in hypoxia. Int J Lab Hematol. 2021;43:1085–92.
Xiao P, Sandhow L, Heshmati Y, Kondo M, Bouderlique T, Dolinska M, et al. Distinct roles of mesenchymal stem and progenitor cells during the development of acute myeloid leukemia in mice. Blood Adv. 2018;2:1480–94.
Borella G, Da Ros A, Borile G, Porcù E, Tregnago C, Benetton M, et al. Targeting the plasticity of mesenchymal stromal cells to reroute the course of acute myeloid leukemia. Blood 2021;138:557–70.
Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018;32:575–87.
Huan J, Hornick NI, Goloviznina NA, Kamimae-Lanning AN, David LL, Wilmarth PA, et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015;29:2285–95.
Scoville SD, Nalin AP, Chen L, Chen L, Zhang MH, McConnell K, et al. Human AML activates the aryl hydrocarbon receptor pathway to impair NK cell development and function. Blood 2018;132:1792–804.
Zhe N, Chen S, Zhou Z, Liu P, Lin X, Yu M, et al. HIF-1α inhibition by 2-methoxyestradiol induces cell death via activation of the mitochondrial apoptotic pathway in acute myeloid leukemia. Cancer Biol Ther. 2016;17:625–34.
Wang B, Wang X, Hou D, Huang Q, Zhan W, Chen C, et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J Cell Physiol. 2019;234:10602–14.
Gomei Y, Nakamura Y, Yoshihara H, Hosokawa K, Iwasaki H, Suda T, et al. Functional differences between two Tie2 ligands, angiopoietin-1 and -2, in regulation of adult bone marrow hematopoietic stem cells. Exp Hematol. 2010;38:82–9.
Schliemann C, Bieker R, Padro T, Kessler T, Hintelmann H, Buchner T, et al. Expression of angiopoietins and their receptor Tie2 in the bone marrow of patients with acute myeloid leukemia. Haematologica 2006;91:1203–11.
Shirzad R, Shahrabi S, Ahmadzadeh A, Kampen KR, Shahjahani M, Saki N. Signaling and molecular basis of bone marrow niche angiogenesis in leukemia. Clin Transl Oncol. 2016;18:957–71.
Reikvam H, Hatfield KJ, Lassalle P, Kittang AO, Ersvaer E, Bruserud Ø. Targeting the angiopoietin (Ang)/Tie-2 pathway in the crosstalk between acute myeloid leukaemia and endothelial cells: Studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-1 release. Expert Opin Investig Drugs. 2010;19:169–83.
Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell. 2017;32:324–41.e6.
Tabe Y, Saitoh K, Yang H, Sekihara K, Yamatani K, Ruvolo V, et al. Inhibition of FAO in AML co-cultured with BM adipocytes: Mechanisms of survival and chemosensitization to cytarabine. Sci Rep. 2018;8:16837.
Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017;129:1320–32.
Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015;126:2404–14.
Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017;130:1649–60.
Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–26.
Brault J, Vigne B, Meunier M, Beaumel S, Mollin M, Park S, et al. NOX4 is the main NADPH oxidase involved in the early stages of hematopoietic differentiation from human induced pluripotent stem cells. Free Radic Biol Med. 2020;146:107–18.
Germon Z P, Sillar J R, Mannan A, et al. Blockade of redox second messengers inhibits JAK/STAT and MEK/ERK signaling sensitizing FLT3-mutant acute myeloid leukemia to targeted therapies. Preprint at bioRxiv https://doi.org/10.1101/2022.03.09.483687 (2022).
Demircan MB, Mgbecheta PC, Kresinsky A, Schnoeder TM, Schröder K, Heidel FH, et al. Combined activity of the redox-modulating compound setanaxib (GKT137831) with cytotoxic agents in the killing of acute myeloid leukemia cells. Antioxidants 2022;11:513.
Al-Aamri HM, Irving HR, Bradley C, Meehan-Andrews T. Intrinsic and extrinsic apoptosis responses in leukaemia cells following daunorubicin treatment. BMC Cancer. 2021;21:438.
Wang X, Dawod A, Nachliely M, Harrison JS, Danilenko M, Studzinski GP. Differentiation agents increase the potential AraC therapy of AML by reactivating cell death pathways without enhancing ROS generation. J Cell Physiol. 2020;235:573–86.
Li L, Liu W, Sun Q, Zhu H, Hong M, Qian S. Decitabine downregulates TIGAR to induce apoptosis and autophagy in myeloid leukemia cells. Oxid Med Cell Longev. 2021;2021:8877460.
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37.
Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724–40.e4.
Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24:1859–66.
Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, et al. Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell. 2020;27:748–64.e4.
Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. 2020;1:1176–87.
Subedi A, Liu Q, Ayyathan DM, Sharon D, Cathelin S, Hosseini M, et al. Nicotinamide phosphoribosyltransferase inhibitors selectively induce apoptosis of AML stem cells by disrupting lipid homeostasis. Cell Stem Cell. 2021;28:1851–67.e8.
Chen F, Licarete E, Wu X, Petrusca D, Maguire C, Jacobsen M, et al. Pharmacological inhibition of Carbonic Anhydrase IX and XII to enhance targeting of acute myeloid leukaemia cells under hypoxic conditions. J Cell Mol Med. 2021;25:11039–52.
Portwood S, Lal D, Hsu YC, Vargas R, Johnson MK, Wetzler M, et al. Activity of the hypoxia-activated prodrug, TH-302, in preclinical human acute myeloid leukemia models. Clin Cancer Res. 2013;19:6506–19.
Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H. et al. Hypoxia-activated prodrug TH-302 targets hypoxic bone marrow niches in preclinical leukemia models. Clin Cancer Res. 2016;22:1687–98.
Papiez MA, Krzyściak W. The dual effect of curcumin on etoposide action in leukemic and healthy bone marrow cells of rats with acute myeloid leukemia. Folia Med Cracov. 2014;54:71–9.
Papież MA. The effect of quercetin on oxidative DNA damage and myelosuppression induced by etoposide in bone marrow cells of rats. Acta Biochim Pol. 2014;61:7–11.
Shah K, Mirza S, Desai U, Jain N, Rawal R. Synergism of curcumin and cytarabine in the down regulation of multi-drug resistance genes in acute myeloid leukemia. Anticancer Agents Med Chem. 2016;16:128–35.
Naimi A, Entezari A, Hagh MF, Hassanzadeh A, Saraei R, Solali S. Quercetin sensitizes human myeloid leukemia KG-1 cells against TRAIL-induced apoptosis. J Cell Physiol. 2019;234:13233–41.
Ivanova D, Zhelev Z, Semkova S, Aoki I, Bakalova R. Resveratrol modulates the redox-status and cytotoxicity of anticancer drugs by sensitizing leukemic lymphocytes and protecting normal lymphocytes. Anticancer Res. 2019;39:3745–55.
Habbel J, Arnold L, Chen Y, Möllmann M, Bruderek K, Brandau S, et al. Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro. Blood Adv. 2020;4:3000–10.
Bajaj J, Konuma T, Lytle NK, Kwon HY, Ablack JN, Cantor JM, et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell. 2016;30:792–805.
Orian-Rousseau V. CD44 acts as a signaling platform controlling tumor progression and metastasis. Front Immunol. 2015;6:154.
Nishioka C, Ikezoe T, Yokoyama A. Blockade of CD 82 by a monoclonal antibody potentiates anti‐leukemia effects of AraC in vivo. Cancer Med. 2015;4:1426–31.
Sison EA, McIntyre E, Magoon D, Brown P. Dynamic chemotherapy-induced upregulation of CXCR4 expression: A mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res. 2013;11:1004–16.
Borthakur G, Ofran Y, Nagler A, Rowe JM, Foran JM, Uy GL, et al. The peptidic CXCR4 antagonist, BL-8040, significantly reduces bone marrow immature leukemia progenitors by inducing differentiation, apoptosis, and mobilization: Results of the dose escalation clinical trial in acute myeloid leukemia. Blood. 2015;126:2546.
Cho BS, Zeng Z, Mu H, Wang Z, Konoplev S, McQueen T, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood. 2015;126:222–32.
Cooper TM, Sison EAR, Baker SD, Li L, Ahmed A, Trippett T, et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03). Pediatr Blood Cancer. 2017. https://doi.org/10.1002/pbc.26414.
Martínez-Cuadrón D, Boluda B, Martínez P, Bergua J, Rodríguez-Veiga R, Esteve J, et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann Hematol. 2018;97:763–72.
Amaya ML, Inguva A, Pei S, Jones C, Krug A, Ye H, et al. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood 2022;139:584–96.
Nieborowska-Skorska M, Kopinski PK, Ray R, Hoser G, Ngaba D, Flis S, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012;119:4253–63.
Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 2008;111:3173–82.
Pua KH, Chew CL, Lane DP, Tergaonkar V. Inflammation-associated genomic instability in cancer. Genome Instab Dis. 2020;1:1–9.
Yu DH, Chen C, Liu XP, Yao J, Li S, Ruan XL. Dysregulation of miR-138-5p/RPS6KA1-AP2M1 is associated with poor prognosis in AML. Front Cell Dev Biol. 2021;9:641629.
Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–65.
Rashidi A, Uy GL. Targeting the microenvironment in acute myeloid leukemia. Curr Hematol Malig Rep. 2015;10:126–31.
Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 2020;11:2042.
Takam Kamga P, Bassi G, Cassaro A, Midolo M, Di Trapani M, Gatti A, et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016;7:21713–27.
Ayyadurai VAS, Deonikar P, McLure KG, Sakamoto KM. Molecular systems architecture of interactome in the acute myeloid leukemia microenvironment. Cancers 2022;14:756.
Venney D, Mohd-Sarip A, Mills KI. The impact of epigenetic modifications in myeloid malignancies. Int J Mol Sci. 2021;22:5013.
Hamidi T, Singh AK, Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015;7:247–65.
Wang J, He N, Wang R, Tian T, Han F, Zhong C, et al. Analysis of TET2 and EZH2 gene functions in chromosome instability in acute myeloid leukemia. Sci Rep. 2020;10:2706.
Wright RL, Vaughan AT. A systematic description of MLL fusion gene formation. Crit Rev Oncol Hematol. 2014;91:283–91.
Pastore F, Levine RL. Epigenetic regulators and their impact on therapy in acute myeloid leukemia. Haematologica 2016;101:269–78.
Adema V, Colla S. EZH2 inhibitors: The unpacking revolution. Cancer Res. 2022;82:359–61.
Lamble AJ, Gerbing RB, Smith JL, Ries RE, Kolb EA, Alonzo TA, et al. Crebbp alterations are associated with a poor prognosis in De Novo AML. Blood 2021;138:3451.
Gutierrez SE, Romero-Oliva FA. Epigenetic changes: A common theme in acute myelogenous leukemogenesis. J Hematol Oncol. 2013;6:57.
Castelli G, Pelosi E, Testa U. Targeting histone methyltransferase and demethylase in acute myeloid leukemia therapy. Onco Targets Ther. 2017;11:131–55.
San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers 2019;11:1794.
Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br J Cancer. 2016;114:605–11.
Wang L, Birch NW, Zhao Z, Nestler CM, Kazmer A, Shilati A, et al. Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat Cancer. 2021;2:515–26.
Acknowledgements
This research was supported by the scientific research project of the Sichuan Province Education Department (No. 16ZA0241), the National Natural Science Foundation of China [82060268], and the Guangxi Natural Science Foundation of China [2020JJA140124].
Author information
Authors and Affiliations
Contributions
Conceptualization, YC and JL; writing—original draft preparation, YC and JL; writing—review and editing, LX, ZZ, and M-AG; prepared figures: YC and JL; supervision, YC and M-AG All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Li, J., Xu, L. et al. The genesis and evolution of acute myeloid leukemia stem cells in the microenvironment: From biology to therapeutic targeting. Cell Death Discov. 8, 397 (2022). https://doi.org/10.1038/s41420-022-01193-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-01193-0
- Springer Nature Limited
This article is cited by
-
Crosstalk between autophagy and metabolism: implications for cell survival in acute myeloid leukemia
Cell Death Discovery (2024)
-
GNF-7, a novel FLT3 inhibitor, overcomes drug resistance for the treatment of FLT3‑ITD acute myeloid leukemia
Cancer Cell International (2023)
-
Spatiotemporal evolution of AML immune microenvironment remodeling and RNF149-driven drug resistance through single-cell multidimensional analysis
Journal of Translational Medicine (2023)
-
NADPH oxidase mediated oxidative stress signaling in FLT3-ITD acute myeloid leukemia
Cell Death Discovery (2023)