
Abstract
Compound heterozygous mutations in SHQ1 have been associated with a rare and severe neurological disorder characterized by global developmental delay (GDD), cerebellar degeneration coupled with seizures, and early-onset dystonia. Currently, only five affected individuals have been documented in the literature. Here, we report three children from two unrelated families harboring a homozygous variant in the gene but with a milder phenotype than previously described. The patients had GDD and seizures. Magnetic resonance imaging analyses revealed diffuse white matter hypomyelination. Sanger sequencing confirmed the whole-exome sequencing results and revealed full segregation of the missense variant (SHQ1:c.833 T > C; p.I278T) in both families. We performed a comprehensive in silico analysis using different prediction classifiers and structural modeling of the variant. Our findings demonstrate that this novel homozygous variant in SHQ1 is likely to be pathogenic and leads to the clinical features observed in our patients.
Similar content being viewed by others


Introduction
Shq1 has been identified in yeast during the biogenesis of H/ACA ribonucleoproteins (RNPs)1 and has been found to interact with one of the catalytic subunits of the RNPs, dyskerin/NAP57. This molecule is a critical assembly factor for H/ACA ribonucleoproteins (RNPs)2. Therefore, it is involved in various important functions, such as telomerase maintenance, ribosomal modifications, protein translation, and pre-mRNA splicing2,3. In humans, SHQ1 has similar functions and stabilizes the accumulation of H/ACA RNPs by binding with NAP57 at an early stage, which promotes the biogenesis of H/ACA RNPs4. Accordingly, loss of SHQ1 will lead to degradation of the RNP assembly5. Mutations in genes encoding components of the H/ACA RNP complex, such as DKC1, can lead to significant effects on neurological development6. Recently, two studies reported patients with pathogenic variants of SHQ1 with neurological disorders; Bizarro and Meier7 reported singletons with intrauterine growth retardation coupled with a severe-onset neurological disease inclusive of cerebellar degeneration, whereas Sleiman et al.6 studied two separate families harboring four patients (two individuals in each family) with early-onset dystonia, hypotonia, seizure disorder, and global developmental delay (GDD)6. Interestingly, all reported variants in both studies were compound heterozygous6,7. Here, we report three individuals from two unrelated families with a novel homozygous variant in SHQ1 and show that the variant is likely to be pathogenic. Informed consent was obtained from all subjects enrolled in the study. The patients were recruited from the pediatric neuroscience clinic. Peripheral blood samples were collected, and genomic DNA was isolated from whole blood using the Gentra® Puregene® DNA Purification Kit (Gentra Systems, Inc. Minneapolis, MN, US). Axiom-based genotyping was conducted using Affymetrix axiom chips followed by autozygome analysis. In addition, whole-exome sequencing (WES) variant calls were performed by an Illumina 2500 platform using libraries that were prepared by a SureSelect kit (Illumina Inc., San Diego, CA, US). WES data were filtered, and low-frequency pathogenic variants were targeted according to previously published protocols. Confirmatory sequencing and segregation were performed by Sanger sequencing using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The structure of the C-terminal domain of SHQ1 was modeled using Swiss-Model based on the 30% identical yeast homolog (PDB ids 3zv0, 3zuz; QMEAN 0.41). The homology model was corroborated by the AlphaFold2 model for this region (RMSD 1.12 Å; pLDDT > 90 for the core region).
Family 1, Patient IV:1
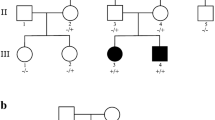
Individual 1 (Fig. 1A, Family 1, Patient IV:1) is a 15-year-old male, who was born after a full-term pregnancy by normal spontaneous delivery, from a first-degree consanguineous Saudi family. He was admitted after birth to the NICU due to jaundice and low birth weight. Patient IV:1 was first diagnosed at 5 years of age with global developmental delay (GDD), ataxia and seizure disorder. Brain MRI showed severely delayed myelination (Fig. 1B). His developmental delay involved motor, cognition, and speech. By age 15, he was able to walk with support and had very limited speech output. His seizures were controlled by one medication. His examination was notable for mild spasticity in the lower limbs and brisk deep tendon reflexes.

A Pedigrees present two unrelated families with carrier parents and three affected individuals in total, in addition to a healthy carrier brother and two normal brothers in Family 1 and four affected deceased parental cousins. Brief chromatograms of the variant are shown below each symbol representing the family members. NT = not tested. Half-filled symbols indicate the carrier status of an individual, whereas completely filled symbols (in black color) refer to the affected individuals. The full chromatograms are presented in Supplementary Fig. 1. The black arrow points to the index case for the study. B Brain MRI of a patient with the SHQ1 variant (Family 1, Patient IV:1) showing severely delayed myelination generally in mildly prominent CSF spaces. C Autozygosity analysis using Agile multi-ideogram revealed a single shared run of homozygosity between the affected individuals on chromosome 3. D The same analysis using AutoSNPa confirmed the shared runs of homozygosity on chromosome 3.
Family 1, Patient IV:2
Individual 2 (Fig. 1A, Family 1, Patient IV:2) is the younger sister of Patient IV:1. She was born after a full-term pregnancy by normal spontaneous delivery and was admitted to the NICU due to jaundice. She had a similar diagnosis as her brother with GDD and seizure disorder. She also had generalized hypotonia and was wheelchair-bound. Brain MRI showed diffuse white matter hypomyelination. Patient IV:2 died at the age of 9 years due to cardiac arrest. The patient’s father reported that Patient IV:2 deteriorated in her last days; she was in a vegetative state and had severe malnutrition.
Individuals 1 and 2 (Fig. 1A, Family 1) had a positive family history of similar illness with four parental cousins diagnosed with GDD, all of whom had passed away.
Family 2, Patient II:1
Individual 3 (Fig. 1A, Family 2, Patient II:1) is a 7-year-old male of a Saudi family who was delivered by C-section late preterm with low birth weight and was admitted to the hospital after birth. He was diagnosed with GDD, seizure disorder and ataxia in addition to frontotemporal atrophy. At approximately 4 years old, the patient developed neutropenia and thrombocytopenia.
WES filtering revealed that the patients in both family 1 and family 2 had the same novel homozygous missense variant (c.833 T > C; p.I278T) in SHQ1 (Fig. 1A). The homozygosity scan using Affymetrix’s axiom SNP arrays showed that SHQ1 is shared in the same runs of the homozygosity block on chromosome 3 (Fig. 1C, D). The variant c.833 T > C has a low frequency (1000 G = 0/GnomAD = 0) with in silico pathogenicity classifiers, indicating that the variant is likely highly pathogenic (SIFT = 0.001, damaging; PolyPhen = 1, probably damaging; VEST = 0.933 and 0.925, deleterious; PP2HVAR (PolyPhen 2) = 0.99, probably damaging; PP2HDIV = 1, probably damaging; MutationTaster = 1,1, disease causing; MutationAssessor, 3.025 medium, predicted functional). The variant is located in a highly conserved region of the C-terminal domain of SHQ1 (Fig. 2A). The three-dimensional structure of this domain can be inferred based on the known structures of the yeast homolog. I278 is buried in the hydrophobic core of this domain (Fig. 2B). Its substitution by a smaller and polar threonine will introduce a polar moiety and a spatial gap in the protein core (Fig. 2C). Hence, the p.I278T mutation will destabilize this domain and affect its function, specifically, acting as an assembly chaperone that protects Cbf5 protein complexes from nonspecific RNA binding and aggregation before assembly of H/ACA RNA8.

A Amino acid sequences of SHQ1 belonging to various species were downloaded from Ensembl9 and aligned using Clustal Omega from EBI. B The modeled structure of SHQ1 residues 143 to 421 is shown as a gray ribbon. I278 is shown in green. C Neighboring side chains are shown as stick models with carbon in gray, oxygen in red and nitrogen in blue. I278 is shown in green, and the corresponding threonine is shown with carbons in yellow and oxygen in red. D Previously reported variants and the novel homozygous p.Ile278Thr variant are presented on a schematic drawing of the gene and protein. Alternating boxes (light green and blue color) represent exons (upper part of the panel). The lower part of the panel includes the protein domains of SHQ1 according to Ensembl9.
The first compound heterozygous variants (SHQ1: c.1003 C > T; p.Arg335Cys/c.1277 C > T; p.Ala426Val) were reported in Bizarro and Meier’s study in a male patient who was delivered by an urgent cesarean section owing to intrauterine growth retardation and sparse fetal movement. The patient was born at gestational week 29 with a body weight of 1000 g7. Since birth, he had GDD in all milestones in addition to enormous feeding difficulties. At early ages, he developed cerebellar hypoplasia with clearly recognizable atrophy and ventricular dilatation in addition to epileptic seizures. On the last examination, he was in a vegetative condition with cortical atrophy and spastic quadriplegia7.
The second report (Sleiman et al.)6 examined four patients in two different families, apparently unrelated6. Although the compound heterozygous variants (SHQ1: c.874 G > A; p.Glu292Lys/c.828_831del; p.Asp277SerfsTer27, Family 1; c.523 G > T; p.Asp175Tyr/c.828_831del; p.Asp277SerfsTer27, Family 2) were different in each family, one of the alleles, a deletion (c.828_831del), was shared6. Interestingly, all the variants were located within the SSD domain of SHQ1. Patients in both families were born at term, and their blood work showed normal biochemistry. In Family 1, both affected individuals had a similar onset that started at birth and was characterized by developmental delay and dystonia, whereas the affected individuals of Family 2 were only described as having a movement disorder. Although the first MRI scans of the patients in both families were normal, a second MRI was performed on Patient IV:1 of Family 1 at the age of 4 years and showed subtle atrophy of the cerebellum accompanied by widening of the cerebellar folia. All patients in this report were treated with levetiracetam in addition to levodopa replacement therapy, but therapy was discontinued in Patient II:1 of Family 2 because it caused dyskinesia in the patient6.
Similarly, our patients were born at term except for Patient II:1 of Family 2 (born late preterm) and had normal biochemistry and low birth weight. However, unlike previously reported cases6,7, our patients showed a homozygous allelic variant in SHQ1. All affected individuals in our study as well as the previously reported cases6,7 were diagnosed with GDD, and most also had a seizure disorder. One of our patients (Patient IV:2) and one patient of Sleiman et al.6 additionally had hypotonia. Table 1 summarizes the clinical features of our patients and the previously reported cases of SHQ1 mutations.
It is noteworthy to mention that our patients presented with a milder phenotype than the patients reported by Sleiman et al.6 and Bizarro and Meier7. Our patients achieved the ability to walk with ataxic gait, were able to produce single but few words and used their hands to play with smartphones. Their understanding was that of a 5- to 6-year-old when they were 14 years old. In addition, they had milder spasticity more noticeable in the lower limbs with brisk reflexes, and the proband with the longest follow-up remained ambulant until the last visit at age 16. The spasticity was very mild and never impaired ambulation. The patients also had limb ataxia. Ataxia was the greatest contributor to recurrent falls. These findings may be explained by the cerebellar atrophy noted during autopsy studies and on MRI showing cerebellar and supratentorial atrophy. Apart from that, there was no clear degenerative course or decline in the proband’s abilities apart from dysphagia. The seizures were easily controlled with a single medication. The movement disorder was characterized by hyperkinetic choreiform movements. Moreover, our patients did not display florid dystonia or ballismus, as reported by Sleiman et al6. Due to the atypical phenotype of our patients, comprehensive evaluation was otherwise unremarkable, including plasma amino acids, renal profile, uric acid, lactate, ammonia, transferrin isoelectric focusing, CK and liver function tests, tandem MS, very long-chain fatty acids, urine organic acids, serum copper and ceruloplasmin. Moreover, the leukodystrophy next-generation sequencing panel and SNP array for cytogenetic analysis were normal. In contrast, the patients reported by Sleiman et al.6 and Bizarro and Meier7 were averbal with severe dystonia and autonomic instability and were wheelchair bound with normal MRI imaging. Hence, our cases represent a milder phenotype. We suspect that such clinical differences may be due to phenotypic variability.
In conclusion, our study reports the first SHQ1 homoallelic variant in three patients from two separate unrelated families who presented with global developmental delay, ataxia, and seizure disorder.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3279.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Yang, P. K., Rotondo, G., Porras, T., Legrain, P. & Chanfreau, G. The Shq1p.Naf1p complex is required for box H/ACA small nucleolar ribonucleoprotein particle biogenesis. J. Biol. Chem. 277, 45235–45242 (2002).
Walbott, H. et al. The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Genes Dev. 25, 2398–2408 (2011).
Machado-Pinilla, R., Liger, D., Leulliot, N. & Meier, U. T. Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18, 1833–1845 (2012).
Grozdanov, P. N., Roy, S., Kittur, N. & Meier, U. T. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 15, 1188–1197 (2009).
Su, H. et al. SHQ1 regulation of RNA splicing is required for T-lymphoblastic leukemia cell survival. Nat. Commun. 9, 4281 (2018).
Sleiman, S. et al. Compound heterozygous variants in SHQ1 are associated with a spectrum of neurological features, including early-onset dystonia. Hum. Mol. Genet. 31, 614–624 (2022).
Bizarro, J. & Meier, U. T. Inherited SHQ1 mutations impair interaction with NAP57/dyskerin, a major target in dyskeratosis congenita. Mol. Genet. Genom. Med. 5, 805–808 (2017).
Li, S., Duan, J., Li, D., Ma, S. & Ye, K. Structure of the Shq1-Cbf5-Nop10-Gar1 complex and implications for H/ACA RNP biogenesis and dyskeratosis congenita. EMBO J. 30, 5010–5020 (2011).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Acknowledgements
We thank the families, the Center for Genomic Medicine Core Laboratories, the Research Advisory Council Committees, and the KFSHRC Purchasing Departments for facilitating and expediting our requests. We extend our special thanks to the King Salman Center for Disability Research (KSCDR; RAC/ORA#2180004, NK) for grant support during the study. For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.
Funding
Funds were received from the King Salman Center for Disability Research (RAC/ORA#2180004) (NK). STA was supported by the King Abdullah University of Science and Technology (KAUST) through the baseline fund and Award No. REI/1/4446-01 from the Office of Sponsored Research (OSR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Ethics approval was obtained from the Institutional Review Board at KFSHRC, Riyadh, Saudi Arabia.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
AlHargan, A., AlMuhaizea, M.A., Almass, R. et al. SHQ1-associated neurodevelopmental disorder: Report of the first homozygous variant in unrelated patients and review of the literature. Hum Genome Var 10, 7 (2023). https://doi.org/10.1038/s41439-023-00234-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00234-z
- Springer Nature Limited