
Abstract
Copper is an essential micronutrient that plays a pivotal role in numerous physiological processes in virtually all cell types. Nevertheless, the dysregulation of copper homeostasis, whether towards excess or deficiency, can lead to pathological alterations, such as atherosclerosis. With the advent of the concept of copper-induced cell death, termed cuproptosis, researchers have increasingly focused on the potential role of copper dyshomeostasis in atherosclerosis. In this review, we provide a broad overview of cellular and systemic copper metabolism. We then summarize the evidence linking copper dyshomeostasis to atherosclerosis and elucidate the potential mechanisms underlying atherosclerosis development in terms of both copper excess and copper deficiency. Furthermore, we discuss the evidence for and mechanisms of cuproptosis, discuss its interactions with other modes of cell death, and highlight the role of cuproptosis-related mitochondrial dysfunction in atherosclerosis. Finally, we explore the therapeutic strategy of targeting this novel form of cell death, aiming to provide some insights for the management of atherosclerosis.
Similar content being viewed by others

Facts
-
1.
Copper is an essential trace element required in various physiological processes in the human body.
-
2.
Dysregulation of copper homeostasis, whether towards excess or deficiency, has been implicated in various health problems, including atherosclerosis.
-
3.
Dysregulation of copper homeostasis and copper-induced cell death (cuproptosis) are acknowledged as potential contributors to the pathogenesis of atherosclerosis.
Open questions
-
1.
What is the safe window for copper levels to avoid the development of atherosclerosis? How can the suitable copper concentration be determined for the treatment of atherosclerosis?
-
2.
What are the potential candidate biomarkers that can reliably and sensitively indicate the occurrence of cuproptosis in the context of atherosclerosis?
-
3.
What are the potential undiscovered roles of copper in mitochondrial function, and is there interplay between copper and mitochondrial dynamics or mitophagy?
Introduction
Atherosclerosis is a chronic, progressive disease characterized by the accumulation of lipids, inflammatory cells, and fibrous elements in the arterial wall, leading to the formation of atherosclerotic plaques [1, 2]. These plaques can lead to serious clinical consequences, such as myocardial infarction (MI) and stroke, which are major causes of morbidity and mortality worldwide [3]. The pathogenesis of atherosclerosis involves multiple genetic, environmental, and metabolic factors [4].
Copper is an essential trace element involved in mitochondrial respiration, antioxidant defense, and neurotransmitter synthesis [5, 6]. Copper homeostasis is tightly regulated. Copper excess or deficiency can lead to pathological alterations, and thus negatively affect human health [7]. Copper dyshomeostasis may contribute to the pathogenesis of atherosclerosis by increasing oxidative stress, inflammation, endothelial dysfunction, and lipid metabolism levels [8,9,10]. Moreover, a novel type of cell death, which is copper-dependent, has recently been described. This copper-induced cell death, termed cuproptosis, may contribute to atherosclerosis development by causing cell death and impairing mitochondrial function [11, 12].
In this review, we explore the role of copper homeostasis and cuproptosis in atherosclerosis. We also discuss potential therapeutic strategies against cuproptosis and provide directions for future research on cuproptosis and atherosclerosis.
Copper metabolism
Systemic copper homeostasis
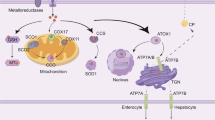
Copper is an essential trace element required in various physiological processes in the human body. Research shows that Cu levels vary across organs and tissues, with values ranging from 3 mg (kidneys) to 46 mg (bone) [13]. Copper serves as a cofactor for numerous enzymes involved in energy metabolism, neurotransmitter synthesis, and antioxidant defense [5, 6, 14]. Excessive or deficient copper levels can, however, lead to cytotoxicity and pathological alterations [7]. Therefore, it is essential to maintain systemic copper levels within a narrow range (Fig. 1).

Copper absorption occurs primarily in the small intestine, a process mediated by SLC31A1. Copper is then transported and exported to the bloodstream through the action of ATP7A, combined with soluble chaperones, and transported through the portal system to the liver, where it is stored and further transported. Excess copper is excreted by the liver into the bile. Figure created with BioRender. SLC31A1 solute carrier family 31 member 1, ATP7A and ATP7B ATPase copper transporters 7 A and 7B, STEAP six-transmembrane epithelial antigen of the prostate, HSA human serum albumin, CP ceruloplasmin, MT metallothionein, HIS histidine, MG macroglobulin.
Copper absorption
Copper is mainly obtained from dietary sources such as organ meat and shellfish [15]. Copper is primarily absorbed by enterocytes in the small intestine, and its uptake is mediated by copper transporter 1 (CTR1), also known as solute carrier family 31 member 1 (SLC31A1) [16]. CTR1 appears to play a primary role in this uptake, as indicated by research showing greatly reduced copper accumulation in the peripheral tissues of neonatal mice lacking CTR1 [17]. The six-transmembrane epithelial antigen of the prostate (STEAP) facilitates this process by reducing divalent Cu2+ to Cu+.
Copper storage and transportation
After uptake by the intestinal epithelial cells, copper is transported and exported into the bloodstream by the ATPase copper transporter 7 A (ATP7A) [18]. Here, copper is transported through the portal system to the liver by binding to soluble chaperones such as human serum albumin (HSA), ceruloplasmin, histidine, and macroglobulin [19, 20]. Copper uptake by the hepatocytes in the liver is also mediated by CTR1. The liver is crucial in regulating copper metabolism by storing and excreting copper. Copper storage is mediated by the copper-binding protein metallothionein (MT), a reducing molecule rich in thiol groups with a high affinity for copper ions [21]. MT plays a crucial role in copper homeostasis by storing and releasing excess copper when required. Copper transport is performed by ATPase copper transporter 7B (ATP7B) in hepatocytes, which pumps copper ions from the liver back into the bloodstream. Here, copper ions bind to their soluble partners and are transported to specific tissues and organs [22].
Copper elimination
Copper is primarily eliminated via biliary excretion [7]. Excess copper is secreted by the liver into the bile and then excreted through feces [23]. The ATP7B transporter regulates the elimination of copper from the liver to bile canaliculi [24]. In cases of ATP7B dysfunction, such as Wilson’s disease, copper accumulates in the liver, leading to liver damage and subsequent health issues [25].
In summary, copper metabolism is a complex process that involves multiple mechanisms to regulate copper absorption, transport, storage, and elimination.
Cellular copper homeostasis
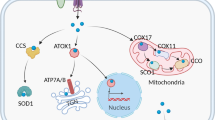
The maintenance of intracellular copper homeostasis is a complex and tightly regulated process involving the coordinated action of copper transporters, chaperones, and cuproenzymes (Fig. 2). Copper concentration is maintained within a narrow range through the collaborative action of these copper-dependent proteins.

The maintenance of intracellular copper homeostasis is a complex and tightly regulated process. Copper ions are primarily taken up by cells via SLC31A1, whereas their export is facilitated by ATP7A/B. Once inside the cell, copper is transported to interact with cytoplasmic copper chaperones, including COX17, CCS, and ATOX1. These chaperones transport copper to specific cellular compartments, such as the mitochondria, TGN, and nucleus. Figure created with BioRender. SLC31A1 solute carrier family 31 member 1, ATP7A and ATP7B ATPase copper transporter 7A and 7B, ATOX1 antioxidant 1 copper chaperone, CCS copper chaperone for superoxide dismutase, COX17 cytochrome c oxidase copper chaperone 17, COX11 cytochrome c oxidase copper chaperone 11, COX cytochrome c oxidase, ROS reactive oxygen species, SCO1 synthesis of cytochrome c oxidase 1, SOD1 superoxide dismutase 1, TGN trans-Golgi network, GSH glutathione, MT1/2 metallothionein 1/2, and STEAP six-transmembrane epithelial antigen of the prostate.
Copper uptake
In mammalian cells, copper uptake is primarily mediated by CTR1 [6], a transmembrane protein that forms a trimeric channel to facilitate the passage of Cu ions across the plasma membrane [26]. CTR1 expression is regulated by copper levels. Low copper levels upregulate CTR1 expression to increase copper uptake, whereas high levels downregulate CTR1 expression to prevent copper cytotoxicity [27, 28]. These findings highlight the importance of CTR1 in copper homeostasis.
Intracellular copper distribution
After entering the cell, copper is first delivered to cytoplasmic copper chaperones, which then transport it to intracellular compartments such as the mitochondria, trans-Golgi network (TGN), and nucleus. In mammalian cells, three major copper chaperones have been identified: antioxidant 1 copper chaperone (ATOX1), copper chaperone for superoxide dismutase (CCS), and cytochrome c oxidase copper chaperone 17 (COX17) [29, 30].
ATOX1 delivers copper to ATP7A and ATP7B in the TGN [31]. Additionally, ATOX1 has been shown to function as a new type of copper-dependent transcription factor that mediates copper-induced cell proliferation [32].
CCS is a soluble cytoplasmic copper-chaperone protein that transfers copper ions to the copper-binding site of superoxide dismutase 1 (SOD1) [33]. SOD1 is a major antioxidant enzyme that catalyzes the conversion of superoxide radicals into oxygen and hydrogen peroxide, thereby maintaining reactive oxygen species (ROS) homeostasis and protecting the cells from oxidative stress damage [34]. This function has been shown in SOD1-knockout mice, where the absence of SOD1 increased oxidative stress and led first to liver cell damage and eventually liver cancer [35].
COX17 is responsible for delivering copper ions to the assembly of cytochrome c oxidase (COX), a key component of the electron transport chain involved in cellular respiration [36]. Subsequently, COX11 and SCO1, which are also important components of the COX assembly, donate copper to Cu(B) and Cu(A) sites in COX2 and COX1 core subunits of COX in the mitochondrial inner membrane, respectively. Additionally, COX17 also acts as a copper donor within the mitochondrial intermembrane space (IMS) [37]. COX17 is thus essential for proper COX assembly, with mutations in COX17 shown to further reduce COX activity, resulting in mitochondrial dysfunction and oxidative stress [38].
Intracellular copper storage and export
Intracellular copper export is mainly mediated by ATP7A and ATP7B, which are copper transporters located in the TGN that regulate copper delivery to secretory pathways and the plasma membrane [39]. Under normal conditions, ATP7A and ATP7B transport copper ions from the TGN to other cellular compartments for various cellular functions. At excessive intracellular copper levels, ATP7A and ATP7B are activated to export excess copper ions from the TGN and sequester them in copper-binding proteins such as metallothionein [40]. These proteins also regulate copper homeostasis by reducing uptake and increasing efflux. Mutations in ATP7A and ATP7B can lead to copper metabolism disorders such as Menkes disease and Wilson’s disease [41, 42].
Evidence linking copper dyshomeostasis to atherosclerosis
Atherosclerosis is a chronic inflammatory disease characterized by plaque accumulation on the arterial walls, leading to narrowing and hardening of the arteries [43]. This process can result in serious complications, including coronary artery disease (CAD), stroke, and peripheral artery disease [44,45,46]. Growing evidence from numerous studies has linked copper dyshomeostasis and the resultant excess or deficiency of copper to atherosclerosis.
Evidence linking copper excess and atherosclerosis
Extensive research has revealed a correlation between elevated copper levels and cardiovascular disease (Table 1). For instance, several prospective cohort studies have shown a significant correlation between elevated serum copper levels and higher mortality rates related to cardiovascular diseases, particularly coronary heart disease [47,48,49]. Elevated copper levels have also been shown to be an independent risk factor for ischemic heart disease [50]. In addition to evidence from prospective studies, Stadler et al. directly detected and quantified transition metal ions in human atherosclerotic plaques and found increased copper levels in the diseased intima [51]. Studies on populations with acute myocardial infarction (AMI) also support these findings, with patients with AMI exhibiting significantly higher serum copper levels than those without AMI [52]. Moreover, an increase in serum copper levels among post-MI patients was found to have considerable diagnostic value for the occurrence of MI [53]. Furthermore, altered copper bioavailability is negatively correlated with carotid intima-media thickness (IMT), which may serve as a reliable predictor for early atherosclerosis in patients with obesity [54]. Furthermore, serum copper concentrations differ among patients with different carotid atherosclerotic plaque morphologies. Specifically, patients with hemorrhagic plaques have significantly higher serum copper concentrations than those with calcified plaques [55]. These findings suggest a potential involvement of elevated copper levels in the pathogenesis of atherosclerosis.
Evidence linking copper deficiency and atherosclerosis
Copper deficiency is also recognized as a major contributing factor to the development of atherosclerosis. This is evidenced by the benefit of high dietary copper and copper supplementation. The Institute of Medicine recommends a daily dietary allowance of 900 ug of copper for adults, with a tolerable upper limit of 10,000 μg/day to prevent liver damage [56]. Although recommendations vary between national authorities, most recommend an intake of 800 to 2400 ug/day [15]. Substantial research suggests that adequate copper intake reduces the risk of atherosclerosis. For example, Rock et al. found that copper supplementation in middle-aged individuals enhanced the antioxidative capacity of cells, which may help prevent vascular damage and thus reduce the risk of atherosclerosis [57]. Another cohort study showed an association between adequate dietary intake of copper (equal to or above the estimated average requirement) and a reduced risk of all-cause- and cardiovascular disease-related mortality. However, this association was limited to copper intake from food sources [58]. Studies on animal models corroborate these findings with Lamb et al., showing that dietary copper deficiency or excess increases susceptibility to atherosclerosis of the aorta in cholesterol-fed rabbits [59]. Similarly, copper supplementation was found to reverse pathological changes induced by dietary iron overload in mice, partially normalizing cardiac hypertrophy [60] and improving cardiac function in pressure overload-induced dilated cardiomyopathy [61]. Nevertheless, the effects of copper supplementation on the cardiovascular system remain controversial. A study by Diaf et al. conducted on middle-aged women from Algeria showed that there is little association between dietary copper intake and atherosclerosis prevalence in diabetes [62]. These conflicting results may be due to differences in study design and the dose and duration of copper supplementation.
Potential mechanisms of atherosclerosis development due to copper dyshomeostasis
The mechanisms of atherosclerosis development due to altered copper levels are not fully understood. Several potential mechanisms include oxidative stress, inflammation, endothelial dysfunction, and lipid metabolism.
Oxidative stress
Oxidative stress is a key factor in atherosclerosis development, which typically involves an imbalance in reactive oxygen species (ROS) production and antioxidant defenses [63]. Since copper is a redox-active metal, changes in copper levels can contribute to the generation of ROS and thereby promote oxidative stress [15, 64] (Fig. 3).

Excessive copper interacts with H2O2 via the Fenton reaction, generating highly reactive hydroxyl radicals. These radicals induce lipid peroxidation, DNA strand breaks, and base oxidation and impair the function of antioxidant enzymes, ultimately leading to increased oxidative stress and potential damage to cells. On the other hand, copper deficiency also impairs the function of certain antioxidant enzymes, causing a decrease in SOD1 and COX activity. This reduced activity results in lowered NO levels, inactivation of complex I, and increased production of ROS, thereby exacerbating oxidative stress within cells. Together, these processes promote the development of atherosclerosis. Figure created with BioRender. SLC31A1 solute carrier family 31 member 1, STEAP six-transmembrane epithelial antigen of the prostate, H2O2 hydrogen peroxide, •OH reactive hydroxyl radicals, ROS reactive oxygen species, ATP7A and ATP7B ATPase copper transporter 7A and 7B, CAT catalase, GSH-Px glutathione peroxidase, CP ceruloplasmin, LOX lysyl oxidase, SOD1 superoxide dismutase 1, COX cytochrome c oxidase, and AS atherosclerosis.
Copper excess and oxidative stress
Elevated levels of free copper ions tend to interact more with hydrogen peroxide through Fenton reactions, leading to the production of highly-reactive hydroxyl radicals [65]. These radicals cause lipid peroxidation, protein oxidation, and DNA damage, ultimately contributing to the initiation and progression of atherosclerosis [66]. Lipid peroxidation is a chain reaction initiated by an attack of ROS on polyunsaturated fatty acids in cell membrane lipids, which then leads to the oxidative damage of lipid molecules. Increased copper levels can promote lipid peroxidation and result in the formation of oxidized low-density lipoprotein (ox-LDL), which is an atherosclerosis risk factor [67]. Furthermore, increased copper levels have been shown to reduce the activity of antioxidant enzymes, such as Cu/Zn-SOD, catalase, and glutathione peroxidase, in the red blood cells, whole blood, live tissue, and brain tissue of rats (67, 68, 69). In rat brain tissue, the decrease in SOD activity and glutathione levels arises because copper overload induces lipid peroxidation [68]. Copper is also capable of causing DNA strand breaks and the oxidation of bases via oxygen-derived free radicals [69]. These findings suggest that copper overload causes impairment of the antioxidant defense system in several tissues.
Copper deficiency and oxidative stress
Copper deficiency has also been suggested to contribute to the development and progression of atherosclerosis via oxidative stress. Copper is an essential cofactor of various antioxidant enzymes, including Cu/Zn-SOD, ceruloplasmin, and lysyl oxidase [70]. Copper deficiency may thus impair the function of these enzymes, leading to an impaired antioxidant defense system and increased susceptibility to oxidative stress [70, 71]. This suggestion is supported by copper deficiency in rats being found to reduce Cu/Zn-SOD activity and increase oxidative damage to various subunits of the erythrocyte spectrin [72]. Decreased activity of SOD1, which encodes Cu/Zn SOD, has also been observed in the liver and red blood cells of copper-deficient rats [73]. The decrease in SOD1 caused by copper deficiency leads to a reduction in NO levels, which may promote endothelial dysfunction, reduce vascular relaxation, and increase oxidative stress, thus ultimately contributing to atherosclerosis development [70]. In addition to affecting antioxidant enzymes, copper deficiency may also reduce COX activity and lead to oxidative inactivation of complex I (NADH: ubiquinone oxidoreductase). This oxidative inactivation may then lead to elevated production of ROS in copper-deficient cells, thereby exacerbating cellular oxidative stress [74].
Inflammation
Copper may also promote atherosclerosis development by modulating inflammatory responses associated with the disease.
Copper excess and inflammation
Excess copper has been implicated in atherosclerosis pathogenesis due to its ability to induce inflammation. High copper levels were previously reported to stimulate pro-inflammatory cytokine production and thereby promote inflammation within arterial walls [75]. For example, in primary cardiac cells, Cu2+ increases the release of interleukin-6 (IL-6) and activates MAP kinases, which are linked to cardiac inflammation and hypertrophy [76]. Copper-induced oxidative stress also contributes to inflammation because excessive copper generates ROS, which causes oxidative damage to lipids, proteins, and DNA and promotes inflammation [15]. Furthermore, increases in ROS levels due to increased copper levels can, in turn, lead to the activation of nuclear factor-κB (NF-κB), a crucial protein involved in regulating the expression of proinflammatory genes [77]. These findings suggest that high copper levels can exacerbate inflammation of the vascular walls and thus promote atherosclerosis development.
Copper deficiency and inflammation
Copper deficiency has also been linked to atherosclerosis development through its impact on inflammation. Copper deficiency may result in decreased expression of adhesion molecules, such as ICAM-1 and VCAM-1, which facilitate leukocyte adhesion onto activated endothelial cells [78]. Evidence from animal studies also supports the role of copper deficiency in inflammation. For example, neutrophil accumulation increases due to increased ICAM-1 expression in the livers of copper-deficient rats following ischemia/reperfusion [79]. Further, this elevated ICAM-1 expression has been shown to activate neutrophils and endothelial cells, as evidenced by F-actin polymerization and increased accumulation of neutrophils within the lung microcirculation [80,81,82]. Pulmonary inflammatory responses are also intensified in copper-deficient animals [83]. Finally, copper deficiency impairs the activity of Cu/Zn-SOD as previously mentioned, leading to an increased accumulation of ROS and exacerbation of oxidative stress, which can further promote inflammation and atherosclerosis development [84].
Endothelial dysfunction
Copper dyshomeostasis has been linked to atherosclerosis, with both an excess and deficiency of copper contributing to endothelial dysfunction, a critical early step in atherogenesis.
Copper excess and endothelial dysfunction
Excessive copper can disrupt the balance between the production and degradation of nitric oxide (NO), a key regulator of vascular tone and endothelial function [85]. Increased copper levels can upregulate inducible NO synthase (iNOS) expression, resulting in excessive NO production and peroxynitrite, a potent oxidant that causes further oxidative damage [86]. Moreover, copper also interacts with atherosclerosis risk factors such as homocysteine and thereby causes increased hydrogen peroxidation and oxidative stress. Specifically, a study shows that incubation with homocysteine and copper for 4 h is able to cause significant damage to endothelial cells [87].
Copper deficiency and endothelial dysfunction
Copper deficiency is associated with increased endothelial dysfunction as well. Endothelial dysfunction can result in decreased production of NO, a vasodilator and inhibitor of platelet aggregation, and thereby promote a proatherogenic environment [88]. Copper deficiency can also lead to decreased NO levels by reducing the levels of SOD1. Reduced NO levels can then inhibit endothelial function and vasodilation, increase oxidative stress, and thereby promote atherosclerosis [70].
Lipid metabolism
Copper excess and lipid metabolism
Excess copper strongly contributes to atherosclerosis development via its effects on the ox-LDL, which plays a central role in atherosclerosis [89]. Specifically, copper participates in the oxidation of LDL particles, and alterations in copper levels may affect the susceptibility of LDL particles to oxidation. Excess copper levels can increase the oxidation of LDL particles and trigger the production of ox-LDL, which then contributes to atherosclerosis development [67].
In addition to its effects on ox-LDL, copper is involved in several forms of lipid metabolism, including fatty acid and cholesterol synthesis and lipoprotein metabolism. Alterations in copper levels may be associated with changes in lipid and lipoprotein concentrations. For example, serum copper and ceruloplasmin levels were found to be positively associated with lipid peroxides, total cholesterol, triglycerides, and apolipoprotein B in healthy individuals [90]. In another study, the serum copper levels of Iranian patients with angiographically defined CAD were found to be positively correlated with fasting serum triglycerides [91]. In vivo, evidence from animal experiments also supports the role of excess copper in lipid metabolism. A study on yellow catfish demonstrated that copper-induced endoplasmic reticulum stress and disrupted calcium homeostasis alter hepatic lipid metabolism, leading to increased lipid accumulation in the liver [92].
Copper deficiency and lipid metabolism
Insufficient copper levels may also contribute to atherosclerosis development by affecting lipid metabolism. Since copper is involved in the synthesis of fatty acids and cholesterol, insufficient copper levels may lead to impaired lipid metabolism and, thus, the development of fatty liver disease. Copper deficiency increases total cholesterol levels as well [7]. Insufficient copper levels can affect the activity of sterol regulatory element-binding proteins 1 and 2 (SREBP-1 and SREBP-2), which are transcription factors involved in fatty acid and cholesterol metabolism [93]. Specifically, both SREBP-1 isoforms (SREBP-1a and SREBP-1c) are involved in the regulation of fatty acid synthesis, whereas SREBP-2 is mainly involved in the regulation of cholesterol biosynthesis [94]. Low copper diets were found to induce accumulation of the mature form of SREBP-1 in the nuclei of rat liver cells, yet no change was observed in the DNA-binding site of SREBP-1 [95]. Hence, copper may play a role in regulating the subcellular localization of SREBP-1, potentially affecting its activity and, in turn, lipid metabolism. Further research is needed to fully understand the mechanisms through which copper affects the activity of SREBP-1 and other transcription factors involved in lipid metabolism.
Other risk factors
Copper levels are also associated with other atherosclerosis risk factors, such as high blood pressure. Copper inhibits the activity of angiotensin-converting enzymes, such that copper deficiency leads to increasing angiotensis levels and, thus, water and sodium retention and hypertension [96]. Moreover, copper is a cofactor of SOD, which is a major player in the antioxidant defense system. Hence, copper deficiency may increase the levels of superoxide free radicals, leading to elevated angiotensin levels and consequent hypertension [97].
Copper-induced cell death and atherosclerosis
From copper-induced cell death to cuproptosis
The discovery of copper-induced cell death dates back to the early 1980s [98]. Increased copper levels were found to promote ROS generation and thereby lead to oxidative stress, DNA damage, and, ultimately cell death [99]. These findings have led to further investigations into the molecular mechanisms underlying copper-induced cell death and their potential implications for human diseases. Indeed, conflicting findings suggest that, in addition to ROS accumulation, excess copper may induce cell death through apoptosis or caspase-independent cell death [100, 101]. Overall, the mechanisms responsible for copper-induced cell death were not well understood.
However, in March 2022, Tsvetkov et al. published a groundbreaking study unveiling the mechanism of copper-induced cell death, which was termed cuproptosis [12]. Cuproptosis represents a unique form of cell death triggered by an excess of copper ions. In their study, Tsvetkov et al. showed that treatment with the copper ionophore elesclomol induced cell death. Remarkably, only the copper chelator can rescue cells from elesclomol-induced cell death. In contrast, rescue is not possible with any of the inhibitors for apoptosis, necroptosis, oxidative stress, ROS induced cell death, or ferroptosis. These findings unequivocally establish that cuproptosis is distinct from other known cell death modalities, underscoring its unique mechanisms and signaling pathways. Notably, cuproptosis is regulated by mitochondrial respiration, as supported by research indicating that mitochondria-dependent cells exhibit a sensitivity to copper ionophores nearly 1,000 times higher than cells undergoing glycolysis [12]. The importance of mitochondrial respiration in cuproptosis has been highlighted in further research, revealing a close correlation between cuproptosis and the tricarboxylic acid (TCA) cycle. During cuproptosis, intracellular copper binds to the lipoylated components of the TCA cycle. This leads to the aggregation of copper-bound lipoylated mitochondrial proteins, which can disrupt the TCA cycle and, therefore, interfere with cellular energy production. The upstream regulatory factors FDX1 and LIAS are critical in this process. Aggregation of these proteins and the subsequent reduction of Fe–S clusters, which are essential cofactors for various cellular processes, including electron transport and enzymatic reactions [102], promote proteotoxic stress and ultimately lead to cell death (Fig. 4).

The copper ionophore ES transports extracellular copper into the cell. Subsequently, intracellular copper binds to lipoylated mitochondrial enzymes (such as DLAT) involved in the TCA cycle. This binding leads to the aggregation of these proteins, which can disrupt the TCA cycle and, therefore, interfere with cellular energy production. The upstream regulatory factors FDX1 and LIAS play a critical role in this process. The aggregation of lipoylated mitochondrial proteins and loss of Fe-S clusters promote proteotoxic stress and ultimately lead to cell death. Figure created with BioRender. SLC31A1 solute carrier family 31 member 1, STEAP the six-transmembrane epithelial antigen of the prostate, ATP7A and ATP7B ATPase copper transporter 7A and 7B, ES elesclomol, α-KG α-ketoglutarate, DLAT dihydrolipoamide S-acetyltransferase, FDX1 ferredoxin-1, Fe–S iron–sulfur, LIAS lipoic acid synthetase, TCA tricarboxylic acid, and GSH glutathione.
Potential interactions between copper-induced cell death and other cell death pathways
Accumulating evidence suggests widespread cross-talk and interactions among the primary initiators, effectors, and executioners involved in pyroptosis, necroptosis, ferroptosis, and cuproptosis.
Pyroptosis
Pyroptosis is a pro-inflammatory programmed cell death that is primarily driven by inflammasome assembly, accompanied by GSDMD cleavage and IL-1β and IL-18 release [103,104,105]. NLRP1, NLRP3, NLRC4, AIM2, and pyrin are well-established inflammasome sensors that assemble canonical inflammasomes in a process induced by inflammatory stimuli such as those resulting from microbial infections [106, 107]. Copper ions, which are essential micronutrients for many physiological processes, have been shown to trigger ROS production and activate the NF-κB signaling pathway. This leads to the upregulation of pro-inflammatory genes and cytokines, potentially influencing the progression of atherosclerosis [108,109,110]. Therefore, there is a plausible suggestion of crosstalk between copper ions and pyroptosis.
Evidence from animal studies demonstrates that copper loading in rat hepatocytes leads to a significant increase in the mRNA expression of pyroptosis-related genes (caspase-1, IL-18, IL-1β, and NLRP3) and the protein expression of caspase-1[111]. Similarly, copper exposure in mouse microglial cells triggers an inflammatory response, resulting in the upregulation of NLRP3/caspase 1/GSDMD axis proteins and subsequent pyroptosis. These effects are likely mediated by the early activation of the ROS/NF-κB pathway and subsequent disruption of mitophagy [77]. Moreover, comparable outcomes were observed in murine macrophages treated with copper oxide nanoparticles. Copper oxide nanoparticle exposure induces oxidative stress and activates NLRP3 inflammasomes, leading to the expression of pro-IL-1β through the MyD88-dependent TLR4 signaling pathway, followed by NF-κB activation in murine macrophages [108].
In summary, these findings demonstrate the presence of crosstalk between copper-induced cell death and pyroptosis. Copper exposure influences gene and protein expression associated with pyroptosis in various cell types, with the underlying mechanisms being ROS/NF-κB pathway activation and the inflammatory response. Further research is needed to elucidate the precise underlying mechanisms and explore the implications of this interaction.
Necroptosis
Necroptosis is a form of programmed necrosis linked to atherosclerosis via its potential involvement in plaque destabilization and rupture [112, 113]. Necroptosis was recently shown to activate the NLRP3 inflammasome by causing potassium efflux through MLKL pores in macrophages [114]. Bioinformatics analysis further indicated an association between ZBP1 and cuproptosis as well as necroptosis [115]. ZBP1 activation led to the recruitment of RIPK3 and caspase-8 to activate the NLRP3 inflammasome, which in turn triggers necroptosis and pyroptosis [116, 117].
Ferroptosis
Ferroptosis, a form of iron-dependent cell death triggered by lipid peroxidation and accumulation of lipid-based reactive oxygen species [118,119,120], appears to be influenced by copper levels due to the redox-active properties of copper ions [121, 122]. Cuproptosis can modulate the expression of key genes involved in ferroptosis regulation, such as glutathione peroxidase 4 (GPX4), by eliminating phospholipid hydroperoxides [123, 124] and acylcoenzyme A synthetase long-chain family member 4 (ACSL4) [125], thereby regulating the sensitivity of cells to ferroptosis inducers. Notably, copper chelators can reduce sensitivity to ferroptosis specifically while leaving other forms of cell death unaffected. In a study by Xue et al., copper was found to play a novel role in promoting ferroptotisis through the degradation of GPX4 via macroautophagy/autophagy [126]. Furthermore, research by Gao et al. reveals an interaction between copper-induced cell death and necroptosis. Their findings indicate that elesclomol administration to colorectal cancer cells increased Cu(II) levels in the mitochondria, downregulated ATP7A expression, and increased ROS accumulation. This process triggered SLC7A11 degradation, intensifying oxidative stress and resulting in ferroptosis [127]. Additionally, copper depletion greatly enhanced ferroptosis through mitochondrial perturbation and the disruption of antioxidant mechanisms [128]. Specifically, copper depletion limits GPX4 protein expression and reduces cellular sensitivity to ferroptosis inducers, establishing a direct link between copper levels and ferroptosis [128].
Based on the aforementioned studies, it is evident that copper is closely linked to necroptosis, pyroptosis, and ferroptosis, suggesting a significant cross-talk between these different cell death pathways. Understanding the underlying mechanisms that connect these modes of cell death is of paramount importance for the development of novel atherosclerotic therapeutic strategies that can target multiple pathways simultaneously.
Cuproptosis-related mitochondrial dysfunction and atherosclerosis
Mitochondria are vulnerable to copper-induced damage, which causes oxidative damage to its membrane [129, 130]. Excessive intracellular copper may also disrupt mitochondrial function by altering the activity of several key enzymes, such as those involved in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation [131]. Severe mitochondrial dysfunction and decreases in the activities of several liver enzymes, including complex I, complex II, complex III, complex IV, and aconitase, were observed in patients with copper overload. These effects are potentially mediated by the accumulation of copper in the mitochondria [132]. Furthermore, copper treatment induces selective changes in metabolic enzymes through lipoylation, which is a highly conserved lysine post-translational modification. Protein lipoylation occurs only on Dihydrolipoamide S-Acetyltransferase (DLAT), Dihydrolipoamide S-Succinyltransferase (DLST), Dihydrolipoamide Branched Chain Transacylase E2 (DBT), and Glycine Cleavage System Protein H (GCSH), all of which are involved in metabolic complexes that regulate carbon entry points to the TCA cycle [133, 134]. Additionally, copper overload may trigger the opening of the mitochondrial permeability transition pore and cause the release of pro-apoptotic factors, ultimately resulting in cell death [135].
Mitochondrial dysfunction is implicated in atherosclerosis development and progression [136]. Impaired mitochondrial function promotes lipid accumulation, oxidative stress, inflammatory responses, and proliferation of vascular smooth muscle cells, all of which contribute to plaque formation and destabilization [137, 138]. Considering its impact on mitochondrial function, cuproptosis may contribute to the progression of atherosclerosis by aggravating mitochondrial dysfunction.
Potential therapeutic strategies targeting cuproptosis in atherosclerosis
Copper chelators
Copper chelators bind and sequester copper ions. Several copper chelators have shown promise in animal studies and clinical trials for atherosclerosis treatment.
Tetrathiomolybdate (TTM) has been shown to be an effective copper chelator with potential therapeutic implications in attenuating atherosclerosis progression, as demonstrated in animal models [139, 140]. In a study using ApoE-/- mice, TTM treatment for 10 weeks significantly reduced aortic lesion development, indicating its potential as an anti-atherosclerotic agent [139]. The beneficial effects of TTM were attributed to its ability to reduce bioavailable copper levels and inhibit vascular inflammation [140]. Copper is involved in vascular inflammation as an etiologic factor of atherosclerotic vascular disease, and by chelating copper, TTM may modulate copper-related pathways involved in atherosclerosis and inflammation. Wei et al. demonstrated that TTM copper chelation inhibited lipopolysaccharide (LPS)-induced inflammatory responses in mice aorta and other tissues. This inhibition may occur by suppressing redox-sensitive transcription factors NF-κB and AP-1, which play crucial roles in inflammation [140]. By targeting copper-related pathways and modulating inflammatory processes, TTM exhibits potential as an anti-atherosclerotic agent.
Ethylenediaminetetraacetic acid disodium salt (EDTA) is a broad-spectrum metal-chelating agent that has also been investigated for its potential to treat atherosclerosis [141]. For instance, a double-blind placebo-controlled trial (N = 1708) shows that chelation therapy with disodium EDTA reduced the risk of adverse cardiovascular outcomes in stable patients with a history of myocardial infarction (MI). The primary endpoint occurred less frequently in the chelation group than in the placebo group (26% vs. 30%) [142]. These findings offer preliminary evidence to direct further studies but do not in themselves provide sufficient evidence to justify the routine use of chelation therapy in patients with MI. Another meta-analysis of five studies covering a total of 1,993 randomized participants failed to identify sufficient evidence to determine the effectiveness of chelation therapy in the treatment of atherosclerotic cardiovascular disease [143]. The contradictory results among these studies may be attributed to variations in research design and/or characteristics of the study populations. Therefore, further research is needed before routine use of chelation therapy can be recommended.
Regulation of copper chaperone protein expression
Copper chaperones play a crucial role in maintaining cellular copper homeostasis by facilitating the transfer of copper ions to target proteins and organelles. Modulating the expression of copper chaperone proteins potentially offers an alternative approach to regulate copper levels and limit the contribution of copper-induced cell death to atherosclerosis.
ATOX1 is a copper chaperone protein that is of major importance in mammalian cells. ATOX1 has been shown to translocate to the nucleus in response to inflammatory cytokines or exogenous copper. Furthermore, ATOX1 is localized in the nucleus of endothelial cells in the inflamed atherosclerotic aorta [144]. The migration of VSMCs is crucial for neointimal formation following vascular injury and atherosclerotic lesion formation. ATOX1 was found to promote VSMC migration and inflammatory cell recruitment to injured vessels [145]. Furthermore, copper-dependent binding of ATOX1 to TRAF4 is required to facilitate nuclear translocation of ATOX1 and ROS-dependent inflammatory responses in TNF-α-stimulated endothelial cells (ECs) [146]. This highlights the potential of targeting the ATOX1-TRAF4 axis as a novel therapeutic strategy for the treatment of atherosclerosis. In summary, ATOX1 represents a promising therapeutic target for inflammation-related vascular diseases such as atherosclerosis.
Copper ionophores
Copper ionophores transport copper into the cells, leading to an increase in intracellular copper levels and subsequent cell death. However, copper chelators can inhibit this process. Several drugs can act as copper ionophores, including disulfiram, pyrithione, chloroquine, and elesclomol [147]. Among these, elesclomol has received the most attention and has been subjected to several clinical trials for use in cancer treatment. Although the majority of these trials have not shown promising results regarding further development of elesclomol as a drug, they have verified its safety [148]. Nanomedicines combining copper ions with copper ionophores are also currently being widely investigated. Recently, the researchers successfully designed multifunctional nanoparticles with pH-responsive and CD44-targeted properties. The utilization of dendritic mesoporous silica nanoparticles capped with copper sulfide and coated with hyaluronic acid enables precise drug delivery and controlled release in the acidic microenvironment of atherosclerotic inflammation [149]. These findings highlight the potential of copper-based nanomedicines in developing innovative approaches for targeted atherosclerosis therapy. Notably, an important aspect to consider in the advancement of copper ionophores for clinical therapeutic applications is the notable impact that slight structural changes can exert on their properties and functions [16]. In addition, copper ionophore-mediated cell death is strongly correlated with mitochondrial metabolism and closely associated with atherosclerosis development. Based on these findings, copper ionophores may represent a novel therapeutic strategy for targeting copper-induced cell death in atherosclerosis.
Conclusions and future perspectives
In conclusion, the multifaceted role of copper in atherosclerosis involves complex cellular and systemic metabolic processes. Copper deficiency or excess promotes atherosclerosis by inducing oxidative stress, inflammation, endothelial dysfunction, and adverse effects on lipid metabolism. Further research is required to elucidate the interactions between these factors and their role in the development of atherosclerotic lesions.
Copper-induced cell death and the subsequent emergence of the concept of cuproptosis have expanded our understanding of the role of copper in atherosclerosis. The involvement of cuproptosis and the associated mitochondrial dysfunction in atherosclerosis highlights the need for further research into the molecular mechanisms underlying these processes. Therapeutic targets and strategies for mitigating the detrimental effects of copper-induced cell death in atherosclerosis have also been identified, including modulating copper levels, inhibiting cuproptosis, or enhancing cellular defenses against cuproptosis. However, potential side effects should also be considered since an excessive reduction in copper levels may impair essential physiological processes or, in itself, lead to atherosclerosis. Additionally, the absence of cuproptosis-specific biomarkers is a significant barrier that hinders further development of clinical applications regarding cuproptosis. Identifying such biomarkers may facilitate risk stratification and the development of personalized therapeutic approaches.
References
Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–33.
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. 2019;5:56.
Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of Atherothrombotic disease. Circ Res. 2016;118:535–46.
Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. 2022;185:1630–45.
Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21:R877–83.
Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85.
Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378.
Chen X, Cai Q, Liang R, Zhang D, Liu X, Zhang M, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. 2023;14:105.
Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Asp Med. 2005;26:268–98.
Fukai T, Ushio-Fukai M, Kaplan JH. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol. 2018;315:C186–c201.
Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32:417–8.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Yang L, Yang P, Lip GYH, Ren J. Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharm Sci. 2023;44:573–85.
Maung MT, Carlson A, Olea-Flores M, Elkhadragy L, Schachtschneider KM, Navarro-Tito N, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021;35:e21810.
Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107–15.
Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22:46.
Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4:235–44.
Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr. 2008;88:846s–50s.
Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134:96–114.
Ramos D, Mar D, Ishida M, Vargas R, Gaite M, Montgomery A, et al. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11:e0149516.
Santoro A, Calvo JS, Peris-Díaz MD, Krężel A, Meloni G, Faller P. The glutathione/metallothionein system challenges the design of efficient O(2) -activating copper complexes. Angew Chem Int Ed Engl. 2020;59:7830–5.
La Fontaine S, Ackland ML, Mercer JF. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int J Biochem Cell Biol. 2010;42:206–9.
Wijmenga C, Klomp LW. Molecular regulation of copper excretion in the liver. Proc Nutr Soc. 2004;63:31–9.
Hernandez S, Tsuchiya Y, García-Ruiz JP, Lalioti V, Nielsen S, Cassio D, et al. ATP7B copper-regulated traffic and association with the tight junctions: copper excretion into the bile. Gastroenterology. 2008;134:1215–23.
Huster D, Kühne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. 2012;142:947–56.e5.
Maryon EB, Molloy SA, Ivy K, Yu H, Kaplan JH. Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J Biol Chem. 2013;288:18035–46.
Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharm. 2012;81:455–64.
Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, Prohaska JR. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J Nutr. 2006;136:21–6.
Muller PA, Klomp LW. ATOX1: a novel copper-responsive transcription factor in mammals? Int J Biochem Cell Biol. 2009;41:1233–6.
Prohaska JR, Gybina AA. Intracellular copper transport in mammals. J Nutr. 2004;134:1003–6.
Yang D, Xiao P, Qiu B, Yu HF, Teng CB. Copper chaperone antioxidant 1: multiple roles and a potential therapeutic target. J Mol Med Berl. 2023;101:527–42.
Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008;283:9157–67.
Rosenzweig AC. Copper delivery by metallochaperone proteins. Acc Chem Res. 2001;34:119–28.
Dong X, Zhang Z, Zhao J, Lei J, Chen Y, Li X, et al. The rational design of specific SOD1 inhibitors via copper coordination and their application in ROS signaling research. Chem Sci. 2016;7:6251–62.
Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–80.
Ding Y, Chen Y, Wu Z, Yang N, Rana K, Meng X, et al. SsCox17, a copper chaperone, is required for pathogenic process and oxidative stress tolerance of Sclerotinia sclerotiorum. Plant Sci. 2022;322:111345.
Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279:35334–40.
Takahashi Y, Kako K, Kashiwabara S, Takehara A, Inada Y, Arai H, et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome C oxidase and embryonic development. Mol Cell Biol. 2002;22:7614–21.
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–46.
La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149–67.
Horn N, Wittung SP. et al. ATP7A-regulated enzyme metalation and trafficking in the menkes disease puzzle. Biomedicines. 2021;9:123–142.
Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflug Arch. 2020;472:1415–29.
Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315–27.
Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–22.
Campia U, Gerhard-Herman M, Piazza G, Goldhaber SZ. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133–41.
Endres M, Moro MA, Nolte CH, Dames C, Buckwalter MS, Meisel A. Immune pathways in etiology, acute phase, and chronic sequelae of ischemic stroke. Circ Res. 2022;130:1167–86.
Reunanen A, Knekt P, Marniemi J, Mäki J, Maatela J, Aromaa A. Serum calcium, magnesium, copper and zinc and risk of cardiovascular death. Eur J Clin Nutr. 1996;50:431–7.
Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151:1182–8.
Kok FJ, Van Duijn CM, Hofman A, Van der Voet GB, De Wolff FA, Paays CH, et al. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am J Epidemiol. 1988;128:352–9.
Salonen JT, Salonen R, Korpela H, Suntioinen S, Tuomilehto J. Serum copper and the risk of acute myocardial infarction: a prospective population study in men in eastern Finland. Am J Epidemiol. 1991;134:268–76.
Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004;24:949–54.
Begum S, Sultana I, Faysal MR, Alam S, Tasnim J, Akter T, et al. Study of changes in serum copper level in patients with acute myocardial infarction. Mymens Med J. 2023;32:39–43.
Nowicki GJ, Ślusarska B, Prystupa A, Blicharska E, Adamczuk A, Czernecki T, et al. Assessment of concentrations of heavy metals in postmyocardial infarction patients and patients free from cardiovascular event. Cardiol Res Pr. 2021;2021:9546358.
Tarantino G, Porcu C, Arciello M, Andreozzi P, Balsano C. Prediction of carotid intima-media thickness in obese patients with low prevalence of comorbidities by serum copper bioavailability. J Gastroenterol Hepatol. 2018;33:1511–7.
Tasić NM, Tasić D, Otašević P, Veselinović M, Jakovljević V, Djurić D, et al. Copper and zinc concentrations in atherosclerotic plaque and serum in relation to lipid metabolism in patients with carotid atherosclerosis. Vojnosanit Pregl. 2015;72:801–6.
Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294–301.
Rock E, Mazur A, O’Connor JM, Bonham MP, Rayssiguier Y, Strain JJ. The effect of copper supplementation on red blood cell oxidizability and plasma antioxidants in middle-aged healthy volunteers. Free Radic Biol Med. 2000;28:324–9.
Chen F, Du M, Blumberg JB, Ho Chui KK, Ruan M, Rogers G, et al. Association among dietary supplement use, nutrient intake, and mortality among U.S. adults: a cohort study. Ann Intern Med. 2019;170:604–13.
Lamb DJ, Avades TY, Ferns GA. Biphasic modulation of atherosclerosis induced by graded dietary copper supplementation in the cholesterol-fed rabbit. Int J Exp Pathol. 2001;82:287–94.
Wang T, Xiang P, Ha JH, Wang X, Doguer C, Flores SRL, et al. Copper supplementation reverses dietary iron overload-induced pathologies in mice. J Nutr Biochem. 2018;59:56–63.
Hughes WM Jr, Rodriguez WE, Rosenberger D, Chen J, Sen U, Tyagi N, et al. Role of copper and homocysteine in pressure overload heart failure. Cardiovasc Toxicol. 2008;8:137–44.
Diaf M, Khaled MB. Associations between dietary antioxidant intake and markers of atherosclerosis in middle-aged women from North-Western Algeria. Front Nutr. 2018;5:29.
Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625–31.
Fang Y, Xing C, Wang X, Cao H, Zhang C, Guo X, et al. Activation of the ROS/HO-1/NQO1 signaling pathway contributes to the copper-induced oxidative stress and autophagy in duck renal tubular epithelial cells. Sci Total Environ. 2021;757:143753.
Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–208.
Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–73.
Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341–90.
Ozcelik D, Uzun H. Copper intoxication; antioxidant defenses and oxidative damage in rat brain. Biol Trace Elem Res. 2009;127:45–52.
Brezová V, Dvoranová D, Zúbor V, Breza M, Mazúr M, Valko M. Photochemical properties of camptothecin in the presence of copper(II) ions: the role of radicals as prospective species in photodynamic therapy. Mol Biotechnol. 2007;37:48–51.
Al-Bayati MA, Jamil DA, Al-Aubaidy HA. Cardiovascular effects of copper deficiency on activity of superoxide dismutase in diabetic nephropathy. N. Am J Med Sci. 2015;7:41–6.
Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63:797s–811s.
Sukalski KA, LaBerge TP, Johnson WT. In vivo oxidative modification of erythrocyte membrane proteins in copper deficiency. Free Radic Biol Med. 1997;22:835–42.
Bertinato J, Iskandar M, L’Abbé MR. Copper deficiency induces the upregulation of the copper chaperone for Cu/Zn superoxide dismutase in weanling male rats. J Nutr. 2003;133:28–31.
Johnson WT, Thomas AC. Copper deprivation potentiates oxidative stress in HL-60 cell mitochondria. Proc Soc Exp Biol Med. 1999;221:147–52.
Malekahmadi M, Firouzi S, Rezayi M, Ghazizadeh H, Ranjbar G, Ferns GA, et al. Association of zinc and copper status with cardiovascular diseases and their assessment methods: a review study. Mini Rev Med Chem. 2020;20:2067–78.
Ansteinsson V, Refsnes M, Skomedal T, Osnes JB, Schiander I, Låg M. Zinc- and copper-induced interleukin-6 release in primary cell cultures from rat heart. Cardiovasc Toxicol. 2009;9:86–94.
Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y, et al. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem Toxicol. 2022;168:113369.
Schuschke DA, Saari JT, Miller FN. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol Lett. 2001;76:139–44.
Sakai N, Shin T, Schuster R, Blanchard J, Lentsch AB, Johnson WT, et al. Marginal copper deficiency increases liver neutrophil accumulation after ischemia/reperfusion in rats. Biol Trace Elem Res. 2011;142:47–54.
Gordon SA, Lominadze D, Saari JT, Lentsch AB, Schuschke DA. Impaired deformability of copper-deficient neutrophils. Exp Biol Med Maywood. 2005;230:543–8.
Schuschke DA, Percival SS, Lominadze D, Saari JT, Lentsch AB. Tissue-specific ICAM-1 expression and neutrophil transmigration in the copper-deficient rat. Inflammation. 2002;26:297–303.
Lominadze D, Saari JT, Percival SS, Schuschke DA. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol. 2004;82:231–8.
Lentsch AB, Kato A, Saari JT, Schuschke DA. Augmented metalloproteinase activity and acute lung injury in copper-deficient rats. Am J Physiol Lung Cell Mol Physiol. 2001;281:L387–93.
Paynter DI, Moir RJ, Underwood EJ. Changes in activity of the Cu-Zn superoxide dismutase enzyme in tissues of the rat with changes in dietary copper. J Nutr. 1979;109:1570–6.
Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14.
Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest. 1986;77:1370–6.
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009;32:S314–21.
Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr Med Chem. 2019;26:1693–700.
Craig WY, Poulin SE, Palomaki GE, Neveux LM, Ritchie RF, Ledue TB. Oxidation-related analytes and lipid and lipoprotein concentrations in healthy subjects. Arterioscler Thromb Vasc Biol. 1995;15:733–9.
Kazemi-Bajestani SM, Ghayour-Mobarhan M, Ebrahimi M, Moohebati M, Esmaeili HA, Parizadeh MR, et al. Serum copper and zinc concentrations are lower in Iranian patients with angiographically defined coronary artery disease than in subjects with a normal angiogram. J Trace Elem Med Biol. 2007;21:22–8.
Song YF, Luo Z, Zhang LH, Hogstrand C, Pan YX. Endoplasmic reticulum stress and disturbed calcium homeostasis are involved in copper-induced alteration in hepatic lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Chemosphere. 2016;144:2443–53.
Morrell A, Tallino S, Yu L, Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life. 2017;69:263–70.
Ye J, DeBose RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 2011;3:96–105.
Tang Z, Gasperkova D, Xu J, Baillie R, Lee JH, Clarke SD. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J Nutr. 2000;130:2915–21.
Loyke HF. Copper and zinc in experimental hypertension. Biol Trace Elem Res. 1991;29:45–9.
Ozumi K, Sudhahar V, Kim HW, Chen GF, Kohno T, Finney L, et al. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension. 2012;60:476–86.
Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14.
Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147–63.
Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, et al. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J Am Chem Soc. 2011;133:6235–42.
Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52:2142–50.
Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. 2020;89:471–99.
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128.
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4.
Nunes T, de Souza HS. Inflammasome in intestinal inflammation and cancer. Mediat Inflamm. 2013;2013:654963.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16:319–26.
Tao X, Wan X, Wu D, Song E, Song Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A.1 macrophage. J Hazard Mater. 2021;411:125134.
Liu H, Guo H, Deng H, Cui H, Fang J, Zuo Z, et al. Copper induces hepatic inflammatory responses by activation of MAPKs and NF-κB signalling pathways in the mouse. Ecotoxicol Environ Saf. 2020;201:110806.
Deng H, Zhu S, Yang H, Cui H, Guo H, Deng J, et al. The dysregulation of inflammatory pathways triggered by copper exposure. Biol Trace Elem Res. 2023;201:539–48.
Liao J, Yang F, Tang Z, Yu W, Han Q, Hu L, et al. Inhibition of caspase-1-dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicol Environ Saf. 2019;174:110–9.
Zhang X, Ren Z, Xu W, Jiang Z. Necroptosis in atherosclerosis. Clin Chim Acta. 2022;534:22–8.
Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X, Liu NF. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022;13:467.
Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1β independently of gasdermin-D. J Immunol. 2017;198:2156–64.
Miao Y, Liu J, Liu X, Yuan Q, Li H, Zhang Y, et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front Genet. 2022;13:951239.
Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:52–68.
Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Publisher Correction: Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:E10.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–43.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Yang F, Pei R, Zhang Z, Liao J, Yu W, Qiao N, et al. Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicol Vitr. 2019;54:310–6.
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982–96.
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15:3527–44.
Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z, et al. Copper depletion strongly enhances ferroptosis via mitochondrial perturbation and reduction in antioxidative mechanisms. Antioxid Basel. 2022;11:652–98.
Arciello M, Rotilio G, Rossi L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem Biophys Res Commun. 2005;327:454–9.
Cobine PA, Moore SA, Leary SC. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochim Biophys Acta Mol Cell Res. 2021;1868:118867.
Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55:645–53.
Gu M, Cooper JM, Butler P, Walker AP, Mistry PK, Dooley JS, et al. Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. Lancet. 2000;356:469–74.
Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293:7522–30.
Rowland EA, Snowden CK, Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018;42:76–85.
Su R, Wang R, Cao H, Pan J, Chen L, Li C, et al. High copper levels promotes broiler hepatocyte mitochondrial permeability transition in vivo and in vitro. Biol Trace Elem Res. 2011;144:636–46.
Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. 2018;50:121–7.
Ciccarelli G, Conte S, Cimmino G, Maiorano P, Morrione A, Giordano A, et al. Mitochondrial dysfunction: the hidden player in the pathogenesis of atherosclerosis? Int J Mol Sci. 2023;24:56–68.
Huynh DTN, Heo KS. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res. 2021;44:1051–61.
Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223:306–13.
Wei H, Frei B, Beckman JS, Zhang WJ. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am J Physiol Heart Circ Physiol. 2011;301:H712–20.
Clarke CN, Clarke NE, Mosher RE. Treatment of angina pectoris with disodium ethylene diamine tetraacetic acid. Am J Med Sci. 1956;232:654–66.
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013;309:1241–50.
Villarruz-Sulit MV, Forster R, Dans AL, Tan FN, Sulit DV. Chelation therapy for atherosclerotic cardiovascular disease. Cochrane Database Syst Rev. 2020;5:Cd002785.
Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T, et al. Whole-transcriptome sequencing analyses of nuclear antixoxidant-1 in endothelial cells: role in inflammation and atherosclerosis. Cells. 2022;11:881–96.
Kohno T, Urao N, Ashino T, Sudhahar V, McKinney RD, Hamakubo T, et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler Thromb Vasc Biol. 2013;33:805–13.
Das A, Sudhahar V, Ushio-Fukai M, Fukai T. Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. Am J Physiol Cell Physiol. 2019;317:C1161–c71.
Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X, et al. Copper metabolism in cell death and autophagy. Autophagy. 2023;12:65–104.
O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31:1211–8.
Liu S, Zhao Y, Shen M, Hao Y, Wu X, Yao Y, et al. Hyaluronic acid targeted and pH-responsive multifunctional nanoparticles for chemo-photothermal synergistic therapy of atherosclerosis. J Mater Chem B. 2022;10:562–70.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81202805, 82074254, 82374281), the Beijing Natural Science Foundation (No. 7172185), and the Science and Technology Innovation Project of China Academy of Chinese Medical Science (C12021A01413).
Author information
Authors and Affiliations
Contributions
MW and LL provided the guidance for this study. SY prepared the original manuscript and the illustrations. YL, LZ, and XW assisted with manuscript review and revision. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., Li, Y., Zhou, L. et al. Copper homeostasis and cuproptosis in atherosclerosis: metabolism, mechanisms and potential therapeutic strategies. Cell Death Discov. 10, 25 (2024). https://doi.org/10.1038/s41420-023-01796-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01796-1
- Springer Nature Limited
This article is cited by
-
Cuproptosis: unveiling a new frontier in cancer biology and therapeutics
Cell Communication and Signaling (2024)
-
Copper’s dual role: unravelling the link between copper homeostasis, cuproptosis, and cardiovascular diseases
Hypertension Research (2024)
-
Cuproptosis and Cu: a new paradigm in cellular death and their role in non-cancerous diseases
Apoptosis (2024)
-
Natural products: A potential immunomodulators against inflammatory-related diseases
Inflammopharmacology (2024)