
Abstract
Transcriptional super-enhancers and the BET bromodomain protein BRD4 are emerging as critical drivers of tumorigenesis and therapeutic targets. Characterized by substantial accumulation of histone H3 lysine 27 acetylation (H3K27ac) signals at the loci of cell identity genes and critical oncogenes, super-enhancers are recognized, bound and activated by BRD4, resulting in considerable oncogene over-expression, malignant transformation, cancer cell proliferation, survival, tumor initiation and progression. Small molecule compound BRD4 BD1 and BD2 bromodomain inhibitors block BRD4 binding to super-enhancers, suppress oncogene transcription and expression, reduce cancer cell proliferation and survival, and repress tumor progression in a variety of cancer types. Like other targeted therapy agents, BRD4 inhibitors show moderate anticancer effects on their own, and exert synergistic anticancer effects in vitro and in preclinical models, when combined with other anticancer agents including CDK7 inhibitors, CBP/p300 inhibitors and histone deacetylase inhibitors. More recently, BRD4 BD2 bromodomain selective inhibitors, proteolysis-targeting chimera (PROTAC) BRD4 protein degraders, and dual BRD4 and CBP/p300 bromodomain co-inhibitors have been developed and shown better anticancer efficacy and/or safety profile. Importantly, more than a dozen BRD4 inhibitors have entered clinical trials in patients with cancer of various organ origins. In summary, super-enhancers and their reader BRD4 are critical tumorigenic drivers, and BRD4 BD1 and BD2 bromodomain inhibitors, BRD4 BD2 bromodomain selective inhibitors, PROTAC BRD4 protein degraders, and dual BRD4 and CBP/p300 bromodomain co-inhibitors are promising novel anticancer agents for clinical translation.
Similar content being viewed by others

Introducton
Transcriptional enhancers are short regulatory DNA elements which bind RNA polymerase II (RNA Pol II), transcription factors and co-regulators, and are characterized by acetylated histone H3 lysine 27 (H3K27ac) and monomethylated H3K4 (H3K4me) signals in chromatin immunoprecipitation sequencing assays [1]. As enhancers can form loops with promoters over a long distance, enhancers augment the transcription of neighboring genes, irrespective of the sense or antisense direction of their target genes [2, 3].
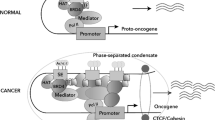
Super-enhancers are large clusters of enhancers that are in close genomic proximity, are densely bound by the BET bromodomain protein BRD4 and master transcription factors, and are characterized by massive H3K27ac and H3K4me signals in ChIP sequencing [4,5,6].
Enhancers activate gene transcription and induce tumorigenesis
Transcriptional enhancers recruit BRD4, transcription factors and cofactors to activate RNA Pol II and gene transcription from gene promoters [7] (Fig. 1A). Transcriptional enhancers have been confirmed to play an important role in the activation and over-expression of oncogenes, such as MYC which is juxtaposed to the immunoglobulin heavy-chain gene enhancer in Burkitt’s lymphoma [8].

A, B The BET bromodomain protein BRD4 recognizes acetylated (Ac) histone H3 lysine 27, binds to and activates enhancers (A) and super-enhancers (B). BRD4 recruits the positive transcription elongation factor b (P-TEFb) and Mediator, leading to RNA Polymerase II (RNA Pol II) activation and binding to enhancer- and super-enhancer-associated gene promoter, transcriptional activation and target gene over-expression. As super-enhancers are bound by much larger clusters of BRD4 proteins, super-enhancer-associated oncogenes are transcribed at substantially higher levels than enhancer-associated genes.
The Hippo pathway transcription coactivators YAP/TAZ form a protein complex with TEAD and AP-1 at distal transcriptional enhancers rather than promoters, located >100,000 base pairs away from transcription start sties. Through chromatin looping, the YAP/TAZ/TEAD/AP-1 transcription cofactor and transcription factor complex activate the transcription of enhancer-associated genes including those controlling S-phase entry and mitosis of the cell cycle, resulting in cell proliferation and skin tumorigenesis [9]. The oncogenic transcription factor FOXA1 is hyperactive in metastatic endocrine-resistant breast cancer cells due to gene amplification or overexpression. FOXA1 induces enhancer reprogramming and transcriptional activation of pro-metastatic oncogenes in endocrine-resistant breast cancer cells [10].
The transcriptional activator NRF2 is frequently activated in non-small cell lung cancer, and NRF2 overexpression results in the accumulation of CCAAT Enhancer Binding Protein Beta (CEBPB) [11]. NRF2 and CEBPB co-operatively induce the establishment of transcriptional enhancers at the loci of oncogenes such as the NOTCH3 gene [11]. Importantly, in mouse models of non-small cell lung cancer, disruption of the NOTCH3 enhancer significantly suppresses tumor progression and augments the anticancer effects of cisplatin, demonstrating the important role of the NOTCH3 enhancer in tumorigenesis and drug resistance [11].
Recent transcriptome profiling has shown that squamous cell lineage markers are present in ~25% of pancreatic ductal adenocarcinoma tumors, and the squamous cell subtype is associated with poorer patient prognosis [12]. Aberrant enhancers have recently been found to be established in the squamous cell subtype of pancreatic ductal adenocarcinoma tumors. Enhancers at the loci of oncogenes, such as MYC and HRAS, play a critical role in pancreatic ductal adenocarcinoma cell transition into squamous cells, cell migration and invasion in vitro, and accelerated tumor growth and metastases in vivo [13].
Enhancers can also activate tumor suppressor gene transcription and suppress tumorigenesis
Enhancers can also activate tumor suppressor gene transcription and thereby suppress tumorigenesis. The N-terminal SNAG domain of the transcriptional repressor GFI1 binds to the CoREST transcriptional complex proteins LSD1 and RCOR1 at the enhancers of transcription factor genes, such as SPI1 (PU.1), CEBPA and IRF8 which are important for acute myeloid leukemia cell differentiation [14]. GFI1 inactivation or LSD1 inhibition with small molecule compound inhibitors disrupts the interaction between GFI1, LSD1 and RCOR1, leading to considerable increase in H3K27ac at enhancer regions of the transcription factor genes, transcriptional activation, acute myeloid leukemia cell differentiation, growth inhibition and clonogenicity reduction [14].
SWI/SNF (mSWI/SNF or BAF) chromatin remodeling complex inactivation contributes to >20% of human cancers. Forced over-expression of the core BAF complex subunit SMARCB1 in sarcoma cells results in the activation of distal typical enhancers and super-enhancers at the loci of genes such as CDKN1A [15]. The activated typical enhancers and super-enhancers play critical roles in sarcoma cell growth arrest, demonstrating a tumor suppression effect [15]. Therefore, enhancers can induce or suppress tumorigenesis, probably depending on cancer subtypes and cellular contexts.
Super-enhancers activate oncogene transcription and induce tumorigenesis
Super-enhancers consist of enhancer clusters, span large genetic regions, and are generally an order of magnitude larger than typical enhancers [4, 5]. Super-enhancers are bound by a large number of BRD4 which recruits the Mediator, a protein complex connecting the transcription factors at the super-enhancers and RNA pol II at the gene promoters (Fig. 1B) [4, 5].
Super-enhancers are emerging as critical regulators of oncogene transcription and tumorigenesis. In glioblastoma cells, super-enhancers have been found to be associated with a number of oncogenic genes, such as RUNX1, BCL3 and FOSL2 [6]. In glioblastoma stem cells isolated from PDX mouse models originally derived from human tumor samples, a subset of super-enhancers at the loci of critical genes, such as CDK6, SOX2, EGFR and BRD4, are shared by the majority of human glioblastoma stem cells [16]. Proximity of the super-enhancers to their associated genes correlates with gene over-expression in glioblastoma stem cells and human tumor samples, and the core glioblastoma stem cell super-enhancer-associated genes are essential for glioblastoma cell proliferation and tumorigenesis [16] (Table 1). In addition, patients with glioblastoma that is enriched of the core glioblastoma stem cell super-enhancer signature show more advanced tumor stage and poorer prognosis [16].
Super-enhancers are extensively reprogrammed during liver cancer tumorigenesis [17]. Liver cancer cells acquire super-enhancers at the loci of critical oncogenic genes, such as SPHK1, MYC, MYCN, SHH and YAP1, to drive their substantial over-expression. The super-enhancer “writer” p300, super-enhancer “reader” BRD4, and super-enhancer activity regulators CDK7 and MED1 are often over-expressed in human liver cancer tissues, and their over-expression predicts poor patient prognosis [17]. Importantly, inhibition of p300, BRD4, CDK7 or MED1 reduces the expression of super-enhancer-associated oncogenes and exerts anticancer effects against liver cancer [17].
The histone demethylase KDM6A gene is often mutated in a variety of human malignancies. Loss of function of KDM6A causes squamous-like metastatic pancreatic cancer through aberrant activation of super-enhancers at the loci of MYC and RUNX3 oncogenes and consequent MYC and RUNX3 over-expression [18]. Treatment with BRD4 inhibitors results in KDM6A mutant pancreatic cancer cell differentiation and tumor growth inhibition in a mouse model [18] (Table 1).
The super-enhancer landscape of small cell lung cancer cells recapitulates embryonic, neural and tumorigenic signatures, as many super-enhancers are associated with lineage-specific transcription factor genes and oncogenes such as MYC, SOX2 and NFIB [19] (Table 1). In a high-throughput compound screening, small cell lung cancer cells have been found to be very sensitive to the CDK7 inhibitor THZ1 which selectively suppresses the expression of super-enhancer associated genes [19].
In chromosome 17q-gained neuroblastoma, the JMJD6 gene is over-expressed due to both gene gain and transcriptional super-enhancers, and suppression of super-enhancer activity reduces JMJD6 gene expression, neuroblastoma cell proliferation in vitro and tumor growth in a mouse model [20] (Table 1). Similarly, in diffuse intrinsic pontine glioma, the expression of critical oncogenic genes such as SOX2 and NOTCH1 is regulated by super-enhancers, and treatment with super-enhancer inhibitors reduces diffuse intrinsic pontine glioma cell proliferation in vitro and tumor progression in mouse models [21] (Table 1).
In clear cell renal cell carcinoma, super-enhancers are formed at the loci of CXC chemokine genes, such as CXCL1, CXCL5 and CXCL8, and induce CXC chemokine gene over-expression and renal cell carcinoma progression and metastasis [22]. Consistent with these findings, suppression of super-enhancer activity reduces CXC chemokine gene expression and renal cell cancer metastasis [22] (Table 1).
Compared with normal counterparts, colon cancer cells gain oncogenic super-enhancers, including super-enhancers associated with ASCL2, a transcription factor for intestinal stem cell fate, and the Wnt target gene MYC [23]. In addition, β-catenin and CTCF up-regulate MYC by connecting nucleoporins to oncogenic super-enhancers, leading to MYC mRNA export to the cytoplasm, stabilization and over-expression [24, 25]. Interestingly, inflammation in the tumor microenvironment results in the formation of super-enhancers at the PDZK1IP1 gene locus, resulting in colon cancer cell proliferation in vitro and tumor progression in a mouse model [23] (Table 1).
Medulloblastoma are divided into 4 distinct groups, WNT, SHH, Group 3, and Group 4 groups, and the 4 different groups show distinct super-enhancer profiles. Association of critical oncogenes with super-enhancers has been found at the ALK gene locus in WNT group, at SMO and NTRK3 gene loci in SHH group, at the LMO1, LMO2 and MYC gene loci in Group 3, and at the ETV4 and PAX5 gene loci in Group 4 [26] (Table 1).
In leukemic stem cells, the MYC gene locus is characterized by super-enhancers which recruit critical transcriptional factors including MYB, RUNX1 and GFI1b to drive MYC over-expression and leukemogenesis [27]. In chronic myelogenous leukemia stem cells, suppression of super-enhancer-driven gene transcription by a CDK7 inhibitor eradicates leukemia stem cells in a mouse model without effects in normal hematopoietic stem cells [28]. In human primary T cell acute lymphoblastic leukemia samples, a topologically associating domain ‘fusion’ event due to CTCF-mediated insulation absence results in the interaction between distal super-enhancers and the MYC gene promoter, leading to MYC over-expression and leukemogenesis [29] (Table 1).
Super-enhancers have also been shown to be important in epithelial-to-mesenchymal transition (EMT) and metastasis. ETS2, JUNB, EGFR and HNF4A genes are associated with super-enhancers in non-small cell lung cancer cells. Suppression of super-enhancer activity reduces the expression of these super-enhancer-associated genes, decreases non-small cell lung cancer cell migration and invasion, and abrogates TGF-β-induced EMT, demonstrating the role of super-enhancers in regulating EMT and tumor metastasis [30].
Super-enhancers can function as tumor suppressors
While generally proven to promote tumor initiation and progression, super-enhancers can also function as tumor suppressors. The histone methyltransferase KMT2D is often inactivated in human lung cancer tissues. Loss of KMT2D reduces the activity of super-enhancers at critical genes, such as the circadian rhythm repressor Per2, resulting in Per2 gene down-regulation, glycolysis and lung cancer tumorigenesis [31]. In breast cancer, loss of the tumor suppressor gene RCAN1.4 augments tumor metastasis. Unexpectedly, RCAN1.4 gene expression is driven by super-enhancers in breast cancer cells, and suppression of super-enhancer activity with BRD4 knockdown or BRD4 inhibitor treatment reduces RCAN1.4 tumor suppressor gene expression [32].
The super-enhancer “reader” BRD4 forms a protein complex with the repressive LSD1/NuRD transcription regulators at super-enhancers to suppress the expression of drug resistance genes in breast cancer cells [33]. Repression of super-enhancer activity with BRD4 inhibitors does not have an immediate effect on the expression of the drug resistance genes, however, long-time treatment with BRD4 inhibitors causes resistance to both BRD4 inhibitors and a broad spectrum of anticancer agents, demonstrating the role of super-enhancers and BRD4 in super-enhancer-mediated transcriptional repression of genes involved in tumorigenesis and chemoresistance [33]. Therefore, long-term treatment with BRD4 inhibitors might promote multidrug resistance and tumor progression, and close monitoring and prompt intervention are required in clinical trials.
The super-enhancer “reader” Brd4 promotes super-enhancer-associated oncogene transcription and tumorigenesis and Brd4 inhibitors exert anticancer effects
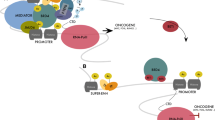
The BET bromodomain protein BRD4 recognizes, binds to and activates super-enhancers and substantially up-regulate the expression of super-enhancer-associated oncogenes (Fig. 1B), and BRD4 inhibitors blocks BRD4 binding and reduce oncogene expression (Fig. 2). In diffuse large B cell lymphoma, approximately one-third of BRD4 protein localizes to super-enhancers which occupy ~1.6% of genes [34]. Treatment with four different BRD4 inhibitors reduces the expression of super-enhancer-associated oncogenes, such as MYC, E2F1, BCL6 and PAX5, and reduces diffuse large B cell lymphoma cell proliferation. Treatment of mice xenografted with diffuse large B cell lymphoma with the BRD4 inhibitor JQ1 suppresses lymphoma progression [34] (Table 1).

The BET bromodomain protein BRD4 recognizes acetylated (Ac) histone H3 lysine 27, binds to and activates super-enhancers, leading to RNA Polymerase II (RNA Pol II) binding to super-enhancer-associated oncogene promoter, gene transcriptional activation and over-expression. Treatment with BRD4 inhibitors displaces BRD4 at super-enhancers, leading to RNA Pol II disassociation from gene promoters and transcriptional suppression.
In estrogen receptor alpha (ERα)-positive breast cancer cells, BRD4 is a master activator of ERα-occupied super-enhancers and the transcription of ERα target genes, such as RET which in turn activates ERα phosphorylation and ERα target gene expression. BRD4 therefore induces breast cancer cell proliferation and tumor progression [35] (Table 1).
In human neck squamous cell carcinoma, BRD4 recruits Mediators and NF-κB at super-enhancers associated with cancer stemness genes such as MET, TP63 and FOSL1. Treatment with BRD4 inhibitors reduces stemness gene expression; suppresses cancer stem cell self-renewal, invasive growth and metastasis; and eliminates tumor cells and cancer stem cells in a mouse model of neck squamous cell carcinoma [36]. In nasopharyngeal carcinoma cells, super-enhancers are enriched of BRD4, NF-κB, IRF1 and IRF2 transcription factors at the loci of critical oncogenes such as ETV6, high expression of which in human nasopharyngeal carcinoma tissues is correlated with poor patient prognosis [37]. Treatment with the BRD4 inhibitor JQ1 significantly suppresses super-enhancer-associated ETV6 gene expression and induces nasopharyngeal carcinoma cell growth inhibition [37] (Table 1).
In diffuse intrinsic pontine glioma, super-enhancers are found at the loci of a number of genes indicating undifferentiation status such as SOX2 and NES as well as oncogenes such EGFR [21]. These super-enhancers are characterized by BRD4 binding, and BRD4 knockdown or inhibition reduces diffuse intrinsic pontine glioma cell proliferation in vitro and tumor progression in mouse models [21] (Table 1).
In rhabdomyosarcoma, super-enhancers are bound by core regulatory transcription factors and are characterized by the highest levels of histone acetylation [38]. Counterintuitively, the super-enhancers are also bound by the most histone deacetylases (HDACs), and HDAC inhibitors augment BRD4, but decreases RNA Pol II and core regulatory transcription factor, binding to the super-enhancers. The data demonstrate super-enhancer-specific requirement to balance histone acetylation and deacetylation for maintaining super-enhancer architecture and gene transcription [38]. In alveolar rhabdomyosarcoma, the chimeric transcription factor PAX3-FOXO1 interacts with the master transcription factors MYCN, MYOG and BRD4 at target gene super-enhancers, resulting in over-expression of SOX8, MYOD1, MYOG and MYCN, alveolar rhabdomyosarcoma tumorigenesis and dependence on BRD4 [39]. Inhibition of BRD4 with the BRD4 inhibitor JQ1 or OTX015 abolishes PAX3-FOXO1 function, suppresses alveolar rhabdomyosarcoma cell proliferation in vitro and induces tumor growth inhibition in mouse models [39] (Table 1).
Melanoma with PGC-1α over-expression is characterized by substantial BRD4 protein binding at the PGC-1α gene super-enhancer [40]. Treatment with the BRD4 inhibitor JQ1 or BAY 1238097 blocks BRD4 binding to the super-enhancer and PGC-1α expression, suppresses melanoma cell proliferation in vitro, and inhibits tumor growth in a mouse model [40] (Table 1).
In multiple myeloma, BRD4 and Mediator are enriched at super-enhancers associated with oncogenes including MYC, BCL-xL and IRF4. Treatment of multiple myeloma cells with the BRD4 inhibitor JQ1 results in BRD4 disassociation from super-enhancers, and reduction in MYC, BCL-xL and IRF4 gene expression and multiple myeloma cell proliferation [6]. In t(4;14)-positive multiple myeloma, BRD4 interacts with the histone lysine methyltransferase NSD2 at the HJURP gene super-enhancers, leading to HJURP gene over-expression, multiple myeloma cell proliferation and survival [41] (Table 1). Taken together, BRD4 promotes super-enhancer-associated oncogene transcription and tumorigenesis, and BRD4 inhibitors exert anticancer effects.
Small molecule compound Brd4 inhibitors and degraders exert promising anticancer effects in pre-clinical models
Small molecule compound BRD4 BD1 and BD2 bromodomain inhibitors in cancer therapy
In the past decade, a number of small molecule compound BRD4 inhibitors have been developed through chemical synthesis, structure-based in silico screen, and wet lab screen of small molecule compound libraries. The majority of the inhibitors, such as JQ1, OTX015, I-BET762, MK-8628, NHWD870, ABBV-744, PLX2853 and INCB054329, target both the BD1 and BD2 bromodomains of BRD4, reduce oncogene expression, and exert anticancer effects in pre-clinical models.
Pancreatic ductal adenocarcinoma, head and neck squamous cell carcinoma and leukemia are characterized by oncogene over-expression due to super-enhancers. Combination therapy with the BRD4 inhibitor JQ1 and the CDK7 inhibitor THZ1 synergistically reduces super-enhancer-associated oncogene expression and exerts synergistical anticancer effects against pancreatic ductal adenocarcinoma and head and neck squamous cell carcinoma in vitro and in mouse models [42, 43]. Interestingly, combination therapy with BRD4 inhibitors and CDK7 inhibitors overcomes resistance to BRD4 inhibitor therapy in leukemia cells and mouse models of leukemia [44]; and nanoparticle-mediated delivery of JQ1 and THZ1, compared with free drug formulation, considerably reduces cytotoxicity to liver cells but synergistically suppresses tumor progression in a mouse model of drug-resistant pancreatic ductal adenocarcinoma [42].
CDK4/CDK6 inhibitors have also been shown to exert synergistic anticancer effects with BRD4 inhibitors in castration-resistant prostate cancer and NUT midline carcinoma [45, 46]. Castration-resistant prostate cancer cells with high levels of the deubiquitinase DUB3 and NUT midline carcinoma cells with high levels of KLF4 are resistant to BRD4 inhibitors, because DUB3 binds to BRD4 and augments its deubiquitination and stabilization and KLF4 up-regulates E2F and MYC gene expression [45, 46]. As DUB3 is activated after phosphorylation by CDK4 and CDK6 and E2F and MYC expression are activated after Rb phosphorylation by CDK4 and CDK6, treatment with the CDK4/CDK6 inhibitor Palbociclib sensitizes prostate cancer and NUT midline carcinoma cells to the BRD4 inhibitor JQ1, and exerts synergistic anticancer effects with JQ1 in vitro and in mouse models of castration-resistant prostate cancer and NUT midline carcinoma [45, 46].
In a high-throughput drug screen, BRD4 inhibitors have been found to be one of the two classes of compounds exerting the best synergistic anticancer effects with the CDK4/CDK6 inhibitor Ribociclib in medulloblastoma cells [47]. A reverse combination drug screen identifies CDK4/CDK6 inhibitors as the compounds exerting the best synergy with the BRD4 inhibitor JQ1 against medulloblastoma cells [47]. Treatment with the orally bioavailable BRD4 inhibitor MK-8628 suppresses medulloblastoma cell proliferation and induces apoptosis by reducing MYC expression, and MK-8628 suppresses medulloblastoma tumor progression in preclinical models [48]. Co-treatment with MK-8628 and the PLK1 inhibitor Volasertib, which targets MYC protein for degradation, shows synergistic anti-medulloblastoma effects in vitro and in preclinical models [48].
Another well-studied anticancer agent for BRD4 inhibitor combination therapy is HDAC inhibitors, particularly the pan-HDAC inhibitor Panobinostat. Combination therapy with the BRD4 inhibitor JQ1 or OTX015 and Panobinostat synergistically reduces the expression of oncogenes, such as MYC, MYCN and LIN28B; suppresses proliferation and induces apoptosis in MYCN gene-amplified neuroblastoma, medulloblastoma and diffuse intrinsic pontine glioma cells; and significantly suppresses neuroblastoma and medulloblastoma tumor progression in mouse models [21, 49, 50]. In neuroblastoma due to TERT gene rearrangement with super-enhancers, BRD4 is required for TERT gene transcription and neuroblastoma cell proliferation [51]. In an unbiased screen of approved oncology drugs, the BRD4 inhibitors I-BET762 and OTX015 exert the best synergistic anticancer effects with the proteasome inhibitor Carfilzomib; and OTX015 and carfilzomib synergistically reduce TERT expression, induces TERT gene-rearranged neuroblastoma cell apoptosis, blocks tumor progression and improves survival in multiple mouse models of TERT gene-rearranged neuroblastoma [51].
Unbiased high-throughput drug combination screens reveal that PI3K-AKT-mTOR pathway inhibitors exert synergistic anticancer effects with BRD4 inhibitors against small cell lung cancer cells, and mTOR inhibitors exhibit the best synergy [52]. Mechanistically, while BRD4 inhibitors up-regulate RSK3 to activate the mTOR pathway, mTOR inhibitors block this cell survival signaling and enhance BRD4 inhibitor-mediated cancer cell apoptosis [52]. In multiple patient-derived xenograft models of small cell lung cancer, combination therapy with the mTOR inhibitor Everolimus and the BRD4 inhibitor NHWD870 synergistically induce cancer cell apoptosis and blocks tumor progression without significantly increasing toxicity to normal tissues in mice [52]. In Ewing sarcoma cell lines and patient-derived xenograft (PDX) lines, AKT pathway activation protects Ewing sarcoma cells against BRD4 inhibitors, and IGF1R inhibitors and mTOR inhibitors suppress AKT pathway activation and synergistically enhance cancer cell sensitivity to BRD4 inhibitors [53]. In PDX models of Ewing sarcoma, treatment with the BRD4 inhibitor NHWD870 and the IGF1R inhibitor BMS754807 results in substantial and durable anticancer effects, while monotherapy was much less effective [53].
Genome-wide loss-of-function clustered regularly interspaced short palindromic repeats (CRISPR) screens identify SPOP gene deficiency as a resistance factor to BRD4 inhibitor therapy in KMT2A gene-rearranged leukemia cells [54]. Kinase vulnerability CRISPR screens identify GSK3 inhibitors as effective agents to overcome SPOP deficiency-induced BRD4 inhibitor resistance. Combination therapy with the BRD4 inhibitor ABBV-744 and the GSK3 inhibitor CHIR-98014 considerably suppresses KMT2A-rearranged leukemia progression in patient-derived xenograft models in mice, confirming ABBV-744 and CHIR-98014 combination therapy as an effective therapeutic strategy [54]. Since it is now clear that targeted therapies need to be combined with other anticancer agents in the clinic to exert better anticancer effects and to reduce toxicity, the other anticancer agents should be identified by unbiased screening of anticancer drug libraries for each cancer subtype.
Small molecule compound BRD4 BD2 selective bromodomain inhibitors in cancer therapy
While the majority of BRD4 inhibitors bind to the BD1 and BD2 bromodomains of BRD4 with similar affinities, the small molecule compound ABBV-744 selectively binds to the BD2 bromodomain [55]. By selectively suppressing the BD2 bromodomain, ABBV-744 induces acute myeloid leukemia and prostate cancer cell growth inhibition, and exhibits significant anticancer effects against acute myeloid leukemia and prostate cancer in mouse models with better toxicity profile and therapeutic index than BRD4 BD1 and BD2 bromodomain inhibitors [55, 56]. In addition, while the BRD4 inhibitors PLX2853 and INCB054329 show synergistic anticancer effects when combined with the BCL2 inhibitor Venetoclax in mouse models of diffuse large B-cell lymphoma and acute myeloid leukemia, ABBV-744 also exerts synergistic anticancer effects with Venetoclax in mouse models of acute myeloid leukemia (Table 2) [56, 57]. Interestingly, GSK620, another small molecule compound BRD4 BD2 bromodomain selective inhibitor, suppresses inflammatory disease in pre-clinical models (Table 2) [58].
Small molecule compound proteolysis-targeting chimera (PROTAC) BRD4 protein degraders in cancer therapy
PROTAC protein degraders are emerging as novel anticancer agents. ARV-771, a small molecule compound PROTAC BRD4 protein degrader, down-regulates the expression of oncogenes such as MYC [59]. ARV-771 reduces cell proliferation and induces apoptosis substantially more effectively than the BRD4 inhibitor JQ1 and OTX015 in castration-resistant prostate cancer and diffuse large B cell lymphoma cells [59, 60]. Importantly, while OTX015 suppresses castration-resistant prostate cancer progression, treatment with ARV-771 results in tumor regression in mice xenografted with castration-resistant prostate cancer cell tumors [60] and growth inhibition in mice xenografted with diffuse large B cell lymphoma cells [59] (Table 2).
A1874 is a nutlin-based small molecule compound PROTAC BRD4 protein degrader. A1874 combines the activities of the BRD4 inhibitor JQ1 and the MDM2 antagonist idasanutlin, degrades BRD4 protein by 98% at nanomolar concentrations and stabilizes p53 protein [61]. Treatment with A1874 more significantly reduces cell proliferation and induces cell death in a variety of cancer cell lines with wild type p53 than PROTAC BRD4 protein degraders [61] (Table 2). PROTAC BRD4 protein degraders are therefore likely to be more effective anticancer agents than BRD4 bromodomain inhibitors.
Small molecule compound dual BRD4 and CBP/p300 bromodomain co-inhibitors in cancer therapy
Another effective approach is to target the bromodomains of the super-enhancer “reader” BRD4 and the “writers” CBP/p300 simultaneously. The dual BRD4 and CBP/p300 bromodomain co-inhibitor XP-524 exhibits higher potency and superior tumoricidal activity than the BRD4 inhibitor JQ-1, and shows anticancer efficacy comparable to combination therapy with high-dose JQ-1 and the CBP/p300 inhibitor SGC-CBP30 in pancreatic ductal adenocarcinoma cells [62]. XP-524 suppresses KRAS activity, blocks KRAS-induced malignant transformation in vivo and improves mouse survival in transgenic mouse models of aggressive pancreatic ductal adenocarcinoma. In addition, XP-524 and an anti-PD-1 antibody exert synergistic anticancer effects and improve survival in two transgenic mouse models of pancreatic ductal adenocarcinoma cells [62] (Table 2).
The other dual BRD4 and CBP/p300 bromodomain co-inhibitor NEO2734 up-regulates the expression of p53 and its target PUMA and induces colorectal cancer cell apoptosis through the intrinsic and extrinsic apoptosis pathways, suppression of the intrinsic or extrinsic apoptosis pathway partly rescues colorectal cancer cells, and NEO2734 represses colon cancer progression by inducing colorectal cancer cell apoptosis in a mouse model [63] (Table 2). In addition, NEO2734 shows more potent anticancer effects than single-agent BRD4 or CBP/p300 inhibitors in lymphoma and acute myeloid leukemia cell lines, and exerts substantial anticancer effects in mouse models of lymphoma and acute myeloid leukemia [64] (Table 2). Dual BRD4 and CBP/p300 bromodomain co-inhibitors are therefore likely to be more effective anticancer agents than BRD4 bromodomain inhibitors.
BRD4 inhibitors show anticancer effects in clinical trials
More than a dozen BRD4 BD1 and BD2 bromodomain inhibitors, including ABBV-075, AZD5153, BAY 1238097, BMS-986158, BMS-986378, CC-90010, CPI-0610, FT-1101, GSK525762 (Molibresib), INCB054329, INCB057643, ODM-207, OTX015 and PLX51107 have been or are currently in clinical trials in patients with cancer from various organ origins. The BRD4 inhibitors show anticancer effects in clinical trials as monotherapy, but it is now clear that BRD4 inhibitors need to be combined with other anticancer agents to effectively treat cancer patients (Table 3).
In a dose-escalation, phase I clinical study in acute myeloid leukemia, lymphoma and myeloma patients, plasma OTX015 concentration increases proportionally up to 120 mg/day [65, 66]. A minority of patients achieve complete remission or partial remission [65, 66]. While minor side effects, including thrombocytopenia, diarrhea, vomiting, fatigue and hyponatraemia occur, OTX015 is well-tolerated and is currently undergoing phase II clinical trials in patients with acute leukemia, lymphoma or myeloma on a 14 days on and 7 days off schedule (Table 3).
In a Phase I clinical trial of the BRD4 inhibitor CC-90010 in 67 solid tumor and 2 lymphoma patients, one patient each with astrocytoma or endometrial carcinoma achieves a complete response or a partial response, and six additional patients experience prolonged stable disease [67]. Side effects including thrombocytopenia anemia and fatigue are well-tolerated, and CC-90010 at 45 mg on a 4 days on and 24 days off schedule has been proposed for Phase II clinical trials [67] (Table 3). In addition, in a Phase Ib clinical trial in glioblastoma patients, CC-90010 in combination with Temozolomide is safe and well tolerated with encouraging anticancer efficacy [68] (Table 3).
ABBV-075 has been tested in 12 patients with prostate cancer, 72 patients with other solid tumors such as melanoma, colorectal, breast and pancreatic cancers, and 44 patients with acute myeloid leukemia [69, 70]. While ABBV-075 monotherapy shows limited anticancer effects in both solid tumor and leukemia patients, combination therapy with ABBV-075 and the BLC2 inhibitor Venetoclax is considerably more effective. Despite adverse events including dysgeusia, loss of appetite, diarrhea, thrombocytopenia, fatigue, nausea and anemia, ABBV-075 has a good safety profile for Phase II studies at the dose of 1.5 mg daily [69, 70] (Table 3).
In two independent Phase I/II dose-escalation, safety and tolerability studies of the BRD4 inhibitors INCB054329 and INCB057643 in patients with solid tumors or lymphoma, 69 and 134 patients have been recruited to INCB054329 (completed) and INCB057643 (ongoing) studies respectively [71]. Two complete responses and four partial responses have been observed in INCB057643 treatment group; INCB057643 shows a more favorable pharmacokinetic profile than INCB054329; and side effects, including thrombocytopenia, nausea, fatigue and decreased appetite, can be safely managed in both INCB054329 and INCB057643 treated patients [71] (Table 3).
The BRD4 inhibitor Pelabresib (CPI-0610) has shown synergistic anticancer effects, when combined with Ruxolitinib, the current standard of care treatment in myelofibrosis patients, in 84 myelofibrosis patients in a Phase II clinical trial [72]. At 24 weeks, 68% patients reached a reduction in spleen volume of ≥35%, and 56% acquired a reduction in total symptom score of ≥50%. Side effects including thrombocytopenia and anemia are not common and are manageable. Importantly, a double-blinded placebo-controlled Phase III clinical trial is currently ongoing to examine the synergistic anticancer effects of Ruxolitinib and CPI-0610 combination therapy in myelofibrosis patients [73] (Table 3).
The BRD4 inhibitor GSK525762 (Molibresib) has shown promising anticancer effects in a Phase I clinical trial in patients with NUT carcinoma [74, 75]. However, in a dose-escalation Phase I clinical trial of GSK525762 in 87 patients with acute myeloid leukemia, non-Hodgkin lymphoma or multiple myeloma and in a Phase II clinical trial in 24 patients with relapsed/refractory myelodysplastic syndrome or cutaneous T-cell lymphoma, only 6 patients achieved complete response and 7 patients partial responses [76]. Adverse effects such as thrombocytopenia, anemia and neutropenia limit dose escalation and anticancer effects [76] (Table 3).
Two other BRD4 BD1 and BD2 bromodomain inhibitors also show significant toxicity to normal tissues. In the first phase I, open-label, non-randomized clinical trial of the BRD4 inhibitor BAY 1238097 in 8 patients with solid tumors, BAY 1238097 shows on-target effects on BRD4-inhibition biomarkers, such as reduction in MYC expression, but results in dose-limiting toxicities including nausea, vomiting, headache, back pain and fatigue, and the study has been terminated [77] (Table 3). In an open-label Phase I clinical trial of the BRD4 inhibitor ODM-207 in 35 patients with solid tumors including castrate-resistant prostate cancer, no complete or partial responses were observed, and side effects such as thrombocytopenia, anorexia, nausea, diarrhea and fatigue were common, indicating that ODM-207 is not efficacious and has a narrow therapeutic window [78] (Table 3).
Importantly, the BRD4 BD2 domain inhibitor ABBV-744, which shows much less toxicity to normal tissues in preclinical models, has also entered a Phase I clinical trial in relapsed or refractory acute myeloid leukemia patients. However, clinical data have not been published.
Conclusions and future perspective
Characterized by massive histone H3K27 acetylation signal at the loci of cell identity genes and critical oncogenes, super-enhancers are recognized by the BET bromodomain protein BRD4; and super-enhancers and BRD4 play critical roles in oncogene transcriptional activation, over-expression, malignant transformation, cancer cell proliferation, survival, tumor initiation, progression and metastasis in a number cancer types. However, it is important to note that super-enhancers and BRD4 can also activate tumor suppressor gene transcription and suppress drug resistance gene expression. While super-enhancers and BRD4 generally promote tumorigenesis, it is imperative to comprehensively investigate the specific scenarios, such as certain sub-types of cancer cells under particular cellular context, in which super-enhancers and BRD4 exert tumor suppressive, rather than tumorigenic, functions.
BRD4 bromodomain BD1 and BD2 inhibitors have been discovered through small molecule compound library screen, in silico compound screen and chemical synthesis. By blocking BRD4 binding to super-enhancers, BRD4 inhibitors suppress oncogene transcription and expression, reduce cancer cell proliferation and survival, and suppress tumor progress in cancers of a variety of organ origins. However, BRD4 inhibitors, like other targeted therapies, show moderate anticancer effects when employed as a monotherapy. Pre-clinical studies have shown that BRD4 inhibitors exert synergistic anticancer effects when combined with other anticancer agents, such as CDK7 inhibitors, CDK4/CDK6 inhibitors, HDAC inhibitors and BCL-2 inhibitors in vitro and in mouse models of various cancers.
More than a dozen BRD4 BD1 and BD2 bromodomain inhibitors, such as OTX015, have been or are currently in clinical trials in patients with cancer of various organ origins. It is now clear that BRD4 BD1 and BD2 bromodomain inhibitors induce weak to moderate anti-cancer effects in patients as a monotherapy and some of the inhibitors cause significant side effects, such as thrombocytopenia, dysgeusia, diarrhea, fatigue, nausea and anemia. More recently, BRD4 BD2 bromodomain selective inhibitor ABBV-744, PROTAC BRD4 protein degraders such as ARV-771 and A1874, and dual BRD4 and CBP/p300 bromodomain co-inhibitors NEO2734 and XP-524 have been developed and have shown better anticancer effects and/or better safety profile in pre-clinical models. In addition, data from clinical trials of ABBV-744 and NEO2734 are expected to be released, and will further shed lights on the utility of the novel BRD4 inhibitors in the clinical setting.
Future endeavors can focus on developing more potent and selective small molecule compound BRD4 BD2 bromodomain inhibitors to reduce cytotoxicity to normal cells, PROTAC BRD4 protein degraders, and dual BRD4 and CBP/p300 bromodomain co-inhibitors through chemical synthesis, structure-based in silico screen, and wet lab screen of small molecule compound libraries. Their safety profile in normal cells and tissues, pharmacokinetics and anticancer effects can be examined both in vitro and in multiple mouse models. Nevertheless, it should be noted that treatment with BRD4 inhibitors can reduce tumor suppressor gene expression under specific conditions, and that long-term treatment with BRD4 inhibitors can result in cancer cell resistance to a broad spectrum of anticancer agents. It is therefore important to investigate the specific scenarios, such as certain sub-types of cancer cells under particular context and chemotherapy-naïve or -exposed cancer cells and mouse models, in which BRD4 inhibitors reduce tumor suppressor gene expression, augment drug resistance gene expression and render cancer cell resistance to anticancer agents.
As all targeted therapies are expected to be employed in the clinic in combination therapies, the other anticancer agents which exert the best synergistic anticancer effects with BRD4 inhibitors should be identified by unbiased screening of approved anticancer drug libraries against each cancer type. Ultimately, the best combination therapies with BRD4 inhibitors and other anticancer drugs are expected to be tested in clinical trials in patients.
References
Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–54.
Andersson R, Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020;21:71–87.
Bartman CR, Hsu SC, Hsiung CC, Raj A, Blobel GA. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell. 2016;62:237–47.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19.
Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47.
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34.
Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483–93.
Croce CM, Erikson J, Huebner K, Nishikura K. Coexpression of translocated and normal c-myc oncogenes in hybrids between Daudi and lymphoblastoid cells. Science. 1985;227:1235–8.
Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–27.
Fu X, Pereira R, De Angelis C, Veeraraghavan J, Nanda S, Qin L, et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc Natl Acad Sci USA. 2019;116:26823–34.
Okazaki K, Anzawa H, Liu Z, Ota N, Kitamura H, Onodera Y, et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat Commun. 2020;11:5911.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52.
Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic Ductal Adenocarcinoma. Cell Rep. 2018;25:1741–55.e7.
Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute Myeloid Leukemia. Cell Rep. 2018;22:3641–59.
Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet. 2017;49:1613–23.
Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J Exp Med. 2019;216:1071–90.
Tsang FH, Law CT, Tang TC, Cheng CL, Chin DW, Tam WV, et al. Aberrant super-enhancer landscape in human hepatocellular Carcinoma. Hepatology. 2019;69:2502–17.
Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell. 2018;33:512–26.e8.
Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–22.
Wong M, Sun Y, Xi Z, Milazzo G, Poulos RC, Bartenhagen C, et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat Commun. 2019;10:3319.
Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31:635–52.e6.
Nishida J, Momoi Y, Miyakuni K, Tamura Y, Takahashi K, Koinuma D, et al. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat Cell Biol. 2020;22:465–75.
Zhou RW, Xu J, Martin TC, Zachem AL, He J, Ozturk S, et al. A local tumor microenvironment acquired super-enhancer induces an oncogenic driver in colorectal carcinoma. Nat Commun. 2022;13:6041.
Scholz BA, Sumida N, de Lima CDM, Chachoua I, Martino M, Tzelepis I, et al. WNT signaling and AHCTF1 promote oncogenic MYC expression through super-enhancer-mediated gene gating. Nat Genet. 2019;51:1723–31.
Chachoua I, Tzelepis I, Dai H, Lim JP, Lewandowska-Ronnegren A, Casagrande FB, et al. Canonical WNT signaling-dependent gating of MYC requires a noncanonical CTCF function at a distal binding site. Nat Commun. 2022;13:204.
Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530:57–62.
Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515–20.
Zhou J, Wang S, Nie D, Lai P, Li Y, Li Y, et al. Super-enhancer landscape reveals leukemia stem cell reliance on X-box binding protein 1 as a therapeutic vulnerability. Sci Transl Med. 2021;13:eabh3462.
Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet. 2020;52:388–400.
Chang H, Liu Y, Xue M, Liu H, Du S, Zhang L, et al. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nuc Acids Res. 2016;44:2514–27.
Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 2020;37:599–617.e7.
Deng R, Huang JH, Wang Y, Zhou LH, Wang ZF, Hu BX, et al. Disruption of super-enhancer-driven tumor suppressor gene RCAN1.4 expression promotes the malignancy of breast carcinoma. Mol Cancer. 2020;19:122.
Liu B, Liu X, Han L, Chen X, Wu X, Wu J, et al. BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc Natl Acad Sci USA. 2022;119:e2109133119.
Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–90.
Zheng ZZ, Xia L, Hu GS, Liu JY, Hu YH, Chen YJ, et al. Super-enhancer-controlled positive feedback loop BRD4/ERα-RET-ERα promotes ERα-positive breast cancer. Nucleic Acids Res. 2022;50:10230–48.
Dong J, Li J, Li Y, Ma Z, Yu Y, Wang CY. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat Commun. 2021;12:3974.
Ke L, Zhou H, Wang C, Xiong G, Xiang Y, Ling Y, et al. Nasopharyngeal carcinoma super-enhancer-driven ETV6 correlates with prognosis. Proc Natl Acad Sci USA. 2017;114:9683–8.
Gryder BE, Pomella S, Sayers C, Wu XS, Song Y, Chiarella AM, et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat Genet. 2019;51:1714–22.
Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–99.
Gelato KA, Schöckel L, Klingbeil O, Rückert T, Lesche R, Toedling J, et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene. 2018;37:512–21.
Jia Y, Zhou J, Tan TK, Chung TH, Chen Y, Chooi JY, et al. Super enhancer-mediated upregulation of HJURP promotes growth and survival of t(4;14)-positive multiple myeloma. Cancer Res. 2022;82:406–18.
Huang CS, You X, Dai C, Xu QC, Li F, Wang L, et al. Targeting super-enhancers via nanoparticle-facilitated BRD4 and CDK7 inhibitors synergistically suppresses pancreatic ductal adenocarcinoma. Adv Sci. 2020;7:1902926.
Zhang W, Ge H, Jiang Y, Huang R, Wu Y, Wang D, et al. Combinational therapeutic targeting of BRD4 and CDK7 synergistically induces anticancer effects in head and neck squamous cell carcinoma. Cancer Lett. 2020;469:510–23.
Guo L, Li J, Zeng H, Guzman AG, Li T, Lee M, et al. A combination strategy targeting enhancer plasticity exerts synergistic lethality against BETi-resistant leukemia cells. Nat Commun. 2020;11:740.
Jin X, Yan Y, Wang D, Ding D, Ma T, Ye Z, et al. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol Cell. 2018;71:592–605.e4.
Liao S, Maertens O, Cichowski K, Elledge SJ. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev. 2018;32:1188–200.
Jonchere B, Williams J, Zindy F, Liu J, Robinson S, Farmer DM, et al. Combination of ribociclib with BET-bromodomain and PI3K/mTOR inhibitors for medulloblastoma treatment in vitro and in vivo. Mol Cancer Ther. 2023;22:37–51.
Han Y, Lindner S, Bei Y, Garcia HD, Timme N, Althoff K, et al. Synergistic activity of BET inhibitor MK-8628 and PLK inhibitor Volasertib in preclinical models of medulloblastoma. Cancer Lett. 2019;445:24–33.
Kling MJ, Kesherwani V, Mishra NK, Alexander G, McIntyre EM, Ray S, et al. A novel dual epigenetic approach targeting BET proteins and HDACs in Group 3 (MYC-driven) Medulloblastoma. J Exp Clin Cancer Res. 2022;41:321.
Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, et al. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clin Cancer Res. 2016;22:2534–44.
Chen J, Nelson C, Wong M, Tee AE, Liu PY, La T, et al. Targeted therapy of TERT-rearranged neuroblastoma with BET bromodomain inhibitor and proteasome inhibitor combination therapy. Clin Cancer Res. 2021;27:1438–51.
Kumari A, Gesumaria L, Liu YJ, Hughitt VK, Zhang X, Ceribelli M, et al. mTOR inhibition overcomes RSK3-mediated resistance to BET inhibitors in small cell lung cancer. JCI Insight. 2023;8:e156657.
Loganathan SN, Tang N, Holler AE, Wang N, Wang J. Targeting the IGF1R/PI3K/AKT Pathway Sensitizes Ewing Sarcoma to BET Bromodomain Inhibitors. Mol Cancer Ther. 2019;18:929–36.
Wright S, Hu J, Wang H, Hyle J, Zhang Y, Du G, et al. Interrogating bromodomain inhibitor resistance in KMT2A-rearranged leukemia through combinatorial CRISPR screens. Proc Natl Acad Sci USA. 2023;120:e2220134120.
Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–10.
Zhang L, Cai T, Lin X, Huang X, Bui MH, Plotnik JP, et al. Selective inhibition of the second bromodomain of BET family proteins results in robust antitumor activity in preclinical models of acute Myeloid Leukemia. Mol Cancer Ther. 2021;20:1809–19.
Cummin TEC, Cox KL, Murray TD, Turaj AH, Dunning L, English VL, et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020;4:3316–28.
Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–94.
Jain N, Hartert K, Tadros S, Fiskus W, Havranek O, Ma MCJ, et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci Transl Med. 2019;11:eaav5599.
Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2016;113:7124–9.
Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79:251–62.
Principe DR, Xiong R, Li Y, Pham TND, Kamath SD, Dubrovskyi O, et al. XP-524 is a dual-BET/EP300 inhibitor that represses oncogenic KRAS and potentiates immune checkpoint inhibition in pancreatic cancer. Proc Natl Acad Sci USA. 2022;119:e2116764119.
Kuang C, Tong J, Ermine K, Cai M, Dai F, Hao S, et al. Dual inhibition of BET and HAT/p300 suppresses colorectal cancer via DR5- and p53/PUMA-mediated cell death. Front Oncol. 2022;12:1018775.
Spriano F, Gaudio E, Cascione L, Tarantelli C, Melle F, Motta G, et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4:4124–35.
Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–95.
Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196–204.
Moreno V, Sepulveda JM, Vieito M, Hernández-Guerrero T, Doger B, Saavedra O, et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann Oncol. 2020;31:780–8.
Vieito M, Simonelli M, de Vos F, Moreno V, Geurts M, Lorenzi E, et al. Trotabresib (CC-90010) in combination with adjuvant temozolomide or concomitant temozolomide plus radiotherapy in patients with newly diagnosed glioblastoma. Neurooncol Adv. 2022;4:vdac146.
Borthakur G, Odenike O, Aldoss I, Rizzieri DA, Prebet T, Chen C, et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer. 2021;127:2943–53.
Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, et al. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin Cancer Res. 2019;25:6309–19.
Falchook G, Rosen S, LoRusso P, Watts J, Gupta S, Coombs CC, et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin Cancer Res. 2020;26:1247–57.
Mascarenhas J, Kremyanskaya M, Patriarca A, Palandri F, Devos T, Passamonti F, et al. MANIFEST: Pelabresib in combination with ruxolitinib for janus kinase inhibitor treatment-naïve myelofibrosis. J Clin Oncol. 2023;28:2201972.
Harrison CN, Gupta VK, Gerds AT, Rampal R, Verstovsek S, Talpaz M, et al. Phase III MANIFEST-2: pelabresib + ruxolitinib vs placebo + ruxolitinib in JAK inhibitor treatment-naive myelofibrosis. Future Oncol. 2022;18:2987–97.
Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectr. 2020;4:pkz093.
Cousin S, Blay JY, Garcia IB, de Bono JS, Le Tourneau C, Moreno V, et al. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of nuclear protein in testis carcinoma and other cancers: Results of a Phase I/II open-label, dose escalation study. Int J Cancer. 2022;150:993–1006.
Dawson MA, Borthakur G, Huntly BJP, Karadimitris A, Alegre A, Chaidos A, et al. A phase I/II open-label study of molibresib for the treatment of relapsed/refractory hematologic malignancies. Clin Cancer Res. 2023;29:711–22.
Postel-Vinay S, Herbschleb K, Massard C, Woodcock V, Soria JC, Walter AO, et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur J Cancer. 2019;109:103–10.
Ameratunga M, Braña I, Bono P, Postel-Vinay S, Plummer R, Aspegren J, et al. First-in-human phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br J Cancer. 2020;123:1730–6.
Acknowledgements
This work was supported by funding from Tongji University, Shanghai.
Author information
Authors and Affiliations
Contributions
L.Y. designed the research. H.Q. and M.Z. prepared the figures and the tables. H.Q., M.Z., X.T., Y.Z., X.L. and L.Y. drafted the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qian, H., Zhu, M., Tan, X. et al. Super-enhancers and the super-enhancer reader BRD4: tumorigenic factors and therapeutic targets. Cell Death Discov. 9, 470 (2023). https://doi.org/10.1038/s41420-023-01775-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01775-6
- Springer Nature Limited
This article is cited by
-
A self-assembled affibody-PROTAC conjugate nanomedicine for targeted cancer therapy
Nano Research (2024)