

Abstract
DESTINY-CRC01 (NCT03384940) was a multicenter, open-label, phase 2 trial assessing the efficacy and safety of trastuzumab deruxtecan (T-DXd) in patients with HER2-expressing metastatic colorectal cancer (mCRC) that progressed after ≥2 prior regimens; results of the primary analysis are published. Patients received T-DXd 6.4 mg/kg every 3 weeks and were assigned to either: cohort A (HER2-positive, immunohistochemistry [IHC] 3+ or IHC 2+/in situ hybridization [ISH]+), cohort B (IHC 2+/ISH−), or cohort C (IHC 1+). Primary endpoint was objective response rate (ORR) by independent central review in cohort A. Secondary endpoints included ORR (cohorts B and C), duration of response, disease control rate, progression-free survival, overall survival, pharmacokinetics, and safety of T-DXd. 86 patients were enrolled (53 in cohort A, 15 in cohort B, and 18 in cohort C). Results of the primary analysis are published, reporting an ORR of 45.3% in cohort A. Here, we report the final results. No responses occurred in cohorts B or C. Median progression-free survival, overall survival, and duration of response were 6.9, 15.5, and 7.0 months, respectively. Overall serum exposure (cycle 1) of T-DXd, total anti-HER2 antibody, and DXd were similar regardless of HER2 status. Most common grade ≥3 treatment-emergent adverse events were decreased neutrophil count and anemia. Adjudicated drug-related interstitial lung disease/pneumonitis occurred in 8 patients (9.3%). These findings support the continued exploration of T-DXd in HER2-positive mCRC.
Similar content being viewed by others


Introduction
Human epidermal receptor growth factor 2 (HER2)-amplified metastatic colorectal cancer (mCRC) comprises ~2–3% of patients with mCRC1, 2 and represents a molecularly distinct subgroup of colorectal cancer that is characterized by a worse prognosis and resistance to anti–epidermal growth factor receptor (EGFR) monoclonal antibodies3,4,5,6. First- and second-line treatment options for patients with mCRC include fluoropyrimidine-based chemotherapy with anti–vascular endothelial growth factor (VEGF) or anti–EGFR agents, depending on RAS mutational status7,8,9. In addition, for patients who have received anti–EGFR in the first-line setting, retreatment with anti–EGFR therapies may be effective10,11. Currently, third-line treatment options are regorafenib and trifluridine/tipiracil, and they have limited antitumor activity—an objective response rate (ORR) of less than 5% and median progression-free survival (PFS) of about 2.0 months7,9,12,13. The observed median overall survival (OS) demonstrated with these therapies is also relatively short (≤7.1 months)12,13. Considering these poor outcomes, there is a high unmet need for HER2-targeted therapies for patients with HER2-amplified and/or HER2-overexpressed mCRC.
T-DXd is an antibody–drug conjugate that consists of a humanized anti-HER2 monoclonal antibody linked to a topoisomerase I inhibitor payload, DXd, through a tetrapeptide-based cleavable linker14,15. The linker is cleaved after internalization by lysosomal enzymes that are upregulated in tumor cells, allowing the release of the cytotoxic payload, an exatecan derivative14,15. Since it is permeable to the cell membrane, a bystander effect on cells near HER2-expressing tumor cells is also achieved15. T-DXd is already approved in several countries for the treatment of patients with metastatic HER2-positive breast and gastric cancers.
T-DXd (6.4 mg/kg, every 3 weeks [Q3W]) administered intravenously in patients with HER2-positive (cohort A) mCRC demonstrated antitumor activity, with a confirmed ORR of 45.3%, in the previously reported primary results of DESTINY-CRC01, an open-label, phase 2 trial16. The median follow-up was 27.1 weeks, with a data cutoff of August 9, 201916. Here we report the final safety and efficacy results, including ORR and survival, in the overall population and subgroups of patients from DESTINY-CRC01 after longer-term follow-up at study completion.
Results
Patients
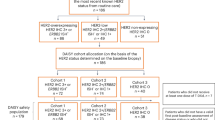
Between February 23, 2018, and November 10, 2020, 86 patients with mCRC were enrolled and received at least 1 dose of T-DXd, including 53 patients in cohort A (HER2-positive, immunohistochemistry [IHC] 3+ or IHC 2+/in situ hybridization [ISH]+), 15 patients in cohort B (HER2 IHC 2+/ISH‒), and 18 patients in cohort C (HER2 IHC 1+; Supplementary Fig. 1). Patients in all cohorts were analyzed for antitumor activity and safety across 25 sites in Asia, Europe, and North America. At the updated data cutoff date of December 28, 2020, no patients remained on treatment in any cohort. The most common reason for discontinuation in all cohorts was disease progression, which occurred in 60 patients (69.8%) overall and in 36 patients (67.9%) in cohort A, 11 patients (73.3%) in cohort B, and 13 patients (72.2%) in cohort C. The median treatment duration was 5.1 months (range, 3.9–7.6), 2.1 months (range, 1.4–2.6), and 1.4 months (range, 1.3–1.5) in cohorts A, B, and C, respectively.
Baseline demographics and disease characteristics were similar among all 3 cohorts (Table 1). The median age was 58.5 years (range, 27–79), and the majority of patients (62.8%) had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0. A left-sided primary tumor, which includes those occurring in the rectum, sigmoid, and descending colon, was observed in 88.7%, 93.3%, and 94.4% of patients in cohorts A, B, and C, respectively. Across all cohorts, most patients (80.2%) had microsatellite stable (MSS) tumors, and none had microsatellite instability–high (MSI-H) tumors. Most cancers were RAS or BRAF wild-type in all cohorts (97.7% and 98.8%, respectively); in cohort A, 1 patient’s tumor had an NRAS mutation, 1 patient’s tumor in cohort B was not examined for RAS, and 1 patient’s tumor in cohort C was not examined for BRAF. Liver metastasis at baseline was present in 66.3% of patients (Table 2). Median duration of follow-up was 14.4 months (range, 1.2–26.8) in cohort A, 6.2 months (range, 0.5–13.8) in cohort B, and 3.9 months (range, 1.1–18.9) in cohort C. Overall, 14 patients (16.3%) had a protocol deviation (Supplementary Table 2).
The median number of prior lines of treatment for metastatic disease was 4 (range, 2–11); prior treatment history for select agents is described in Table 2. All patients had previously received irinotecan therapy and in cohorts A and B, all patients had prior treatment with cetuximab and/or panitumumab. In cohorts A and C, all patients had prior treatment with fluorouracil and oxaliplatin. More than 70.0% of patients in all cohorts received prior treatment with bevacizumab. In cohort A, 30.2% of patients had previously received anti-HER2 therapy.
Efficacy
In cohort A (HER2-positive, IHC 3+ or IHC 2+/ISH+), confirmed ORR based on independent central review (ICR) was 45.3% (95% CI, 31.6–59.6), all of which were partial responses (PR) and included patients previously treated with anti-HER2 therapy (Table 3; Fig. 1A). Disease control rate (DCR) was 83.0% (95% CI, 70.2–91.9). Changes in tumor size from baseline over time are shown in Fig. 1B. The median duration of response (DoR) in cohort A was 7.0 months (95% CI, 5.8–9.5), with several patients maintaining a response until the end of the follow-up period (Supplementary Fig. 2). Median time to response (TTR) was 2.2 months (95% CI, 1.4–2.8) and the median best percentage change from baseline in target lesions was −35.0% (range, −100 to 33). Median PFS and OS were 6.9 months (95% CI, 4.1–8.7) and 15.5 months (95% CI, 8.8–20.8), respectively (Fig. 2).

A Waterfall plot showing the greatest percentage change from baseline in the sum of diameters of measurable tumors in patients with HER2-positive mCRC (cohort A). Each bar represents a patient. The line at 20% indicates progressive disease. The line at −30% indicates partial response. B Spider plot showing change over time from baseline in the sum of diameters of measurable tumors in cohorts A, B, and C. aFour patients from the full analysis set were excluded; 1 patient had no measurable target lesion and 3 patients had no postbaseline data. bBy local assessment. cOne patient from cohort B and 5 patients from cohort C had missing postbaseline data. HER2 human epidermal growth factor receptor 2, IHC immunohistochemistry, ISH in situ hybridization.

Kaplan–Meier curves representing (A) progression-free survival and (B) overall survival. Marks indicate where data were censored. HER2 human epidermal growth factor receptor 2, IHC immunohistochemistry, ISH in situ hybridization, NE not evaluable.
No objective responses were observed in cohorts B or C. The confirmed DCR was 60.0% (95% CI, 32.3–83.7) in cohort B and 22.2% (95% CI, 6.4–47.6) in cohort C. Median PFS was 2.1 months (95% CI, 1.4–4.1) in cohort B and 1.4 months (95% CI, 1.3–2.1) in cohort C (Fig. 2A). Median OS was 7.3 months (95% CI, 3.0–NE) in cohort B and 7.7 months (95% CI, 2.2–13.9) in cohort C (Fig. 2B).
Safety
All patients in the safety analysis set experienced at least one treatment-emergent adverse event (TEAE) (Table 4), of which 96.2%, 100%, and 83.3% were drug-related in cohorts A, B, and C, respectively. Grade ≥3 TEAEs were observed in 35 patients (66.0%) in cohort A, 7 patients (46.7%) in cohort B, and 14 patients (77.8%) in cohort C. Serious TEAEs were observed in 20 (37.7%), 6 (40.0%), and 9 patients (50.0%) in cohorts A, B, and C, respectively.
The most common TEAEs (reported in ≥20% of patients in any cohort) were predominantly gastrointestinal and hematologic events, mostly grade 1 or 2 (Table 5). Across all cohorts, the most common grade ≥3 TEAEs were decreased neutrophil count (22.1%) and anemia (14.0%). TEAEs associated with study drug discontinuation, dose reduction, or dose interruption were reported in 13 (15.1%), 15 (17.4%), and 34 patients (39.5%), respectively, in all cohorts. The TEAE most commonly associated with drug discontinuation was interstitial lung disease (ILD; 7.0%) and the TEAE most commonly associated with dose reduction or dose interruption was decreased neutrophil count (4.7% and 9.3%), respectively. Overall, 9 patients (10.5%) had TEAEs associated with death and 3 (3.5%) were drug-related, of which all were adjudicated as ILD. No patients experienced decreased left ventricular ejection fraction TEAEs.
Across all cohorts, adjudicated drug-related ILD/pneumonitis occurred in 8 patients (9.3%), including four grade 2, one grade 3, and three grade 5 events (Table 6). Per the study protocol, all patients received steroids. All 4 patients with grade 2 ILD/pneumonitis recovered; 1 patient with grade 3 ILD/pneumonitis did not recover and died due to disease progression. The remaining 3 patients experienced grade 5 ILD/pneumonitis. The median time to onset of the adjudicated ILD/pneumonitis events was 66.5 days (range, 7–165 days). The median duration of the adjudicated ILD/pneumonitis events was 23.0 days (range, 7–172 days; the event for 1 patient [grade 3] was ongoing at the time of data cutoff). In the 3 fatal cases adjudicated as drug-related ILD/pneumonitis, the median time to onset was 22 days (range, 7–120 days), and median time to death from diagnosis of adjudicated ILD/pneumonitis was 8 days (range, 7–19 days).
Pharmacokinetics of T-DXd, anti-HER2 antibody, and DXd
All 86 patients were included in the pharmacokinetic (PK) analysis set of T-DXd (cohort A, n = 53; cohort B, n = 15; cohort C, n = 18) for cycle 1. Overall, the serum exposure of T-DXd, total anti-HER2 antibody, and DXd in cohort A was similar to the values observed in cohorts B and C (Table 7). The terminal half-life (t1/2) was similar among the 3 cohorts. Serum exposure parameters assessed for T-DXd and anti-HER2 antibody were comparable, whereas the serum exposure of DXd was lower than the exposure of T-DXd.
Exploratory analysis of ORR and PFS according to patient subgroups
In cohort A, response rates were similar across subgroups stratified by age, sex, region, and prior anti-HER2 treatment (Fig. 3A). Responses were higher in patients with baseline HER2 IHC 3+ (57.5% [95% CI, 40.9–73.0]) compared to those with HER2 IHC 2+/ISH+ (7.7% [95% CI, 0.2–36.0]). The ORR was also higher in patients with ECOG PS of 0 compared with patients with ECOG PS of 1 (54.1% [95% CI, 36.9–70.5] vs 25.0% [95% CI, 7.3–52.4], respectively). Patients with left-sided tumors had a higher ORR (46.8% [95% CI, 32.1–61.9] vs 33.0% [95% CI, 4.3–77.7], respectively) than those with right-sided tumors, which includes those occurring in the cecum, ascending, and transverse colon. Patients with liver metastasis at baseline had a lower ORR than those without (39.4% [95% CI, 22.9–57.9] vs 55.0% [95% CI, 31.5–76.9], respectively).

A, B Forest plot of subgroups for (A) objective response ratea. Data are presented as the point estimate of ORR with its exact 95% CI. The dotted line represents the ORR of patients in the HER2 + cohort A (45.3%). Forest plot of subgroups for (B) progression-free survival. Data are presented as median PFS with its exact 95% CI. The dotted line represents the median PFS of patients in the HER2 + cohort A (6.9 months). aReprinted from ref. 16, with permission from Elsevier. bLeft: rectum, sigmoidal, descending; right: cecum, ascending, transverse. ECOG PS Eastern Cooperative Oncology Group performance status, HER2 human epidermal growth factor receptor 2, IHC immunohistochemistry, ISH in situ hybridization, mCRC metastatic colorectal cancer.
In the subgroup analysis of PFS in cohort A, the median PFS was similar in subgroups stratified by age and sex (Fig. 3B), and a marked difference in median PFS was observed in subgroups of ECOG PS of 0/1, HER2 status, prior anti-HER2 treatment, primary tumor site, and liver metastasis at baseline. Patients with HER2 IHC 3+ mCRC demonstrated improved median PFS versus HER2 IHC 2+/ISH + mCRC (8.3 months [95% CI, 5.4–10.9] vs 4.1 months [95% CI, 1.3–5.5]), which was associated with an improvement in median OS (19.9 months [95% CI, 8.8–25.3] vs 11.0 months [95% CI, 4.2–14.4]). Patients without prior anti-HER2 treatment had median PFS of 7.3 months (95% CI, 4.1–10.8), compared with 3.8 months (95% CI, 2.2–14.4) in those with prior anti-HER2 treatment. In the subgroup of patients with ECOG PS of 0 or 1, median PFS was 8.3 months (95% CI, 5.5–11.2) and 3.8 months (95% CI, 1.4–4.8), respectively. Patients with left-sided tumors had a longer median PFS (7.3 months [95% CI, 4.1–8.7] vs 3.5 months [95% CI, 1.4–11.2]) than those with right-sided tumors. Patients with liver metastasis at baseline had shorter median PFS than those without (6.9 months [95% CI, 3.5–8.7] vs 8.3 months [95% CI, 3.8–12.6], respectively).
Discussion
This longer-term follow up of DESTINY-CRC01 supports the durable antitumor activity of T-DXd in patients with HER2-positive mCRC. In this updated analysis, the confirmed ORR was 45.3%, DCR was 83.0%, and median PFS was 6.9 months. Importantly, the median OS was 15.5 months, which far exceeds the current standard of care7–9. Responses were also observed in patients across subgroups in cohort A, including those who had previously received anti-HER2–targeted therapy, although a shorter PFS was observed compared with those patients who did not receive anti-HER2 therapy. This updated analysis confirmed the lack of responses in patients with HER2-low mCRC (cohorts B and C).
Treatment selection for patients with mCRC are dependent on the tumor molecular profile, tumor location, mismatch repair status, and prior therapies received7,8,17. Currently approved third-line therapies for patients with mCRC demonstrate limited benefit7,9,12,13, with median OS of 6.4 months for regorafenib compared with 5.0 months for placebo and 7.1 months for trifluridine/tipiracil compared with 5.3 months for placebo12,13. The antitumor activity of T-DXd in mCRC, including a median OS of 15.5 months, observed in the present study in a heavily treated patient population appears promising and warrants further study.
In DESTINY-CRC01, patients had a median of 4 (range, 2–11) prior lines of therapy, which included oxaliplatin, irinotecan, fluoropyrimidines, and anti–EGFR treatments. Response rates of anti–EGFR therapies in patients with KRAS wild-type mCRC that progressed on or following chemotherapy (eg, irinotecan and oxaliplatin) were 22% with panitumumab and 19.8% with cetuximab18. HER2 has emerged as a negative predictor of response to EGFR-targeted therapy4,6,19,20,21,22, and HER2 amplification has been shown to drive primary resistance to anti–EGFR treatment7. Therefore, it is plausible that targeting HER2 may be beneficial for a fraction of patients with anti–EGFR resistant colorectal cancer23.
The primary results (data cutoff August 9, 2019) of DESTINY-CRC01 led to the recommendation of T-DXd in patients with HER2-positive mCRC in the United States24. Trastuzumab in combination with either lapatinib or pertuzumab are also included as guideline-recommended therapies24. The combination of trastuzumab plus lapatinib in HERACLES and HERACLES-A yielded ORRs of 30% and 28%, respectively, in patients with HER2-positive mCRC that progressed while on or after standard treatments25,26. Similarly, the combination of trastuzumab plus pertuzumab has demonstrated ORRs that range from 28 to 32% in patients with HER2-amplified CRC that progressed on or after standard treatments in the MyPathway and TRIUMPH studies27,28. In contrast to the HERACLES and MyPathway studies, wherein prior anti-HER2 therapies were excluded, in DESTINY-CRC01, 30.2% of patients in cohort A were previously treated with HER2-targeted therapies. In addition, patients were required to have received at least 2 prior regimens. Despite this heavy pretreatment, the antitumor activity as evidenced by ORR, PFS, and OS in DESTINY-CRC01 appears compelling.
Additional anti-HER2 therapies under investigation for HER2-positive mCRC include pertuzumab plus trastuzumab emtansine (T-DM1, an antibody–drug conjugate) and tucatinib plus trastuzumab, neither of which are included in the guideline-recommended therapy for HER2-positive mCRC. In the HERACLES-B clinical study, pertuzumab plus T-DM1 yielded a much lower ORR of 9.7% and a median PFS of 4.1 months than DESTINY-CRC0129. However, in an interim analysis of the MOUNTAINEER study, patients with HER2-positive mCRC (IHC 3+ or IHC 2+/ISH+) treated with tucatinib and trastuzumab had an ORR of 55% (17/22) and a median OS of 17.3 months30. Unlike DESTINY-CRC01, the MOUNTAINEER study excluded patients having prior anti-HER2 therapy30.
According to the subgroup analysis, we observed clinical benefit of T-DXd regardless of tumor location. Patients with right-sided tumors may have a poor prognosis31. In DESTINY-CRC01, T-DXd demonstrated activity in patients who were heavily pretreated and who had right-sided tumors, with an ORR of 33.3% and a median PFS of 3.5 months; however, the number of patients with right-sided tumors was low (n = 6). This is in contrast to anti–EGFR therapies, which have demonstrated little-to-no benefit in patients with right-sided tumors32,33,34.
Patients with HER2-low expressing mCRC (cohorts B and C) did not experience a response to T-DXd, in contrast to demonstrated efficacy in patients with HER2-low expressing breast and gastric cancers35,36,37,38,39,40. In addition, in the subgroup analysis of HER2 expression levels in cohort A, a greater proportion of patients with high levels of HER2 expression (IHC 3+) had an objective response than did patients with tumors that had moderate HER2 expression with HER2 gene amplification (HER2 IHC 2+ and ISH+); however, the number of patients with HER2 IHC 2+/ISH+ was small. These findings are consistent with the findings of the MyPathway trial, in which some patients with both HER2 gene amplification and HER2 overexpression (IHC 3+) experienced a response (ORR of 32%; 13/34 patients), including 1 CR; whereas the 8 patients without HER2 overexpression, but with HER2 amplification, did not experience a response27. Indeed, a higher IHC score also correlated with longer PFS and a greater ORR in the HERACLES-B trial29. Preclinical work in Takegawa et al. demonstrated that T-DXd was effective in both HER2-expressing CRC cells without HER2 amplification and HER2-amplified gastric cancer cells, with different mechanisms of action. However, the activity of T-DXd in HER2-amplified gastric cancer cells was dependent on HER2 signaling, whereas that in CRC cells was not. These data suggest that variation in the mechanism of action of T-DXd between cells with and without HER2 amplification or differences in intrinsic cancer cell characteristics (e.g., HER2 expression) may help to explain differences in outcomes observed in this study41. Further studies are warranted to define the patient population most likely to benefit from HER2-blockade.
The data presented here support HER2-positive (HER2 IHC 3+ and IHC 2+/ISH+) mCRC as a distinct molecular subtype compared with tumors of lower HER2 expression5,42,43. Distinctive biological differences between these subtypes may impact pathophysiology and response to treatment42. Although the exact mechanism remains unclear44, response to treatment may predominantly depend on the extent of amplification and HER2 gene copies that drive mCRC progression and may reduce the efficacy of T-DXd in patients with HER2-low mCRC42. Further assessment, through blood analysis and tumor biopsies, on the lack of antitumor activity in patients with HER2-low expressing mCRC is warranted.
The safety profile was consistent with the known safety profile of T-DXd. The most common adverse events (AEs) were mainly low-grade gastrointestinal and hematologic AEs. Overall rates of grade ≥3 (65.1%) and drug-related grade ≥3 TEAEs (48.8%) were consistent with the known AE profile of T-DXd and were comparable to rates reported in prior studies37,45,46. Dose modifications or delays in treatment were used to manage TEAEs, with the exception of grade 1 or grade 2 AEs, unless specified in the protocol. A dose could be delayed up to 28 days (49 days from the last infusion date) from the planned date of administration, and 2 dose reductions were allowed. TEAEs leading to dose reductions and discontinuations were consistent with those previously reported for T-DXd in patients with HER2-positive gastric cancer47.
PK parameters assessed for T-DXd, total anti-HER2 antibody, and DXd were generally consistent with a previous report of patients with breast cancer, with an observed lower serum exposure of DXd than T-DXd48. Overall serum exposure of T-DXd, total anti-HER2 antibody, and DXd were similar regardless of HER2 status at cycle 1.
ILD/pneumonitis is an important identified risk that requires careful monitoring and prompt intervention and is associated with T-DXd, irinotecan, and other HER2-targeted therapies. In the present study, 8 patients (9.3%) had adjudicated drug-related ILD/pneumonitis and 3 were grade 5 (3.5%), which was generally similar to the observed incidence across other tumor types37,45,47. ILD/pneumonitis was actively managed per the study protocol and all patients were treated with steroids promptly, resulting in 4 patients who recovered from ILD/pneumonitis by the time of data cutoff. Since this trial was completed, updated guidelines for monitoring and managing ILD/pneumonitis have been implemented; awareness efforts and research on risk factors for interstitial lung disease/pneumonitis are ongoing. Notably, use of T-DXd in earlier lines of therapy and proactive monitoring is recommended to manage the risk of ILD associated with T-DXd49.
A limitation of the present study is that it is not a randomized controlled trial and comparator data are needed. However, the OS benefit observed here was 15.5 months, which is 5 months greater than the reported standard of care in third-line or later mCRC9,12,13,18, and merits future studies in this patient population. Furthermore, the results of the study should be interpreted with caution given the limited sample size, and validation of these results in further studies is warranted. Additional investigation is also needed to evaluate risk factors that may increase the chance of developing ILD/pneumonitis.
T-DXd demonstrated strong and durable antitumor activity in patients with HER2-positive mCRC after 2 or more previous therapies. Responses were observed across various subgroups and in patients with previous HER2-targeted therapy. The safety profile was consistent with previous reports, and ILD/pneumonitis remains an important risk requiring monitoring and quick intervention. These promising results support the investigation of T-DXd in earlier lines of therapy and the continued exploration of T-DXd in patients with HER2-positive mCRC (DESTINY-CRC02; NCT04744831).
Methods
Study design and patients
DESTINY-CRC01 was a multicenter, open-label, 3-cohort, phase 2 trial of T-DXd in patients with HER2-positive and HER2-low advanced CRC. Independent ethics committees or institutional review boards at each study site reviewed and approved the protocol (Supplementary Table 1). The study was registered at Clinicaltrials.gov (NCT03384940) on December 28, 2017. The first patient was enrolled on February 23, 2018, and the last patient on November 10, 2020. The protocol and statistical analysis plan are available in the Supplementary Information.
Patients were eligible for the study if they had pathologically documented unresectable, recurrent, or metastatic colorectal adenocarcinoma, the presence of at least 1 measurable lesion as assessed by the investigator based on Response Evaluation Criteria in Solid Tumours (RECIST) v1.1, and an ECOG PS of 0 to 1. In addition, patients with RAS wild-type who received at least 2 prior regimens of standard treatment, including fluoropyrimidine, irinotecan, or oxaliplatin and an anti-EGFR antibody, were eligible for the study. Patients were required to provide an adequate archival tumor sample to confirm HER2 status by central laboratory; in patients with anti-HER2 therapies previously received, tumor samples used were from after anti-HER2 therapy.
Patients were excluded from the study if they had spinal cord compression or clinically active central nervous system metastases; patients with clinically inactive brain metastases or treated brain metastases that were no longer symptomatic and required no treatment with steroids or anticonvulsants were allowed in the study if they had recovered from the acute toxic effect of radiotherapy. Patients were also excluded if they had a history of ILD/pneumonitis that required steroids, had current ILD/pneumonitis, or had suspected ILD/pneumonitis that could not be ruled out by imaging at screening.
Patients were allocated to 3 separate cohorts based on their centrally confirmed HER2 status. Cohort A included patients with HER2-positive (IHC 3+ or IHC 2+/ISH+) advanced CRC. Cohort B included patients with HER2 IHC 2+/ISH− advanced CRC. Cohort C included patients with HER2 IHC 1+ advanced CRC.
All patients received 6.4 mg/kg of T-DXd administered intravenously Q3W until the occurrence of disease progression according to investigator assessment by RECIST v1.1, clinical progression, withdrawal of patient consent, unacceptable AEs, pregnancy, or death. All lesions were assessed at screening according to RECIST v1.1. Tumor assessments were conducted with computed tomography (CT) or magnetic resonance imaging (MRI) scans of the chest, abdomen, pelvis, and any other sites of disease. A CT or MRI of the brain was included for all patients.
The study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation guidelines for Good Clinical Practice, and other local regulations where applicable. Written, informed consent was provided by all patients before enrollment; participants were not compensated.
Assessments and endpoints
Efficacy assessments were based on tumor assessments performed at screening and every 6 weeks while the patient remained on T-DXd. The primary endpoint was ORR (defined as the proportion of patients who achieved a best overall response of CR or PR) assessed by independent central review based on RECIST v1.1 in cohort A. Secondary endpoints included ORR based on RECIST v1.1 in cohorts B and C; DoR, DCR, and ORR assessed by the investigator based on RECIST v1.1; PFS; OS; and pharmacokinetics of T-DXd. Safety endpoints included serious adverse events (SAEs), TEAEs, physical examination findings, vital sign measurements, standard clinical laboratory parameters, electrocardiogram parameters, echocardiogram/multigated acquisition findings, ophthalmologic findings, and anti-drug antibodies. Exploratory endpoints included TTR, best percentage change in the sum of the longest diameter of measurable tumors, and subgroup analysis of ORR and PFS.
Safety was assessed as the incidence of TEAEs, and SAEs graded based on Common Terminology Criteria for Adverse Events, version 5.0. Patients with suspected ILD/pneumonitis had treatment interrupted until further evaluation, which included high-resolution CT, pulmonologist assessment, pulmonary function tests, pulse oximetry, and other tests as needed. ILD/pneumonitis events were carefully monitored until complete resolution, including after drug discontinuation. Cases of suspected ILD/pneumonitis events were adjudicated by an external independent adjudication committee.
Two dose reductions of T-DXd were permitted, to 5.4 mg/kg and 4.4 mg/kg. Dose reductions related to toxicity were made based on investigator assessment and all cycles after a dose reduction were administered at the lower dose. Patients requiring more than 2 dose reductions were withdrawn from the study. A dose could be delayed for up to 28 days (49 days from the last infusion date) from the planned date of administration. Dose interruptions related to toxicity were made based on investigator assessment. Future cycles of T-DXd were scheduled according to the date of the last dose.
Pharmacokinetic analysis
Starting day 1 of cycle 1, blood samples were collected between 8 h to 0 h before infusion, within 15 min after the end of infusion, or at 4 h (±15 min) or 7 h (±2 h) after the start of drug administration. Samples were also collected on days 8 and 15 (7 and 14 days after the start of drug administration [±1 day]) of cycle 1, and on day 22 (±2 days) of cycle 1. If the schedule on day 1 of the following cycle was delayed for 3 days or more, including if the patient could not continue onto the next cycle, a sample would be collected. For cycles 2, 3, 4, and 6, blood samples were collected up to 8 h before infusion and within 15 min after the end of infusion. For cycle 3, blood samples were also collected at 4 h (±15 min) or 7 h (±2 h) after the start of drug administration. The serum PK parameters assessed included the maximum observed concentration (Cmax), the time to reach Cmax (Tmax), the mean area under the serum concentration-time curve to the time of the last quantifiable concentration (AUClast), the mean area under the concentration-time curve up to day 21 (AUC21d), and if appropriate, the area under the concentration-time curve up to infinity (AUCinf), t1/2, total body clearance, and volume of distribution at steady state in cycle 1 for T-DXd, total anti-HER2 antibody, and DXd for each patient. Serum PK parameters were calculated using the actual time of blood collection.
Statistical analysis
Antitumor activity analyses were assessed in the full analysis set and safety was assessed in the safety analysis set. Both sets of analyses comprised all patients in cohorts A, B, and C who had received at least 1 dose of T-DXd. The Clopper-Pearson method was used for the point estimate of ORR and its two-sided exact 95% CI by cohort. The Kaplan-Meier method was used to summarize the DoR, PFS, OS, and TTR with median event time and two-sided 95% CI for the median using Brookmeyer and Crowley method by cohort. Subgroup analyses of age, sex, region, ECOG PS, HER2 status, prior anti-HER2 treatment, and primary tumor site for ORR and PFS were carried out for cohort A using the same methodology for the overall analysis of the corresponding endpoint. These results were performed only if there were at least 10 patients in each of the categories and considered exploratory due to smaller sample sizes that could not be prespecified.
For cohort A, a sample size of 48 patients provided a 90% probability of achieving a lower limit of 95% CI for the ORR that exceeded 15% under the expected ORR of 35% and enabled a statistical comparison with a historical control on PFS. For cohorts B and C, with a sample size of 20 patients each, the probability that more than 4 responders out of 20 patients are observed will be less than 5% under the threshold ORR of 10%, but more than 75% under the expected ORR of 30%.
Descriptive statistics were used for the best (minimum) percentage change from baseline in the sum of diameters and presented as waterfall and spider plots for each cohort. Only patients with measurable tumors at baseline were included. The data was collected via Medidata Classic Rave 2019.2.1. All analyses were performed using SAS version 9.4.
Analysis of PK parameters was based on the PK analysis set, which included all patients who received at least 1 dose of the study drug and had measurable serum concentrations of T-DXd. Serum concentrations of T-DXd, total anti-HER2 antibody, and DXd were analyzed using non-compartmental analysis with the validated computer program Phoenix® WinNonlin® 6.4 or higher.
Data availability
Anonymized individual participant data (IPD) on completed studies and applicable supporting clinical trial documents may be available upon request at the Vivli website (https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/). In cases where clinical trial data and supporting documents are provided pursuant to our company policies and procedures, Daiichi Sankyo Companies will continue to protect the privacy of our clinical trial participants. Details on data sharing criteria and the procedure for requesting access can be found at Vivli’s Daiichi Sankyo web page (https://vivli.org/ourmember/daiichi-sankyo). Individual participant data, including data dictionaries, will be available. Documents that will be available include the clinical trial protocol, statistical analysis plan, informed consent form, and clinical study report. Data may be requested after the indication has been approved by major health authorities and the study results are published. The data will be made available to qualified science and medical researchers upon formal request and submission of a research proposal detailing planned analyses. De-identified IPD and relevant clinical trial documents will be shared for the purpose of conducting legitimate research as specified in an approved formal research proposal and may be available upon request via the Vivli Data Sharing Platform at https://vivli.org/.
References
Ross, J. S. et al. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer 124, 1358–1373 (2018).
Siena, S. et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 29, 1108–1119 (2018).
Jeong, J. H. et al. HER2 amplification and cetuximab efficacy in patients with metastatic colorectal cancer harboring wild-type RAS and BRAF. Clin. Colorectal Cancer 16, e147–e152 (2017).
Sartore-Bianchi, A. et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist 24, 1395–1402 (2019).
Sawada, K. et al. Prognostic and predictive value of HER2 amplification in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 17, 198–205 (2018).
Raghav, K. et al. Validation of HER2 amplification as a predictive biomarker for anti–epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. JCO Precision Oncol. 3, 1–13 (2019).
Van Cutsem, E. et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 27, 1386–1422 (2016).
Vogel, A., Hofheinz, R. D., Kubicka, S. & Arnold, D. Treatment decisions in metastatic colorectal cancer - Beyond first and second line combination therapies. Cancer Treat. Rev. 59, 54–60 (2017).
Yoshino, T. et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann. Oncol. 29, 44–70 (2018).
Liu, X. et al. Retreatment with anti-EGFR based therapies in metastatic colorectal cancer: impact of intervening time interval and prior anti-EGFR response. BMC Cancer 15, 713 (2015).
Cremolini, C. et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 5, 343–350 (2019).
Grothey, A. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013).
Mayer, R. J. et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl. J. Med. 372, 1909–1919 (2015).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Ogitani, Y., Hagihara, K., Oitate, M., Naito, H. & Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 107, 1039–1046 (2016).
Siena, S. et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol. 22, 779–789 (2021).
Di Nicolantonio, F. et al. Precision oncology in metastatic colorectal cancer - from biology to medicine. Nat. Rev. Clin. Oncol. 18, 506–525 (2021).
Price, T. J. et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 15, 569–579 (2014).
Bertotti, A. et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 1, 508–523 (2011).
Bertotti, A. et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 526, 263–267 (2015).
Martin, V. et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br. J. Cancer 108, 668–675 (2013).
Yonesaka, K. et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 3, 99ra86 (2011).
Xie, Y. H., Chen, Y. X. & Fang, J. Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct. Target. Ther. 5, 22 (2020).
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Colon Cancer. Version 3.2021. National Comprehensive Cancer Network (2021).
Sartore-Bianchi, A. et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 17, 738–746 (2016).
Tosi, F. et al. Long-term clinical outcome of trastuzumab and lapatinib for HER2-positive metastatic colorectal cancer. Clin. Colorectal Cancer 19, 256–262.e252 (2020).
Meric-Bernstam, F. et al. Pertuzumab and trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 20, 518–530 (2019).
Nakamura, Y. et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: a phase 2 trial. Nat. Med. 27, 1899–1903 (2021).
Sartore-Bianchi, A. et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open 5, e000911 (2020).
Strickler, J. H. et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): Initial results from the MOUNTAINEER trial. Ann. Oncol. 30, Abstract 527PD (2019).
Loupakis, F. et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J. Natl. Cancer Inst. 107, dju427 (2015).
Brulé, S. Y. et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur. J. Cancer 51, 1405–1414 (2015).
Moretto, R. et al. Location of primary tumor and benefit from anti-epidermal growth factor receptor monoclonal antibodies in patients with RAS and BRAF wild-type metastatic colorectal cancer. Oncologist 21, 988–994 (2016).
Chen, K. H. et al. Primary tumor site is a useful predictor of cetuximab efficacy in the third-line or salvage treatment of KRAS wild-type (exon 2 non-mutant) metastatic colorectal cancer: a nationwide cohort study. BMC Cancer 16, 327 (2016).
Modi, S. et al. Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a phase Ib study. J. Clin. Oncol. 38, 1887–1896 (2020).
Doi, T. et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 18, 1512–1522 (2017).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020).
Toshinari, Y. et al. A phase 1, multicenter, open-label study to assess the effect of [fam-] trastuzumab deruxtecan (T-DXd; DS-8201a) on QTc and pharmacokinetics in subjects with HER2-expressing metastatic and/or unresectable breast cancer. Can. Res. 80, 18–12 (2020).
Yamaguchi, K. et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients with HER2-low, advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma: Results of the exploratory cohorts in the phase II, multicenter, open-label DESTINY-Gastric01 study. Ann. Oncol. 31, S899–S900 (2020).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Takegawa, N. et al. [fam-] trastuzumab deruxtecan, antitumor activity is dependent on HER2 expression level rather than on HER2 amplification. Int. J. Cancer 145, 3414–3424 (2019).
Yagisawa, M. et al. Prognostic value and molecular landscape of HER2 low-expressing metastatic colorectal cancer. Clin. Colorectal Cancer 20, 113–120.e111 (2021).
Nam, S. K. et al. BRAF, PIK3CA, and HER2 oncogenic alterations according to KRAS mutation status in advanced colorectal cancers with distant metastasis. PLoS ONE 11, e0151865 (2016).
Siravegna, G. et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin. Cancer Res. 25, 3046–3053 (2019).
Li, B. T. et al. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N. Engl. J. Med. 386, 241–251 (2022).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med. 386, 1143–1154 (2022).
Shitara, K. et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 382, 2419–2430 (2020).
Shimomura, A. et al. Effect of trastuzumab deruxtecan on QT/QTc interval and pharmacokinetics in HER2-positive or HER2-low metastatic/unresectable breast cancer. Clin. Pharmacol. Ther. https://doi.org/10.1002/cpt.2757 (2022).
Powell, C. A. et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open. 7, 100554 (2022).
Acknowledgements
This study was sponsored by Daiichi Sankyo and funded by both Daiichi Sankyo and AstraZeneca. The sponsor was involved in data collection, analysis, interpretation, and preparation of the manuscript. We thank the patients who participated in this study, as well as their families and caregivers. We also thank the staff and investigators at all the study sites. We thank Masato Fukae, PhD, and Emi Kamiyama, PhD, for the analysis of the pharmacokinetic parameters, both of whom are employed by Daiichi Sankyo. Under the guidance of the authors, assistance in medical writing and editorial support was provided by Cindy M. Rigby, PhD, and Marianna B. Johnson, PhD, of ApotheCom, and was funded by Daiichi Sankyo.
Author information
Authors and Affiliations
Consortia
Contributions
All authors were involved in the design or conception of the study, in the drafting and revision of the manuscript for publication, in interpretation of the data, and the approval of the final manuscript. T.Y., A.G., and S.S. were members of the study steering committee. T.Y., K.S., A.G., K.K., E.B., and S.S. contributed to the development of the study protocol. T.Y., M.D.B., K.R., T.M., F.L., H.K., K.Y., T.N., Z.W., E.E., J.R., M.F., F.C., A.G., K.K., E.B., and S.S. were involved in data collection. Y.O., K.K., E.B., and G.M. were involved in data analysis. K.S., K.K., E.B., and Y.O. verified the data. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: T.Y. has received support for the present manuscript from Daiichi Sankyo and has received institutional grants or contracts from Taiho Pharmaceutical, Sumitomo Dainippon, Ono Pharmaceutical, Chugai Pharmaceutical, Amgen, Parexel International MSD, and Sanofi KK. M.D.B. has received research funding, provision of study materials, medical writing, and article processing charges from Daiichi Sankyo. K.R. has received institutional research funding from Daiichi Sankyo; has received grants or contracts from Roche/Genentech; and has received honoraria for lectures, presentations, speaker bureaus, manuscript writing, or educational events from Daiichi Sankyo. T.M. has received institutional grants or contracts from MSD, Daiichi Sankyo, Ono Pharmaceuticals, and Novartis, and has received honoraria from Takeda, Chugai Pharmaceuticals, Merck Bio Pharma, Taiho, Bayer, Eli Lilly Japan KK, Yakult Honsha, Sanofi, Daiichi Sankyo, Ono Pharmaceuticals, and BMS. H.K. has received grants or contracts from Chugai Pharmaceuticals, Taiho Pharmaceuticals, and Eisai Co. Ltd; has received consulting fees from BMS, Eli Lilly Japan KK, Ono Pharmaceuticals, Daiichi Sankyo, and Taiho Pharmaceuticals; and has received honoraria from BMS, Bayer Yakuhin Ltd, Eli Lilly Japan KK, MSD KK, Ono Pharmaceuticals, Chugai Pharmaceuticals, Daiichi Sankyo, Takeda Pharmaceuticals, and Taiho Pharmaceuticals. K.Y. has received institutional support for the present manuscript from Daiichi Sankyo and has received honoraria for lectures from Daiichi Sankyo. T.N. has received institutional support for the present manuscript from Daiichi Sankyo and has received honoraria from Ono Pharmaceuticals, Taiho Pharmaceuticals, Takeda Pharmaceuticals, Chugai Pharmaceuticals, and Daiichi Sankyo. Z.W. has received consulting fees from Merck, Ibsen, Eli Lilly, Five Prime, QED, Molecular Templates, Daiichi Sankyo, AstraZeneca, Bayer, Novartis; has received support for attending meetings and/or travel from Lilly, Merck, Novartis, and Daiichi Sankyo; and has participated on a data safety monitory board or advisory board for Array. E.E. has received grants or contracts from F. Hoffman La-Roche, BMS, Servier, Amgen, Merck Serono, Array Biopharma, Sanofi, and Bayer; institutional funds for grants from Array Biopharma, MSD, AbbVie, Amgen, GlaxoSmithKline, AstraZeneca, Merck Sharpe and Dohme Corp, BMS, Novartis, Boehringer Ingelheim, Hoffman La-Roche, MedImmune, Pierre-Fabre, and Sanofi Aventis; has received consulting fees from Hoffman La-Roche, BMS, Servier, Amgen, Merck Serono, Array Biopharma, Sanofi, Bayer; has received honoraria from F. Hoffman La-Roche, BMS, Servier, Amgen, Merck Serono, Array Biopharma, Sanofi, Bayer; has received payment for expert testimony from F. Hoffman La-Roche, BMS, Servier, Amgen, Merck Serono, Array Biopharma, Sanofi, Bayer; and has received support for attending meetings and/or travel from F. Hoffman La-Roche, BMS, Amgen, Merck Serono, Array Biopharma, Sanofi, Bayer. M.F. has received institutional grants or contracts from AstraZeneca, Novartis, and Amgen; has received consulting fees from HalioDx, Incyte Corporation, Mirati, Pfizer, Taiho, and Zhuhai Yufan Biotech; has received honoraria from Amgen and Guardant360; and has participated on a data safety monitoring board or advisory board for Amgen, Seattle Genetics, GlaxoSmithKline, Array Biopharma, Mirati, and Bayer. F.C. has served as an advisor and speaker for Roche, Amgen, Merck Serono, Pfizer, Sanofi, Bayer, Servier, Bristol Myers Squibb, Celgene, and Eli Lilly; and received institutional research grants from Bayer, Roche, Merck Serono, Amgen, AstraZeneca, and Takeda. A.G. has received institutional medical writing support from Daiichi Sankyo; has received consulting fees from Daiichi Sankyo, Bayer, Merck/MSD, Genentech/Roche, Natera, and BMS; has received honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from Bayer, Genentech/Roche, and Merck/MSD; and has participated on a data safety monitoring board or advisory board for Regeneron. K.S., K.K., E.B., Y.O., and G.M. are full-time employees of Daiichi Sankyo. F.L., J.R., and S.S. have nothing to declare.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoshino, T., Di Bartolomeo, M., Raghav, K. et al. Final results of DESTINY-CRC01 investigating trastuzumab deruxtecan in patients with HER2-expressing metastatic colorectal cancer. Nat Commun 14, 3332 (2023). https://doi.org/10.1038/s41467-023-38032-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38032-4
- Springer Nature Limited
This article is cited by
-
HER2-Positive Metastatic Colorectal Cancer
Current Treatment Options in Oncology (2024)
-
Trastuzumab deruxtecan in patients with locally advanced or metastatic HER2-positive gastric cancer: a multicenter, open-label, expanded-access study
International Journal of Clinical Oncology (2024)
-
Trastuzumab-Deruxtecan: Redefining HER2 as a Tumor Agnostic Biomarker
Targeted Oncology (2024)
-
Die Bedeutung Entitäten-agnostischer Zulassungen im klinischen Alltag
InFo Hämatologie + Onkologie (2023)