

Abstract
Houseflies provide a good experimental model to study the initial evolutionary stages of a primary sex-determining locus because they possess different recently evolved proto-Y chromosomes that contain male-determining loci (M) with the same male-determining gene, Mdmd. We investigate M-loci genomically and cytogenetically revealing distinct molecular architectures among M-loci. M on chromosome V (MV) has two intact Mdmd copies in a palindrome. M on chromosome III (MIII) has tandem duplications containing 88 Mdmd copies (only one intact) and various repeats, including repeats that are XY-prevalent. M on chromosome II (MII) and the Y (MY) share MIII-like architecture, but with fewer repeats. MY additionally shares MV-specific sequence arrangements. Based on these data and karyograms using two probes, one derives from MIII and one Mdmd-specific, we infer evolutionary histories of polymorphic M-loci, which have arisen from unique translocations of Mdmd, embedded in larger DNA fragments, and diverged independently into regions of varying complexity.
Similar content being viewed by others


Introduction
Sex determination mechanisms are highly diverse and undergo rapid turnover in evolution. In insects, sex is determined by a hierarchical cascade in which upstream genes regulate the activity of downstream genes. New sex determination genes can be added sequentially or emerge to replace old sex-determining genes at the top of the cascade1. Several primary signal genes have been characterized in insects (reviewed in ref. 2). These genes share remarkably little homology, suggesting that they have arisen independently. As of yet, we know very little about how novel sex-determining genes evolve, both in terms of neofunctionalization of existing sequences and the associated genomic rearrangements.
The emergence of a novel sex determination gene will affect its genomic surroundings. A dominant male- or female-determining gene will always be hemizygous. A specific prediction of the canonical sex chromosome evolution model is that a sex-determining region will undergo progressive recombination suppression3,4,5,6,7. Suppressed recombination is predicted to prevent gene flow between proto-sex chromosomes so that the sex-determining region can be sex-limited and thus effectively hemizygous. This leads to mutation accumulation, transposon insertion, and other structural rearrangements that increase the sequence divergence between the sex chromosome pair6. Validation of this model requires more detailed knowledge of the genomic organization of sex determination loci as well as their neighboring regions.
The housefly (Musca domestica) has a polymorphic sex determination system8,9 that has been instrumental for investigating early processes of sex chromosome evolution10,11,12. A male development trajectory can be induced by a dominant male-determining locus M on the Y chromosome8,13. However, an M-locus can also be present on any of the five autosomes or on the X chromosome14,15,16,17,18,19,20. All chromosomes carrying an M-locus appear to be of recent origin21, suggesting that they are “proto-Y” chromosomes. M is needed to break the autoregulatory splicing loop of the female-promoting transformer (Mdtra) gene to allow for male development. We previously identified Musca domestica male determiner (Mdmd), which is a paralogue of the generic splice factor gene nucampholin (Mdncm), as a male-determining gene of the housefly13. Mdmd is present in M-loci on chromosomes II (MII), III (MIII), and V (MV) and the Y chromosome (MY). However, the structures of the various M-loci are both diverse and complex13, providing a unique opportunity to investigate the primary evolution of sex-determining regions and sex chromosomes.
Here, we show the genomic organization of Mdmd-containing M-loci on various proto-Y chromosomes in the housefly. We find different levels of complexity for these loci, reflected in the number of Mdmd copies and intervening sequences. MV contains only two expressed Mdmd copies in palindromic structure. In contrast, MIII contains numerous Mdmd copies of which only one is functional, and some intervening sequences that represent non-male-specific repeats. MII and MY share MIII-like architecture albeit with fewer repeats. Together, our genomic and cytogenetic results point to a common origin but distinctive evolution of M-loci.
Results
In the following text, genomic regions with a dominant male-determining locus are referred to as M-loci with a Roman numeral superscript indicating on which chromosome the locus is found, i.e., MIII is the M-locus on chromosome III. Non-italic letter M with an Arabic number is used to describe housefly strains or genomic datasets (e.g., M5 is a strain with MV and females without M). Mdmd is the male-determining gene within all of the M-loci investigated.
Complexity and chromosomal location of M-loci
Previous comparison of MII, MIII, and MY revealed that they all contain at least one complete Mdmd gene and various incomplete copies13. In order to estimate structural divergence between M-loci, we performed Illumina sequencing on strains M3 (males that carry MIII and females without an M), M5 (males that carry MV), and M2 (males that carry MII). We also used published Illumina reads of three MY strains of different geographical origin21, namely aabys (laboratory generated strain with MY), A3 (strain with MY that was derived from a collection in Marshall County, Alabama, USA in 1998), and LPR (strain with MY that was originally collected near Horseheads, New York, USA). See Table 1 in Methods for an overview of the strains used and type of genomic data analyzed in this study. We determined the read mapping coverage per base pair of Mdmd relative to that of three single-copy reference genes: Mdtra, yellow (MdY), and asense (Mdase), based on the Illumina sequence data. Such coverages essentially represent Mdmd copy numbers in the tested M-loci and, therefore, are indicative of differences in the sizes of M genomic loci. The two M3 male datasets had the highest average coverage (~41.44 and ~41.88) indicating the highest copy number of Mdmd in MIII, whereas these were lowest in the M5 male dataset (~2.38, Fig. 1). Coverages in the M2 male dataset (~18.58) and two MY datasets (aabys-male, ~19.62; A3-male, ~19.74) were approximately half of the MIII value. Interestingly, one MY dataset, LPR-male, had higher average coverage (~34.78) than the other two MY datasets, and almost as large as the MIII coverage. Taken together, these data reveal that the number of Mdmd sequences vary considerably both between and within M-containing chromosomes.

Coverage rates in female genomes are included to account for off-target mapping to the paralogous gene Mdncm and the calculated average coverages in two M3 female Illumina datasets turned out to be negligible. Average coverages demonstrate that the number of Mdmd sequences are highest in MIII, intermediate in MY and MII, and lowest in MV. Source data are provided as a Source Data file.
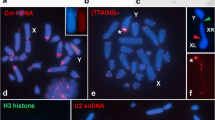
To identify the cytogenetic localization of M-loci on the male-determining chromosomes of various housefly strains, we performed fluorescence in situ hybridization (FISH) with an Mdmd-specific probe and karyogram obtained from the brain tissues of third-instar larvae. M-loci on chromosome II and III as well as on the X and Y chromosomes were successfully localized by detecting a single signal, indicating the presence of clustered Mdmd sequences on these chromosomes (Fig. 2a, b; Supplementary Fig. 1). The MII, MIII, and MY-loci were all located in the pericentromeric regions on the short arm of the chromosomes. M on the X (MX) was located on one arm of the chromosome but was not pericentromeric. Using samples from multiple laboratory strains, as well as wildtype strains from Spain, Italy, and the Netherlands, the M-loci were localized at the same position on their respective chromosomes, regardless of strain origin (Supplementary Fig. 1), suggesting a single evolutionary origin of each of these M-loci. In the M5 samples, we did not detect a hybridization signal for MV (Fig. 2c) although PCR assays were positive for the presence of Mdmd. This is likely due to the low resolving power of the Mdmd-specific probe, which is insufficient to generate a detectable signal if few Mdmd copies are present.

a, b Using an Mdmd-specific probe, MY and MIII were localized to pericentromeric regions of the Y chromosome and chromosome III respectively. c MV was not detected by the Mdmd probe due to insufficient gene copy numbers. d–i Using a probe containing a mixture of amplified MIII sequences including Mdmd and non-Mdmd intervening sequences, the M-locus and the M and sex chromosome-located (MAS) regions of the XY chromosome pair were localized. The signals of the mixed probe on the Y chromosome cover most parts of the short arm and merge with the MY signal. The signal of the mixed probe on the X chromosome mark at the ends of both arms. Positive signals are shown in red and indicated by open triangles, chromosomes are indicated by arrows. Metaphase chromosomes are shown in blue. Signals were only considered as a successful hybridization if they were observed with consistent chromosomal locations on at least 20 metaphase nuclei on each slide. For each strain, 2–3 individuals were tested to ensure reproducibility. Scale bar: 10 μm.
As the results indicated that the genomic sizes of MIII and MV were the most distinct, we proceeded with these two Ms. The housefly reference genome was generated from female genomic DNA22, and we therefore assembled male genomes from Pacbio SMRT sequencing of the strains M3 (~116× total coverage) and M5 (~161× total coverage) in order to obtain genomic sequences of MIII and MV. Both of the assembled genomes were ~1.3 Gb in size; the M3 genome assembly consists of 11,176 contigs with an N50 of ~617.5 kb, and the M5 genome assembly contains 4327 contigs and has an N50 of ~7800.3 kb. The haploid housefly genome is estimated to be ~1 Gb23, suggesting that our assemblies either contain unresolved allelic variation or phased assembly of the proto-X and proto-Y chromosomes. According to BUSCO analysis, both genomes have ~99% complete matches to 3285 universal single‐copy orthologs in dipteran lineages. In addition, we investigated an MY of the aabys strain, in order to compare autosomal M-loci to M from a morphologically differentiated XY pair. We obtained MY sequences by generating an assembly (aabys-male) with Pacbio sequencing data, which was polished with Illumina sequencing data of males from the same strain (~13× coverage). Details of the three assemblies can be found in Supplementary Table 1.
Genomic structure of the M V
We first screened the M5 genome for Mdmd-containing contigs. We identified one ~4 Mb contig (tig00004758; Fig. 3a, Supplementary Table 2, referred to as MV-contig) that contained two intact copies of Mdmd in opposing orientation, approximately 4.7 kb apart (Fig. 3a). This is in line with the estimated ~2× coverage of Mdmd for the M5 genome. Only a single synonymous nucleotide substitution, located in exon 2, was found between these two Mdmd copies. Based on this nucleotide difference, we identified transcripts of both Mdmd copies (Supplementary Fig. 2), demonstrating that both are expressed. The MV-contig is the only contig of the M5 genome that harbors Mdmd sequences, which indicates that MV has a compact architecture.

a Dotplot visualizations of Mdmd presence in inverse orientation on MV-contig indicated by blue (forward) and orange (reverse) lines that represent longer stretches of sequence similarity. b Alignments between MV-contig and non-MV-contigs show MV was inserted in a tandem repetitive region, indicated by the gap. c Self-alignment of MV demonstrates that the main part of MV is a palindrome with a non-palindromic spacer sequence of 3046 bp in the middle. Two blocks of tandem repeats exist on each end of MV (indicated by red arrows). d Schematic drawing indicates the sequence contents of MV and percentage identities between palindromic arms. The spacer sequence shows homology to a reverse transcriptase sequence and an ncRNA. The red blocks and the missing parts represent insertions/deletions. The small green blocks indicate the existence of 9 bp direct repeats at the MV borders. e Single-copy BUSCOs in the MV-contig mainly correspond to those on chromosome 2 R (Muller element C, chromosome V in the housefly) in Drosophila melanogaster. f The 9-bp direct repeats (TTTTAGGTT) are found with one copy in non-MV-contigs at insertion sites, indicated by the red dashed-line box. In non-MV-contigs, the upstream and downstream sequences of TTTTAGGTT match with the upstream and downstream sequence of MV (indicated by the blue and orange shading). g Two examples of other genomic regions that contain the same tandem repeat blocks as MV. The self-alignment figures demonstrate MV-like palindromic structures in Contig1514 and Contig1321. Alignment between Contig1514 and Contig1321 shows sequence similarity only for the palindromic region but not for the palindrome flanking sequences. The TIRs in MV are also found in Contig1514 and Contig1321 palindromes, and direct repeats with the same 9-bp length are flanking them.
To determine the borders of MV, we examined whether parts of the MV-contig were covered by sequences derived from the non-M-containing chromosome V of the M5 and M3 genomes. We identified one such contig in the M5 genome (tig00002184, referred to as non-MV-contigM5) and one in the M3 genome (contig7533, referred to as non-MV-contigM3). Alignment of the MV-contig and both non-MV-contigs revealed the sequences shared between chromosome V with and without the M-locus (Fig. 3b). The ~31 kb sequence, which exists only on the MV-contig and includes the two opposing Mdmd copies, can thus be considered as the complete MV locus. MV is integrated in a tandem repeat block with a repeat unit ~10 kb shared between the MV-contig and both non-MV-contigs.
MV has a palindromic structure (Fig. 3c, d) with the two arms separated by a 3046 bp spacer sequence. Part of the spacer sequence shows homology to reverse transcriptase in Lasius niger (Accession: KMQ86458) and Drosophila simulans (Accession: AAS13459), and partially overlaps with a predicted housefly non-coding RNA (Accession: LOC109613599). At each end of the spacer, mariner-like terminal repeats are present and extend into the palindrome arms. Although some small variations and a few deletions/insertions were found, high sequence identity was observed between the palindromic arms. Based on the distribution of single-copy BUSCOs, similar synteny was observed between the MV-contig and Drosophila melanogaster chromosome 2 R (Muller element C), which corresponds to chromosome V in the housefly21, confirming the chromosomal location of MV (Fig. 3e).
RepeatModeler recognized large blocks of tandem repeats (Fig. 3c, d) located palindromically at the distal parts of MV as interspersed repeats, reminiscent of transposable elements. Moreover, at the ends of the MV locus, we identified Terminal Inverted Repeats (TIRs) and a 9-bp long direct repeat (TTTTAGGTT), which flanks the TIRs and is present as a single copy in the non-MV-contigs (Fig. 3f). This direct repeat sequence thus resembles a target site duplication of a transposition event. Interestingly, by examining 16 independent genomic regions containing a similar stretch of interspersed repeats and palindromic structures, we could identify almost identical TIRs to MV and respective target site duplications (Fig. 3g; Supplementary Fig. 3).
The complex structure of M III
The architecture of MIII is distinctive from MV, MIII contains only a single functional Mdmd gene, and also a high number of additional truncated copies of Mdmd. We identified two contigs in the M3 genome carrying Mdmd sequences (Contig6762, ~202 kb, referred to as MIII-contig-1; Contig7871, ~389 kb, referred to as MIII-contig-2; Fig. 4a, Supplementary Table 2). The Mdmd sequences scattered across both contigs indicate the large size of MIII. We could not determine the exact borders of MIII because we did not find corresponding sequences derived from the non-M-containing chromosome III when performing a BLAST search with the terminal sequences of both MIII-contigs against the M3 and M5 genomes. Thus, the MIII locus might extend beyond the length of the two MIII-contigs. As these two MIII-contigs share high sequence similarity at one end of each contig (Fig. 4c), they are presumably connected via the overlap. We, therefore, consider both MIII contigs as part of one continuous locus encompassing in total ~591 kb, which is more than twenty times larger than MV.

a Visualization of Mdmd sequences distribution in MIII-contigs. Only one complete Mdmd copy is found (blue line indicated by the red arrow). b Self-alignments of MIII-contigs show many tandem duplications clustering together (short blue lines, indicated by solid-lined boxes). Most of the duplications are Mdmd copies and their flanking sequences (red solid-lined boxes). Duplications of non-Mdmd sequences also exist (indicated by gray solid-lined boxes). c The end part of MIII-contig-1 shares homology to the beginning part of MIII-contig-2 (indicated by the red dashed-lined box). d Visualization of Mdmd sequences distribution in MY-contigs. Only one complete Mdmd copy is found in MY-contig-4 (blue line indicated by the red arrow). e MY-contig-4 show homology to regions of MIII- and MV-contigs that contain intact Mdmd gene which is indicated by red shading. f Schematic drawing of homologous regions that contain intact Mdmd in MIII, MV and MY. MV and MY both have a block of tandem repeats is located downstream (~4 kb apart) of the intact Mdmd. g MY-contigs show homology to various parts of MIII-contigs. h Tandem repeats that are adjacent to intact Mdmd in MV and MY are also found in MIII, but exist near one end of MIII-contig-2. i The coverage for MIII-specific regions in various male genomic datasets that contain sequences of MII, MIII, MV, and MY loci respectively. Schematic drawing shows Mdmd distribution (complete copy blue, truncated copies orange) in MIII-contigs. Other M-loci show various similarities to the MIII. I: Specific to MIII; II: Shared between MIII and LPR MY; III: Shared among MIII and all three MY; IV: Shared among MII, MIII and all MY; V: Shared among all tested M-loci.
Unlike MV, the MIII-contigs do not have a palindromic structure, but instead, contain highly replicated sequences that mostly occur in a tandem (head-to-tail) fashion and largely cluster together. Even though the majority of the repetitive sequences are truncated Mdmd copies (approximately 13% of MIII-contig-1 and 26% of MIII-contig-2), non-Mdmd-associated repeats were also identified (Fig. 4b, gray boxes). The Mdmd copies and the additional repeats do not show any obvious replication pattern, as the repeated sequences vary in length as well as start and end points (Supplementary Fig. S4).
In MIII, we identified 88 Mdmd copies, of which only one represents an intact open reading frame (ORF) (Supplementary Data 1, No. 36). To identify genes in MIII other than Mdmd, we used sequences of MIII-contigs as queries in a BLAST search against the NCBI M. domestica (Taxid: 7370) Nucleotide Collection database. We found many matches to uncharacterized mRNA and ncRNA sequences as well as 17 matches to predicted genes (Supplementary Table 3). For each of these partially matched genes, we could identify MIII-independent contigs with higher sequence similarity, which indicates that the non-Mdmd genes in MIII are likely degenerated pseudo-copies of genes present elsewhere in the genome. None of these genes have been reported to be involved in sex-determination. Using RepeatModeler, we identified 136 instances of known transposable elements in MIII-contig-1 and 196 in MIII-contig-2 (Supplementary Table 4). In nine cases, the transposable element resides within Mdmd copies (Supplementary Data 2), which indicates that some transposons accumulated after Mdmd replication in MIII.
M Y shows homology to both M III and M V
In the aabys-male genome, we retrieved 4 contigs, MY-contig-1 (contig_6317_pilon), MY-contig-2 (contig_2268_pilon), MY-contig-3 (contig_2269_pilon), and MY-contig-4 (contig_12930_pilon), that contain Mdmd sequences, which were considered as MY sequences (Fig. 4d, Supplementary Table 2). Note that because of the low coverage of our Pacbio dataset, we likely did not capture all sequences of MY. The MY-contigs are informative as one of them (MY-contig-4) appears to contain an intact copy of Mdmd although with several indels that may be due to the low quality of the assembly. Upon examining all four MY-contigs, many truncated Mdmd copies are observed in a tandem fashion similar to MIII (Fig. 4d). Further homology is found for MY-contigs to various parts of MIII-contigs (Fig. 4e, f, g). MY-contig-1 and MY-contig-2 align with two separate regions on MIII-contig-1 which are ~50 kb apart (Fig. 4g). MY-contig-3 and MY-contig-4 align to a continuous region on MIII-contig-2, which cover both upstream and downstream sequences of the intact Mdmd gene. Thus, MY shares a similar sequence architecture with MIII, which is also demonstrated via independent alignment of Illumina aabys male reads to MIII (see below, Fig. 4i).
In all three investigated loci, MIII, MV and MY, homology is observed for upstream and downstream sequences of the intact Mdmd gene (Fig. 4f). The Mdmd flanking region described as the spacer in MV, which includes the partial ncRNA, reverse transcriptase-like sequences and mariner-like terminal repeats, is found in all three M-loci (Fig. 4f). Interestingly, MY harbors sequence arrangements that are found in MV but not in MIII. In MY, a block of tandem repeats is located downstream (~4 kb apart) of the intact Mdmd. Similar arrangements of the same repeats exist on both palindrome arms of MV (Fig. 4e, f). Although these repeats are also found in MIII, they exist near one end of MIII-contig-2 and are not adjacent to the intact Mdmd copy (Fig. 4h). Thus, MY has a MIII-like structure but also shares sequence characteristics with MV.
Structures of M II and M Y based on Illumina read mapping
Given the distinct architectures of MIII, MV, and MY, we further examined the structures of the M-loci by mapping Illumina reads originating from males against the MIII-contigs. As Illumina reads are short and MIII contigs contain many repetitive sequences, we first mapped female reads against the MIII-contigs to identify the repetitive regions in MIII that are not M-locus-specific (Supplementary Fig. 5). When subsequently mapping the male reads, we subtracted the regions that showed female coverage. Thus, coverages only for MIII-locus-specific regions were retained. The Illumina reads from M3 males cover the entire MIII-locus, whereas sequences from male M5 cover only the region of the functional Mdmd (Fig. 4e). The coverage patterns of the three MY datasets are quite similar, though LPR-male showed additional coverage of MIII-specific sequences containing Mdmd copies (section II in Fig. 4e). This is consistent with the aforementioned results that the MY-locus in LPR male contains more Mdmd copies than the other MY-loci. M2-male sequences, for which we did not produce any PacBio sequencing data, show less coverage of MIII than MY but still cover about 300 kb of the MIII-locus (roughly 50%). The similar coverage patterns of MII, MIII, and MY datasets indicate the presence of highly similar sequence regions in these M-loci, which suggests an already complex architecture prior to the origin of these individual loci. Additionally, differences observed among MY imply that the duplication events within the M-loci happened gradually in sequential steps, or that there was a large ancestral MY-locus, which was degraded differently in various strains.
Sequence divergence of intact Mdmd copies in different M-loci
To examine sequence divergence across in intact Mdmd copies of MII, MIII, MV and MY, we drew the Mdmd consensus sequence for the ORF based on our data on MIII and MV as well as previously published sequences13. Mdmd in MIII contains the highest number (8) of nucleotide differences from the consensus, whereas the two copies in MV have the fewest (1 in MdmdA, 2 in MdmdB, Supplementary Table 5). The number of divergent sites in Mdmd in MII and MY is 4 and 6 respectively. Besides one divergent site that is shared between MV Mdmd copies (nucleotide position 527, G), different Mdmd sequences all possess unique sets of divergent sites, indicating these mutations arose independently in those M-loci. These few nucleotide differences did not allow for a reliable reconstruction of the ancestry of the Mdmd gene when using Mdncm as an outgroup.
Chromosomal localization of M-loci and M-associated repeats on the X and Y chromosome
Alongside the aforementioned Mdmd-specific probe, we applied another probe, referred as the Mix probe, for FISH. The Mix probe was generated from PCR products that were specifically amplified from the genomic DNA of MIII-locus. The PCR products contains Mdmd fragments and interveing non-Mdmd sequences within truncated Mdmd copies in MIII. The Mix probe localized to the MIII-locus in male samples from the M3 strain as expected. Interestingly, two additional large signals at both ends of the X chromosomes that do not carry M-loci were observed in both male and female samples of the M3 strain (Fig. 2e, h). We further tested the Mix probe with samples from strains aabys (males with MY), M5 (males with MV), and two Spanish strains (SPA1 and SPA4 in which samples possess MX). The large signals on the X chromosomes were also detected in female aabys, and in both M5 female and male samples (Fig. 2f, g, i). When using the Mix probe on male samples from the aabys strain that carry a Y chromosome, we observed similar hybridization signals on the non-M-possessing X chromosome (Fig. 2d). In addition, a Mix probe hybridized region, much larger than the Mdmd-specific signal, covers almost the entire short arm of the Y chromosome. As the MY-specific signal cannot be distinguished from additionally detected Y chromosome regions, it indicates they are either overlapping or closely located. In SPA1 and SPA4 samples, the MX-specific signal also cannot be distinguished from additionally detected X chromosome regions (Supplementary Fig. 1i, j).
The Mix probe likely detected large terminal regions of the sex chromosomes by hybridizing to repeats shared by the XY chromosomes. We refer to these repeats as M And Sex chromosome located (MAS) repeats. Note that those detected terminal regions likely contain sequences other than MAS repeats as well. The MAS repeats seem to be universally present on the X and Y chromosomes regardless of the strain of origin and the presence or absence of M. However, MIII also contains MAS repeats as the Mix probe originated from MIII genomic DNA.
Discussion
In order to compare genomic structures of Mdmd-containing loci, we assembled three male housefly genomes, one with the male-determining locus on chromosome V (MV), one on chromosome III (MIII), and one on the Y chromosome (MY). The M-loci differ considerably in size, sequence composition and structure. MV is the simplest and contains only two intact Mdmd copies with minimal sequences in between. In contrast, MIII and likely MY contain a single complete Mdmd ORF, many truncated Mdmd copies, and other pseudogenes that are absent in MV. The male-determining capacity of MV demonstrates the importance of the intact Mdmd copy as the male-determining factor, and suggests that the remaining sequences in other M-loci are dispensable for sex determination.
We find that the Mdmd gene can be embedded in very different genomic regions on the chromosomes on which it is located. Palindromes are often found in regions on Y/W sex chromosomes that contain genes with sex-determining function, such as the Y chromosome of mammals24,25,26,27,28, European rabbits29, and the W chromosome of the white-throated sparrow30, where they appear to facilitate concerted evolution via gene conversion24,29. The palindromic MV has the fewest nucleotide changes between the two Mdmd copies and the Mdmd consensus may reflect a similar process of concerted evolution. In contrast, Mdmd on chromosome III points to a very different genomic dynamic. MIII contains many repeated but degenerated sequences, which is consistent with the model of junk DNA and mutational degeneration of sex-determining loci6. At this stage, we have no evidence for a functional role of the duplicated sequences and the Mdmd truncated copies.
Different mechanisms likely underlie the formation of distinctive M architectures. MV seems to have been produced by transposase activity as we found sequence signatures of TIRs and DRs as well as highly similar palindromic structures in other regions with the same TIRs. As the TIRs are not present in the corresponding non-MV-contigs, their insertion likely has occurred together with the Mdmd gene(s). An intriguing possibility is that the TIRs are involved in translocation of Mdmd, and MV potentially gained the ability to change its genomic location via nonautonomous translocation mediated by the TIRs. Other genomic processes may be responsible for the complex architecture of MIII. The multiple Mdmd copies in MIII could be the result of double-strand breakage and homologous repair that are known to generate tandem duplications31. According to the duplication-dependent strand annealing model31, several traces are characteristic of such duplications, i.e., microhomology in template and duplicated sequences that allow reinvasion during homology repair. Upon examination of duplicated sequences in MIII contigs we indeed found such signatures (Supplementary Discussion, Supplementary Fig. 6) supporting the occurrence of these genomic processes.
There is another process that may have affected the genomic evolution of M-loci. Populations with multiple M loci often carry the Mdtra gene variant, MdtraD, which could have also affected M structure. MdtraD is epistatically dominant over M and can promote female development even in the presence of M, which allows females to carry M-loci8,32. Although recombination in males may be reduced, in females it is not. Hence, if a female is homozygous for an M-locus, unequal crossover within the M-locus may cause expansion or reduction of M, although we currently do not have evidence to support this hypothesis. In addition, it is presently unknown if the MdtraD allele originated before, during, or after the evolution of the various M-loci, which could affect the plausibility of this model.
From our results, we infer the complex structure of M formed via gradual duplication on the Y chromosome. During this process, not only did the copy number of Mdmd increase, but the M-surrounding sequences likely also became a part of M, by getting intercalated by Mdmd copies. Thus, non-Mdmd sequences in MY are expected to show homology to the X chromosome. We tested this hypothesis by mapping Pacbio reads of aabys-male and M5-male to the MY-contigs. Indeed, we observed some regions intervening Mdmd copies showed coverage to non-MY reads (Supplementary Fig. 7). M And Sex chromosome located (MAS) repeats detected by the Mix probe indicate abundant repetitive sequences that are prevalent on the X and Y chromosomes but not on other chromosomes. This is consistent with previous reports21,33 that housefly XY chromosomes mostly consist of highly repetitive sequences that are unique to them. MIII also contains MAS repeats given the fact that the Mix probe was derived from MIII DNA but there are not MAS repeats on the non-M third chromosome. This suggests that MIII has originated from the translocation of a DNA segment from the Y chromosome that contained Mdmd and MAS repeats.
MY contains sequences characteristic shared with both MIII and MV, which points to two possible evolutionary routes for how the housefly M-loci arose. The first is that Mdmd originated on chromosome V and then translocated to an X chromosome (according to current karyotype numbering), which converted the X into what we refer to as the housefly Y chromosome now. M on the Y then evolved a complex, tandem duplicated structure, which later translocated to other chromosomes. A second possibility is that M first established on the Y, and subsequently translocated to other chromosomes embedded in DNA fragments that vary in size, ranging from a single copy when transposing to chromosome V to many copies when transposing to chromosome III. Although the first scenario appears more intuitive as it follows a “simple to complex” order, we consider the second scenario more plausible for several reasons. Based on cytogenetic data the XY is the only morphologically differentiated chromosome pair (Y being smaller than the X chromosome), whereas other M-carrying chromosomes (e.g., MIII and MV) do not visibly differ from their non-M-carrying counterparts. This observation is consistent with the hypothesis that the Y chromosome is “older” than other M-carrying chromosomes, whereas the other proto-Y chromosomes have not experienced substantial degeneration and remained intact. An indication for the “young” status of MV is that high sequence identity is observed between homologous sequences of non-MV-contigs and MV-contigs except for the small MV-locus region. This suggests a minimal degree of divergence for the M-surrounding regions on the chromosome V pair. With our current data, we cannot entirely discern the evolutionary histories of the various M loci as we lack information regarding the X chromosome region that is homologous to the MY locus. This would require constructing chromosomal-level assemblies of different M-carrying chromosomes and comparing the sequence divergence and recombination rates between M-carrying chromosomes and their non-M-carrying homologs in future studies.
Our study sheds light on the complex evolution of the polymorphic sex determination system of the housefly. Mdmd originated as a copy of the Mdncm gene, which was followed by duplication events generating multiple, incomplete Mdmd copies13. Our results imply the intact Mdmd was translocated from one chromosome to the others embedded in large DNA fragments which varied in size and often contained incomplete Mdmd copies. Transposable elements were likely involved in the translocation events, such as the establishment of MV resulting in a distinctive palindromic structure. An interesting finding is that even M-loci with comparable structures (MII, MIII and MY) show signs of diversification. For example, MIII contains specific genomic regions that are not found in other investigated M-loci. Even MY from different populations vary in M-locus sequence, indicating that M-loci are independently evolving in separate populations. In summary, our study demonstrates that nascent sex determination regions can be subject to different genomic processes leading to diverse genomic architectures.
Methods
Data type and housefly strains used in each analysis are listed in Table 1. Notably, males and females in M2, M3 and M5 strains have two X chromosomes and no Y chromosomes. Strains established from collections of wild populations contain various combinations of MI, MII, MIII, MY, or MX. Both hemizygous and homozygous M-loci were found in these strains.
Analysis of Mdmd copy number variation
The copy number of Mdmd in different M-loci was determined by mapping the raw reads to the published Mdmd sequence13 (Accession: KY020049.1). The mapping and coverage analysis was done with Burrows-Wheeler Aligner34 mem (BWA, v0.7.17) and SAMtools35,36 (v1.10) using the default parameters. The average coverage of Mdmd was calculated for each base. The Mdmd coverage was standardized among datasets by calculating a relative coverage which is a ratio of Mdmd coverage dividing by the coverage of a single copy autosomal reference gene. To minimize potential errors, three reference genes were selected, i.e., Mdtra35 (Accession: GU070694.1), yellow (MdY, Accession: KY979110.1) and asense (Mdase, Accession: XM_005176302.3). The final Mdmd copy numbers for each dataset were calculated by taking the average relative coverage for each of the three reference genes and multiplying by two as autosomal genes have two alleles but the M-locus is hemizygous. Sequence depth files that are generated by SAMtools and are used to calculate coverages are provided as a Source Data file.
Chromosome preparations
Chromosome slides were prepared from the brain tissues of third instar larvae. Spreads of mitotic chromosomes were made according to the method of ref. 37. with slight modification. In short, larval brains were dissected in Ringer’s solution, pre-treated in hypotonic solution (75 mM KCl) for 10 min and then fixed in Carnoy’s fixative (ethanol:acetic acid, 3:1) for 10 min. Fixed tissues were then transferred to glass slides (Thermo Fisher Scientific SuperFrost Microscope Slides) with a drop of 60% acetic acid and spread with a tungsten needle on a 45 °C heating plate. Slides were examined under a phase contrast microscope (Carl Zeiss Axio Lab.A1) to check whether the nuclei were appropriately spread before FISH.
The remaining larval tissue was used for DNA extraction using a high salt protocol38 followed by PCR with primers (Mdmd_1as, GATTGGCTCAGATCGGCGTA and Mdmd_6as, GGTTGACGCGGACAATCAAC) designed on Mdmd specific sequences according to ref. 13. to determine whether the larva possessed the Mdmd sequences. PCR was conducted with Platinum II Taq Hot-Start DNA Polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions. The thermocycling program was as follows: initial denaturation at 94 °C for 2 min; 30 cycles of 15-s denaturation at 94 °C, 15-s annealing at 60 °C, 1 min 15-s extension at 72 °C, with a final extension of 3 min at 72 °C. PCR products were visualized on a 1% agarose gel in TAE buffer to evaluate the presence of Mdmd in the samples.
Probe preparation
To prepare probes for FISH experiments, DNA fragments of MIII were amplified with Mdmd-specific primer pair Mdmd_FISHs, GGAAGTCGTATTGGAAGTAGT and Mdmd_FISHa, ATTTGGTGCGCCCTTCT using Platinum II Taq Hot-Start DNA Polymerase according to the manufacturer’s instructions. The PCR product contains a mixture of Mdmd fragments and non-Mdmd fragments as many intergenic sequences exist in MIII such as repeats and transposable elements. A Mix probe was prepared directly from purified PCR products by labeling with digoxigenin (DIG)−11-deoxyuridine triphosphate (dUTP) using the DIG-Nick Translation Mix (Roche) according to the manufacturer’s instructions. Labeled in the same way, an Mdmd-specific probe was made from a cloned Mdmd gene which sequence was confirmed by Sanger sequencing.
Fluorescence in situ hybridization
The FISH procedure was adapted from ref. 39. with minor modifications. Chromosome slides were pretreated with 100 μg/ml RNase A in 1×PBS for 1 h at 37 °C, followed by washing three times with 2 × SSC at room temperature for 5 min each. Subsequently, the slides were denatured in 2 × SSC containing 70% formamide at 68 °C for 3.5 min, dehydrated by passing them through an ice-cold ethanol series (70%, 90%, 100%; 5 min each) and air-dried. The 20 μl probe mixture contained 200–300 ng digoxigenin-labeled DNA probe, 50% (v/v) deionized formamide, 10% (v/v) dextran sulfate in 2 × SSC. The probe was denatured at 90 °C for 5 min and rapidly cooled on ice for 10 min. The denatured probe mixture was then applied to the slides and left to hybridize at 37 °C for at least 14 h.
After hybridization, slides were washed with 2 × SSC, 50% formamide at 42 °C for 10 min, followed by three washes with 2 × SSC at 42 °C for 5 min each. Slides were blocked with 3% (w/v) bovine serum albumin blocking buffer (dissolved in 4 × SSC with 0.1% Tween 20). Probes were detected with Anti-Digoxigenin-Rhodamine (Roche) by incubating at 37 °C for an hour. Slides were then washed three times with washing buffer (4 × SSC with 0.1% Tween 20) at 37 °C for 5 min each. After washing, slides were shortly rinsed with 2 × SSC and air-dried. Chromosomes were counterstained with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Signals were detected with a Leica epifluorescence microscope (DMI6000 B) equipped with a Leica CCD camera (DFC365 FX) and analyzed with Leica Application Suite X (3.4.2.18368.1.2). Chromosomes were numbered according to ref. 33. Signals were only considered as a successful hybridization if they were observed with consistent chromosomal locations on at least 20 metaphase nuclei.
Genome sequencing
We employed Pacbio sequencing to generate datasets for genome assembly. Two datasets were generated for M3 genome assembly. For Dataset1, 25 adult males from a single pair of parents were pooled for DNA extraction using Genomic-tip 100/G (Qiagen) according to the instruction manual. A DNA library was constructed with SMRTbell at the Leiden Genome Technology Center in the Netherlands and BluePippin was used for size selection of >10 kb fragments. For Dataset2, 20 non-related adult males from the M3 strain were pooled for DNA extractions using Nucleo Bond AXG columns (Macherey Nagel) according to the instruction manual. A DNA library was constructed at the Functional Genome Center Zürich (FGCZ), Switzerland and >20 kb fragments were selected for sequencing. Two datasets together correspond to an approximate genome sequencing coverage of ~116× (~84× for Dataset1 and ~32× for Dataset2). For the M5 genome, genomic DNA of 3 adult M5 males was pooled and extracted using Nucleo Bond AXG columns (Macherey Nagel) according to the instruction manual. A DNA library was constructed at the FGCZ and sequenced on three Pacbio Sequel IIe cells generating HiFi reads, with an approximate coverage of ~161×. For the aabys-male genome, DNA was extracted from flash frozen house fly male heads using the Qiagen Blood and Cell culture DNA MIDI Kit. High molecular weight DNA extraction was prepared for input of PacBio library prep according to ref. 40. A DNA library was constructed at the Clemson University Genomics and Computational Biology Facility (Clemson, SC, USA) and sequenced on Pacbio RSII cells using P6-C4 chemistry, generating a dataset with an approximate coverage of ~13×.
For comparing genomic sequences of different M-loci, sequence data were obtained at the FGCZ, on the Illumina HiSeq2500 platform, generating 101 bp paired-end reads for M2 males and M5 males, 126 bp paired-end reads for M3 males or 151 bp paired-end reads for M3 females. All the genomic DNA was isolated using NucleoBond AXG 20 (MACHEREY-NAGEL) according to the instruction manual. For each DNA sample, the DNA was extracted from a pool of 5 flies. Separate libraries were prepared from each pool of DNA. The genomic sequences for MY, published in ref. 21., were downloaded from the NCBI database.
Genome assembly
We performed M3 genome assembly using Canu41 (v1.8) with the following parameter settings: corOvlErrorRate = 0.24, obtOvlErrorRate = 0.045, utgOvlErrorRate = 0.045, corErrorRate = 0.3, obtErrorRate = 0.045, utgErrorRate = 0.045, cnsErrorRate = 0.075, genomeSize = 900,000,000. The assembled M3 genome was then error-corrected with Quiver42 (v2.2.1). The M5 genome was assembled using a newer Canu43 version (v2.2) but with the same settings as for M3. A Quiver correction was not necessary, because Pacbio HiFi reads were used. The aabys-male genome was assembled using Flye44 (v2.9.3) with the parameter “--no-alt-contigs”. Male illumina reads of the same strain were then used to polish the genome assembly with Pilon45 (v1.24). The summary statistics of the assembled genomes were obtained with QUAST46 (v4.6.3). BUSCO47 (v5.0.0) was used to estimate the completeness of the genomes by estimating the percentage of assembled universal protein-coding genes in dipteran lineages. Furthermore, the repeat content including interspersed repeats and tandem repeats of the M3 genome was analyzed with RepeatModeler (v1.0.11) and RepeatMasker (v4.0)48.
Genomic analysis of M III, M V and M Y
To identify sequences spanning the M region, we performed a search with BLAST using the published Mdmd sequence13 against our newly assembled genomes and identified Mdmd-containing contigs. To exclude Mdncm sequences that shared a significant degree of sequence similarity with Mdmd, contigs that contain a single-copy sequence with over 95% identity to Mdncm were removed from the Mdmd contig pool. Non-MV-contigs were identified by using MV-contigs to search for sequence similarity against the genomes by BLAST. Synteny analysis was based on single-copy BUSCO information in MV-contig and D. melanogaster and plotted by R package RIdeogram49. TIRs and DR were manually checked based on alignments.
All the Mdmd sequences in MIII-contigs were manually checked based on the previous BLAST search and grouped into different Mdmd copies based on the sequence continuity and position on the contigs. MIII-contigs were screened for annotated sequences by using BLASTto compare the contigs against the NCBI Musca domestica (Taxid: 7370) Nucleotide Collection database. Obtained hits of annotated genes were subsequently used to search for the presence of these genes on other contigs in the M3 genome by BLAST.
Dotplot visualization of the alignments were done via Flexidot50 (v1.06) with different wordsize setting, i.e., 15 for alignments of Mdmd sequences in MV-contig and MIII-contigs, 10 for alignments of Mdmd sequences in MY-contigs, 100 for alignments between MV-contig and non-MV-contigs, 20 for alignments between MIII-contigs and MY-contigs, 50 for the rest.
Analysis of M-locus coverage
The female Illumina reads were mapped to MV-contigs and MIII-contigs to detect sequences that are specific to MV and MIII using BWA mem with adjusted parameters, i.e., -t 16 -M -P -c 5000 -k 65 -B 7 -w 10 -d 60. The BWA output was used to calculate read depth for each nucleotide position using SAMtools with the “depth” function.
To detect sequence content variation between different M-loci, male Illumina reads were mapped to MIII-contigs using the same pipeline with BWA and SAMtools as described above. To minimize the false positive alignments, a minimum coverage value of 5 was set when plotting with R package ggplot251 as dotplot.
Minimap252 (v2.26) was applied to map Pacbio raw reads of aabys-male and M5-male to MY-contigs. Integrative Genomics Viewer53 (v2.17.4) was used to visualize the mapping results.
Verification of MdmdA and MdmdB transcription
RNA and DNA were simultaneously isolated from individual M5 pupae using TRIzol reagent (Thermo Fisher Scientific). RNA was isolated according to the manufacturer’s instructions whereas gDNA was isolated from the organic phase using a back extraction protocol as described in ref. 54. RNA samples were DNase-treated with the Invitrogen TURBO DNA-free kit (Thermo Fisher Scientific), and approximately 2 μg RNA was converted to cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with the oligo(dT)18 primer included in the kit in a total reaction volume of 20 μL. The sex of the samples (using gDNA template) as well as the transcription of MdmdA and MdmdB (using cDNA template) were tested using primers that amplify a region of Mdmd that includes the intron and the SNP between MdmdA and MdmdB, Mdmd_4F (TTGCATCAAGGCAAGTTGGA) and Mdmd_4R (TCTGAATCACTTGAAGAATCGT). PCR was carried out in 20 μL reaction volumes consisting of 1× DreamTaq buffer, 0.2 mM dNTPs, 0.2 μM of each primer, 0.5 U DreamTaq DNA polymerase (Thermo Fisher Scientific) and 1 μL 10× diluted cDNA (or 50–100 μg gDNA). The thermocycling program was as follows: initial denaturation at 94 °C for 3 min; 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at 59 °C, 1 min 15 s extension at 72 °C, with a final extension of 3 min at 72 °C. Amplification was verified by gel electrophoresis using a 1% agarose gel in 1× TAE buffer. To remove contaminants before sequencing, 5 μL of the amplified samples was combined with 1.6 U exonuclease I and 0.12 U FastAP (both Thermo Fisher Scientific) in a total volume of 9 μL, and incubated at 37 °C for 30 min. The reactions were inactivated at 80 °C for 15 min after which the samples were sent for Sanger sequencing (Eurofins).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All genomic data supporting the findings of this study are available under BioProject: PRJNA1013067 and PRJNA1072234 in the NCBI database. The M3, M5, and aabys-male assemblies were deposited at NCBI GenBank under accession JAVQME000000000, JAVVNY000000000, and JAZGUT000000000, respectively. Illumina reads generated from this study were deposited at NCBI Sequence Read Archive (SRA) under accession numbers: SRX21801162 (M2-male), SRX21801164 (M3-male_1), SRX21801165 (M3-male_2), SRX21801166 (M3-female_1), SRX21801167 (M3-female_2), SRX21801163 (M5-male). Illumina reads of MY samples were from the previous publication21 and were downloaded from SRA (accession numbers: SRX2154714- SRX2154719). Reference gene sequences, Mdmd (accession: KY020049.1), Mdtra (accession: GU070694.1), MdY (accession: KY979110.1) and Mdase (accession: XM_005176302.3), were obtained from GeneBank. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Wilkins, A. S. Moving up the hierarchy: a hypothesis on the evolution of a genetic sex determination pathway. Bioessays 17, 71–77 (1995).
Saccone, G. A history of the genetic and molecular identification of genes and their functions controlling insect sex determination. Insect Biochem. Mol. Biol. 151, 103873 (2022).
Steinemann, M. & Steinemann, S. Enigma of Y chromosome degeneration: neo-Y and neo-X chromosomes of Drosophila miranda a model for sex chromosome evolution. Genetica 102, 409 (1998).
Charlesworth, B. & Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 1563–1572 (2000).
Carvalho, A. B., Koerich, L. B. & Clark, A. G. Origin and evolution of Y chromosomes: drosophila tales. Trends Genet. 25, 270–277 (2009).
Bachtrog, D. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat. Rev. Genet. 14, 113–124 (2013).
Nei, M. Linkage modification and sex difference in recombination. Genetics 63, 681 (1969).
Dübendorfer, A., Hediger, M., Burghardt, G. & Bopp, D. Musca domestica, a window on the evolution of sex-determining mechanisms in insects. Int. J. Dev. Biol. 46, 75–79 (2002).
Hamm, R. L., Meisel, R. P. & Scott, J. G. The evolving puzzle of autosomal versus Y-linked male determination in Musca domestica. G3 5, 371–384 (2015).
Meisel, R. P., Scott, J. G. & Clark, A. G. Transcriptome differences between alternative sex determining genotypes in the house fly, Musca domestica. Genome Biol. Evol. 7, 2051–2061 (2015).
Son, J. H. et al. Minimal effects of proto-Y chromosomes on house fly gene expression in spite of evidence that selection maintains stable polygenic sex determination. Genetics 213, 313–327 (2019).
Adhikari, K. et al. Temperature‐dependent effects of house fly proto‐Y chromosomes on gene expression could be responsible for fitness differences that maintain polygenic sex determination. Mol. Ecol. 30, 5704–5720 (2021).
Sharma, A. et al. Male sex in houseflies is determined by Mdmd, a paralog of the generic splice factor gene CWC22. Science 356, 642–645 (2017).
Franco, M. G., Rubini, P. G. & Vecchi, M. Sex-determinants and their distribution in various populations of Musca domestica L. of Western Europe. Genet. Res. 40, 279–293 (1982).
Denholm, I., Franco, M. G., Rubini, P. G. & Vecchi, M. Identification of a male determinant on the X chromosome of housefly (Musca domestica L.) populations in South-East England. Genet. Res. 42, 311–322 (1983).
Tomita, T. & Wada, Y. Multifactorial sex determination in natural populations of the housefly (Musca domestica) in Japan. Jpn. J. Genet. 64, 373–382 (1989).
Hamm, R. L., Shono, T. & Scott, J. G. A cline in frequency of autosomal males is not associated with insecticide resistance in house fly (Diptera: Muscidae). J. Econ. Entomol. 98, 171–176 (2005).
Kozielska, M., Feldmeyer, B., Pen, I., Weissing, F. J. & Beukeboom, L. W. Are autosomal sex-determining factors of the housefly (Musca domestica) spreading north? Genet. Res. 90, 157–165 (2008).
Feldmeyer, B. et al. Climatic variation and the geographical distribution of sex-determining mechanisms in the housefly. Evol. Ecol. Res. 10, 797–809 (2008).
Li, X., Lin, F., van de Zande, L. & Beukeboom, L. W. Strong variation in frequencies of male and female determiners between neighboring housefly populations. Insect Sci. 29, 1470–1482 (2022)
Meisel, R. P., Gonzales, C. A. & Luu, H. The house fly Y Chromosome is young and minimally differentiated from its ancient X Chromosome partner. Genome Res. 27, 1417–1426 (2017).
Scott, J. G. et al. Genome of the house fly, Musca domestica L., a global vector of diseases with adaptations to a septic environment. Genome Biol. 15, 466 (2014).
Picard, C. J., Johnston, J. S. & Tarone, A. M. Genome sizes of forensically relevant Diptera. J. Med. Entomol. 49, 192–197 (2012).
Rozen, S. et al. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423, 873–876 (2003).
Hughes, J. F. et al. Chimpanzee and human y chromosomes are remarkably divergent in structure and gene content. Nature 463, 536–539 (2010).
Hughes, J. F. et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus y chromosomes. Nature 483, 82–87 (2012).
Tomaszkiewicz, M. et al. A time- and cost-effective strategy to sequence mammalian Y chromosomes: an application to the de novo assembly of gorilla Y. Genome Res. 26, 530–540 (2016).
Trombetta, B. & Cruciani, F. Y chromosome palindromes and gene conversion. Hum. Genet. 136, 605–619 (2017).
Geraldes, A., Rambo, T., Wing, R. A., Ferrand, N. & Nachman, M. W. Extensive gene conversion drives the concerted evolution of paralogous copies of the SRY gene in European rabbits. Mol. Biol. Evol. 27, 2437–2440 (2010).
Davis, J. K., Thomas, P. J. & Thomas, J. W. AW-linked palindrome and gene conversion in New World sparrows and blackbirds. Chromosome Res. 18, 543–553 (2010).
Fiston-Lavier, A.-S., Anxolabehere, D. & Quesneville, H. A model of segmental duplication formation in Drosophila melanogaster. Genome Res. 17, 1458–1470 (2007).
Hediger, M. et al. Molecular characterization of the key switch F provides a basis for understanding the rapid divergence of the sex-determining pathway in the housefly. Genetics 184, 155–170 (2010).
Hediger, M., Niessen, M., Müller-Navia, J., Nöthiger, R. & Dübendorfer, A. Distribution of heterochromatin on the mitotic chromosomes of Musca domestica L. in relation to the activity of male-determining factors. Chromosoma 107, 267–271 (1998).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Carabajal Paladino, L. Z., Nguyen, P., Šíchová, J. & Marec, F. Mapping of single-copy genes by TSA-FISH in the codling moth, Cydia pomonella. BMC Genet. 15, S15 (2014).
Aljanabi, S. M. & Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693 (1997).
Li, X. et al. Chromosomal mapping of tandem repeats in the yesso scallop, patinopecten yessoensis (jay, 1857), utilizing fluorescence in situ hybridization. Comp. Cytogenet. 10, 157–169 (2016).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563 (2013).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. in Gene prediction: methods and protocols (Springer, 2019).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2013–2015).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, 1–11 (2020).
Seibt, K. M., Schmidt, T. & Heitkam, T. FlexiDot: highly customizable, ambiguity-aware dotplots for visual sequence analyses. Bioinformatics 34, 3575–3577 (2018).
Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 3, 180–185 (2011).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Robinson, J. T., Thorvaldsdóttir, H., Turner, D. & Mesirov, J. P. igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 39, btac830 (2023).
Visser, S., Voleníková, A., Nguyen, P., Verhulst, E. C. & Marec, F. A conserved role of the duplicated Masculinizer gene in sex determination of the Mediterranean flour moth, Ephestia kuehniella. PLoS Genet. 17, e1009420 (2021).
Acknowledgements
We thank Anna Rensink, Peter Hoitinga, Ljubinka Francuski Marcetic, Marloes van Leussen, Dré Kampfraath, Jan Keijser, Jacopo Martelossi, and Alexander Suh for their advice and assistance. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Hábrók high performance computing cluster. This work was completed in part with resources provided by the Research Computing Data Core at the University of Houston. X.L. is supported by China Scholarship Council Scholarship no. 201606330077.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
X.L., L.W.B., D.B., L.v.d.Z., and E.W. designed the project; L.W.B., D.B., L.v.d.Z., E.W., and R.P.M. supervised and funded the project; X.L., S.V., Y.W., and F.M. performed molecular experiments and analyzed data; X.L., S.V., J.H.S., E.G., E.N.K., S.Y.M., M.P., S.A., M.A.S., M.D.R., and R.P.M. contributed to genomic data collection, genome assembly and genomic analysis. X.L., S.V., L.W.B., D.B., L.v.d.Z., and E.W. designed the figures in the manuscript. The manuscript was written by X.L., S.V., L.W.B., D.B., L.v.d.Z., and E.W. All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Giuseppe Saccone and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Visser, S., Son, J.H. et al. Divergent evolution of male-determining loci on proto-Y chromosomes of the housefly. Nat Commun 15, 5984 (2024). https://doi.org/10.1038/s41467-024-50390-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50390-1
- Springer Nature Limited