

Abstract
Multiple bacterial genera take advantage of the multifunctional autoprocessing repeats-in-toxin (MARTX) toxin to invade host cells. Secretion of the MARTX toxin by Vibrio vulnificus, a deadly opportunistic pathogen that causes primary septicemia, the precursor of sepsis, is a major driver of infection; however, the molecular mechanism via which the toxin contributes to septicemia remains unclear. Here, we report the crystal and cryo-electron microscopy (EM) structures of a toxin effector duet comprising the domain of unknown function in the first position (DUF1)/Rho inactivation domain (RID) complexed with human targets. These structures reveal how the duet is used by bacteria as a potent weapon. The data show that DUF1 acts as a RID-dependent transforming NADase domain (RDTND) that disrupts NAD+ homeostasis by hijacking calmodulin. The cryo-EM structure of the RDTND-RID duet complexed with calmodulin and Rac1, together with immunological analyses in vitro and in mice, provide mechanistic insight into how V. vulnificus uses the duet to suppress ROS generation by depleting NAD(P)+ and modifying Rac1 in a mutually-reinforcing manner that ultimately paralyzes first line immune responses, promotes dissemination of invaders, and induces sepsis. These data may allow development of tools or strategies to combat MARTX toxin-related human diseases.
Similar content being viewed by others

Introduction
Multifunctional autoprocessing repeats-in-toxin (MARTX) toxins, secreted by multiple Gram-negative bacteria, are primary virulence factors that promote initial colonization, dissemination, and lethality in a wide range of hosts, including humans1,2,3,4. Among bacterial genera, Vibrio is the main genus of bacteria that secrete MARTX toxins1,5. These single-polypeptide exotoxins are equipped with a diverse repertoire of enzymatically functional effector domains arranged in a modularly structured fashion within their central regions6. Once MARTX toxins translocate into host cells, the toxins are cleaved to release functionally independent effector domains that target specific host substrates1,2,7,8.
Vibrio vulnificus is the most deadly of the foodborne pathogens, with a case fatality rate of 50%9. Vibrio spp. survive within a hostile environment and expand their virulence by exchanging or rearranging the genetic information encoding the effectors within MARTX toxins2,10,11. By doing this, the clinical strain V. vulnificus MO6-24/O generates a MARTX toxin (MARTXVv) containing four effectors, which are arranged in the following order: domain of unknown function in the first position (DUF1), the Rho inactivation domain (RID), the α/β hydrolase domain (ABH), and a Makes caterpillars floppy-like effector (MCF) with a cysteine protease domain (CPD). The latter is activated by binding to host inositol hexakisphosphate, resulting in cleavage of the linkers that connect the effectors12. The functions of most of the effectors within the MARTX toxins of all biotypes of V. vulnificus are known; the exception is DUF1. Previously, we reported that DUF1 and RID within MARTXVv are not uncoupled by activated CPD in host cells; rather, they are discharged as a duet (it is assumed that the DUF1-RID effector module acts as a functional unit1).
Successful host invasion by pathogenic bacteria such as V. vulnificus relies on the ability of their MARTX toxins to counteract host defense mechanisms, although it is not clear how this is achieved3,12,13. The effector RID, which is shared by the MARTX toxins of human pathogens, is an Nε-fatty acyl-transferase that modifies the lysine residues within the C-terminal polybasic region (PBR) of the small GTPase Rac1 at the cell membrane, thereby inhibiting cellular processes such as phagocytosis and production of reactive oxygen species (ROS)14. A previous study shows that RID appears to block Rac1-dependent mitogen-activated protein kinase (MAPK) signaling to suppress pro-inflammatory responses prior to induction of actin cytoskeleton collapse by a V. cholerae MARTX toxin (MARTXVc) effector called the actin cross-linking domain (ACD)13.
ROS production is important in that it activates innate immune responses early in the course of infection. An isoform of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, called NOX, is the multicomponent enzyme responsible for ROS generation mainly in macrophages and neutrophils15. The GTP-bound (active) form of Rac1 or Rac2 acts as a critical regulator of NOX2 activation15 by interacting with p67phox through its Switch I region16,17,18. The participation of small GTPases in regulating ROS generation plays an important role in innate immune responses against invading microbes19,20,21,22. It is assumed that MARTX toxin-secreting pathogens utilize the effector RID to disrupt first line immune responses triggered by the Rac GTPase-NOX2 signaling axis; however, we do not know how RID affects this signaling axis, nor do we know anything about the interrelated roles of DUF1 and RID during/after cell invasion.
In this study, we performed interactome analyses and found that the DUF1-RID effector duet hijacks calmodulin (CaM) and Rac1 in infected host cells. Crystal and cryo-EM studies of the effector duet complexed with CaM and Rac1, together with biochemical and immunological analyses, provide key insight into a concerted invasion strategy in which pathogenic bacteria coordinate the DUF1-RID duet, the components of which act synergistically to suppress ROS generation and promote dissemination of pathogens, ultimately resulting in sepsis.
Results
The DUF1-RID effector duet hijacks calcium-free CaM
A previous study used mass spectrometry analysis to identify the Rho family small GTPase Rac1 as a target for RID14. In separate experiments, we performed an interactome analysis using ectopically expressed inactive RIDC/A (residue C2838 mutated to alanine, corresponding to the active residue in the previous study14) in HEK293T cells. Note that we constructed RID based on the inositol hexakisphosphate-activated CPD-mediated cleavage sites between effectors within MARTXVc23. Unlike in the previous study14, we found that CaM showed the highest intensity ratio (relative to the control) among the potential RID interactors, although Rac1 was also detected as an interactor (Supplementary Fig. 1a).
We previously reported that MARTXVv discharges the effectors DUF1 and RID as a duet (Fig. 1a)1. Further analysis of DUF1-RID duet- or RID-carrying MARTX toxins secreted from different bacterial genera revealed a tendency towards the composition of the effectors with genus type (Fig. 1b). Among the different genera, Vibrio spp. is the primary genus that secretes MARTX toxins. Bacteria belonging to this genus secreted a similar proportion of DUF1-RID duet- or RID only-containing toxins (Fig. 1b). V. cholerae and V. vulnificus are the main species within this genus, and the former tended to largely secrete RID-containing toxins while the latter mainly secreted DUF1-RID duet-containing toxins (Fig. 1b). Additionally, opportunistic human pathogens belonging to Proteus spp. secreted MARTX toxins carrying RID or its N-terminal domain-deleted form (Fig. 1b). Importantly, the MARTX toxins of most clinical V. vulnificus strains carried the DUF1-RID duet, while clinical V. cholerae strains contained mainly RID (Fig. 1c). Consistent with CPD-mediated cleavage site-based analysis23, we found that nearly all RIDs from Vibrio spp. comprise an N-terminal domain (ND, RIDND), a membrane localization domain (MLD)-containing domain (MLD_C, RIDMLD_C), and a catalytic domain (CD, RIDCD), with few exceptions (Fig. 1a, b). Thus, our previous results, coupled with those obtained herein, led us to question whether CaM is a host target of RID alone, or also a target of DUF1.

a Schematic diagram showing the domains of the DUF1-RID module within the MARTX toxin secreted by V. vulnificus MO6-24/O strain. DUF1 domain of unknown function in the first position, RID Rho inactivation domain, ND N-terminal domain (RIDND), MLD_C membrane localization domain-containing domain (RIDMLD_C), CD catalytic domain (RIDCD). b Analysis of the DUF1-RID module, and its variants containing RID, within the MARTX toxin from different bacterial genera. c Characteristics of effectors within the MARTX toxin from clinical V. cholerae and V. vulnificus strains. Not aligned, not aligned with the DUF1-RID module. d Cytopathogenicity of the indicated proteins. GFP or GFP-fused proteins were expressed in HEK293T cells and analyzed by confocal imaging. Note that the expression level of DUF1-RID, DUF1-RIDC/A, and RID was much lower than that of RIDC/A due to their inherent cytotoxicity. e In vitro pull-down assay showing the interactions between DUF1-RID and its variants with calmodulin (CaM). The data shown in d and e are representative of three independent experiments, each with similar results. Source data are provided as a Source Data file.
First, we evaluated the cytopathogenicity of MARTXVv DUF1 coupled to inactive RIDC/A (i.e., DUF1-RIDC/A). The results showed that DUF1-RIDC/A, but not DUF1 alone or RIDC/A, caused marked morphological changes in HEK293T cells, suggesting that DUF1 induces cytotoxicity in a RID-dependent manner that is unrelated to the Rac1 modification function of RID (Fig. 1d).
Subsequently, the interaction between DUF1-RID and CaM was validated by performing in vitro pull-down assays using purified proteins. The duet interacted with CaM, regardless of the presence of Ca2+; indeed, it interacted more readily with Ca2+-free CaM (Fig. 1e and Supplementary Fig. 1b). Consistent with the pull-down results, isothermal titration calorimetry (ITC) analysis revealed that the effector duet binds much more strongly to Ca2+-free CaM (dissociation constant (KD) = 80 nM) than to Ca2+-bound CaM (CaM/Ca2+, KD = 1.93 μM), with a 1:1 binding stoichiometry (Supplementary Fig. 1c–e). It should be mentioned that the binding affinity of MARTXVc RID (KD = 4 μM) and MARTXVv RID (KD = 20 μM) for Rac1 was much weaker14, suggesting that CaM is the primary host target of the effector duet. Next, we showed that the CaM-binding domain of RIDND is the most critical part that interacts with CaM (Fig. 1e). The interaction between DUF1-RIDND and CaM was comparable with that of DUF1-RID, while in the absence of RIDND, DUF1 failed to interact with CaM. Thus, we named RIDND the “CaM-binding domain (RIDCBD)”. Collectively, these data suggest that the DUF1-RID duet hijacks host CaM via RIDCBD to facilitate infection.
DUF1 is transformed into a NAD(P)+ hydrolase upon RID-dependent CaM-binding
To elucidate the relevance of CaM binding by the effector duet, we first tried to solve the structure of the MARTXVv DUF1-RID duet in the presence/absence of CaM; however, we were unable to initially generate crystals. Later, we did manage to generate crystals of DUF1-RIDCBD (residues, 1959–2374) in the absence of CaM, and determined the structure at a resolution of 3.38 Å (Supplementary Figs. 2a and 3a, and Supplementary Table 1). Subsequently, we determined the structure of DUF1-RIDCBD in complex with Ca2+-bound CaM or Ca2+-free CaM at a resolution of 2.82 Å and 2.90 Å, respectively (Supplementary Fig. 2b, c and 3b–d, and Supplementary Table 1). The structures of both complexes are identical (RMSD of 1.323 Å) (Supplementary Fig. 2d). Thus, from now on we describe the structural details of DUF1-RIDCBD complexed with Ca2+-bound CaM, unless stated otherwise.
DUF1 within the DUF1-RIDCBD structure comprises the N-terminal domain (DUF1ND, residues 1959–2227), formed by a bundle of eight α helices (α1–α8), three 310 helices (310h1–310h3), and four short β strands (β1–β4), as well as a small lid domain (DUF1Lid, residues 2228–2264) formed by an α helix (α9), a 310 helix (310h4), and two short β strands (β5 and β6) (Fig. 2a and Supplementary Fig. 2a). RIDCBD composes three helices (α10−α12) and a mixed parallel and anti-parallel beta sheet (β7−β12) (Fig. 2a and Supplementary Fig. 2a). Of note, the two effectors are connected by α10, which forms part of the RIDCBD.

Overall structures of DUF1-RIDCBD (a) and DUF1-RIDCBD complexed with CaM (b). The structures are shown as cartoon diagrams. c Superimposition of the DUF1-RIDCBD structure (gray) onto the CaM-binding DUF1-RIDCBD structure. The arrows indicate significant conformational changes induced in DUF1-RIDCBD upon interaction with CaM. d Structural comparison between DUF1 within DUF1-RIDCBD complexed with CaM, human CD38 (PDB ID 1YH3), and A. californica ADP-ribosyl cyclase (PDB ID: 1R12). The catalytic residues in each protein are represented by sticks. e Switching of non-functional catalytic residues (gray) within DUF1 to functional residues (yellow) within DUF1-RIDCBD complexed with CaM. f Superimposition of the functional catalytic site of DUF1 onto that of human CD38 or A. californica ADP-ribosyl cyclase. Catalytic residues are displayed. g In vitro NAD+-hydrolyzing assay for DUF1-RID and its truncates, with or without CaM. Levels of NAD+ relative to PBS are represented (n = 3 per group). In vitro NAD+- or NADP+-hydrolyzing assay for DUF1-RID and its mutants complexed with CaM or Ca2+-bound CaM. NAD+ (h) or NADP+ (i) levels relative to PBS are represented (n = 3 per group). Data shown in g–i are representative of three independent experiments, each with similar results (mean ± SEM). P-values were calculated using one-way ANOVA with multiple comparisons. ns, not significant. Source data are provided as a Source Data file.
Upon interaction with CaM, DUF1-RIDCBD undergoes a marked conformational change (Fig. 2b, c). Although Ca2+-free CaM interacts much more strongly with the effector duet than Ca2+-bound CaM (Supplementary Fig. 1e), the structure of both CaM proteins complexed with DUF1-RIDCBD is identical, and resembles the Ca2+-free CaM structure24 that adopts two N- and C-lobes separated by a flexible linker (Supplementary Fig. 2e). The C-lobe (residues, 81–148) forms a hydrophobic groove and tightly grips the RIDCBD α10 (Fig. 2b, c and Supplementary Fig. 2f). The hydrophobic face of α10 comprises L2266, I2269, M2272, and L2273, which form extensive nonpolar interactions with the C-lobe groove. The C-lobe αF and αG are sandwiched between DUF1 α1 and α8 and RIDCBD α10, and stabilize the complex structure via intensive hydrophobic and hydrophilic interactions (Fig. 2b, c and Supplementary Fig. 2g). The N-lobe (residues, 4–80) αA and αB form strong interactions with the α11, α12, and C-terminus loop of RIDCBD through multiple hydrophobic and hydrophilic contacts (Fig. 2b, c and Supplementary Fig. 2g). These interactions result in notable conformational changes, leading to a large rotation of α10 (~67.4°) and the rest region (~93.4°) of the RIDCBD (Fig. 2c). Along with the large structural movement of the RIDCBD, helices α1, α8, and α9 move toward the center of DUF1 by 8.2 Å, 8.9 Å, and 11 Å, respectively (Fig. 2c). In detail, 46 residues in DUF1-RIDCBD, and 40 residues in CaM, are involved in maintaining the structure of the complex via formation of 242 hydrophobic contacts, 25 hydrogen bonds, and eight salt bridges (Supplementary Fig. 2g, h). Collectively, these data suggest that hijacking host CaM triggers marked rearrangement of the domains within DUF1-RIDCBD.
Unexpectedly, the DUF1 moiety within the duet-CaM complex showed the highest structural similarity (Z-score = 6.9) to human CD38 (cluster of differentiation 38, PDB ID: 6VUA), known primarily as a nicotinamide adenine dinucleotide (NAD+) hydrolase (Fig. 2d). A structural comparison revealed that residues W2082, D2107, and E2186 in the duet-CaM complex, which are shifted by 12.5 Å, 2.3 Å, and 10.0 Å (Fig. 2e), respectively, upon CaM binding, adopt the same conformations as catalytically important residues W125, E146, and E226 in CD38, respectively (Fig. 2d, f). In addition to CD38, the structure of the DUF1-CaM complex is similar to that of Aplysia californica ADP-ribosyl cyclase (Fig. 2d, f).
Subsequently, we assessed the NAD+-hydrolyzing activity of the DUF1-RID duet using purified proteins. An in vitro NAD+-hydrolyzing assay clearly showed that the effector duet hydrolyzed NAD+ in a CaM-dependent manner (Fig. 2g). The DUF1-RIDCBD, but not DUF1 alone, showed NAD+-hydrolyzing activity comparable with that of the effector duet, indicating that the RIDCBD is critical for the enzymatic function of DUF1 via interaction with CaM, as shown by the structure of the complex (Fig. 2g).
We then used DUF1-RIDC/A containing an inactive RID to further validate the enzymatic activity of DUF1. The effector duet showed full NAD+-degrading activity, indicating that enzymatic activity is independent of the RID function that inactivates Rho GTPases (Fig. 2h). Substitution of the active residue E2186 in DUF1 (corresponding to the active residue E226 in CD38) with glutamine (DUF1E/Q-RIDC/A) completely abolished the NAD+-degrading activity (Fig. 2h). The DUF1-RIDC/A was furthermore mutated at residues critically important for the interaction with CaM (i.e., W2183L, R2195L, E2285L, and R2328L), thereby generating DUF14mt-RIDC/A (Supplementary Fig. 2i). This mutant also lacked NAD+-hydrolyzing activity (Fig. 2h). Since CD38 also has NADP+-hydrolyzing activity25, we tested whether DUF1 has the same activity and found that DUF1 does indeed hydrolyze NADP+ (Fig. 2i). Consistent with the biochemical and structural analysis, these data suggest that the enzymatic activity of CaM-mediated DUF1 is independent of calcium (Fig. 2h, i).
Next, we used nuclear magnetic resonance (NMR) spectroscopy and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to examine the by-products generated in vitro by DUF1-RIDC/A-mediated hydrolysis of NAD(P)+. The results showed that DUF1-RIDC/A hydrolyzes the NAD+ to yield nicotinamide (NAM) and ADP-ribose (ADPR) (Supplementary Fig. 4a–c). In addition, the effector duet hydrolyzed NADP+ to yield NAM and ADP-ribose 2′-phosphate (Supplementary Fig. 4c). Taken together, these data demonstrate that DUF1 is transformed into a NAD(P)+-hydrolyzing enzyme by RID-dependent CaM-binding. Thus, we renamed DUF1 the “RID dependently transforming NADase domain (RDTND)”.
Molecular details of RDTND-RIDCBD/CaM complexed with NAD+
While CD38 is responsible for >99% of NAD+ hydrolysis to generate ADPR and NAM, it also functions as a very inefficient ADP-ribosyl cyclase to synthesize cyclic ADPR (cADPR) from NAD+ (cADPR comprises less than 1% of reaction products)26,27,28,29,30. To provide structural insight into the enzymatic activity and potential substrate promiscuity of RDTND, we next determined the structure of enzymatically inactive RDTNDE/Q-RIDCBD/CaM in complex with NAD+ (Fig. 3a and Supplementary Table 1). Overall, the NAD+-bound structure is similar to the RDTND-RIDCBD/CaM structure (root mean square deviation of 0.97 Å); however, the RDTNDLid shows marked movement when bound to NAD+ (Fig. 3b). In detail, the adenine group of NAD+ forms a cation-π interaction with R2234, hydrogen bonds with G2179, and hydrophobic contacts with F2001, F2150, and Y2181 (Fig. 3a and Supplementary Fig. 5). In addition, the 2″-hydroxyl group in the 2R-ribose moiety interacts with (Fig. 3a and Supplementary Fig. 5). These interactions rotate the α9 helix by 42°, bringing RDTNDLid closer to the catalytic site (Fig. 3b), suggesting that RDTNDLid may play a role in recruiting substrate. The di-phosphate group of NAD+ forms hydrogen bonds with S2083, G2084, R2177, N2180, Y2181, and F2182 (Supplementary Fig. 5). The 1R-ribose group forms a hydrophobic contact with W2082, and hydrogen bonds with D2107 and E2186Q (Supplementary Fig. 5). The nicotinamide group interacts with W2082, I2117, T2153, and Y2181 via hydrophobic contacts, and with D2107 through hydrogen bonding (Supplementary Fig. 5). In particular, residues W2082, D2107, and E2186Q are critically involved in maintaining the stable conformation of the nicotinamide and 1R-ribose sugar (Fig. 3a). These structural data suggested that W2082, D2107, E2186, and R2234 are critical for substrate interaction and/or catalytic activity. Data from the in vitro NAD+-hydrolyzing assay clearly show that mutations at these residues completely abolish the enzymatic function of RDTND (Fig. 3c).

a Overall structure of RDTND-RIDCBD/CaM complexed with NAD+, and the binding mode of NAD+ at the catalytic site (magnified panel). The mFo-DFc omit electron density map of NAD+ contoured at 3.0 σ is shown as a mesh (red). b Superimposition of the NAD+-bound RDTND-RIDCBD/CaM structure onto the RDTND-RIDCBD/CaM structure (gray). Significant conformational changes in RDTNDLid are shown (magnified panel). c In vitro NAD+-hydrolyzing assay for CaM-bound RDTND-RIDC/A and its RDTND mutants. The NAD+ levels relative to the PBS control are presented (n = 3 per group). Data are representative of three independent experiments, each with similar results (mean ± SEM). P-values were calculated using one-way ANOVA with multiple comparisons. ns, not significant. d Structural comparison of active NAD+-bound RDTND with that of NAD+-bound human CD38 (PDB ID 2I65) and NAD+-bound A. californica ADP-ribosyl cyclase (PDB ID: 3ZWM). Structures are shown as cartoon (upper panel) or surface diagrams (lower panel). Source data are provided as a Source Data file.
To assess the possibility that RDTND plays a role in the synthesis of cADPR from NAD+, we compared the structures of RDTND-RIDCBD/CaM/NAD+, CD38/NAD+, and Aplysia californica ADP-ribosyl cyclase/NAD+ (Fig. 3d). The mode by which RDTND interacts with NAD+ is similar to the way in which CD38 and Aplysia californica ADP-ribosyl cyclase interact with NAD+ at its catalytic site; however, the NAD+ binding pocket within the catalytic domain is significantly different between the three enzymes. Unlike CD38 and Aplysia californica ADP-ribosyl cyclase, in which the catalytic site pocket has no spatial restriction with respect to cyclization of ADPR after hydrolysis of NAD+, RDTND has a smaller and narrower pocket due to the bulky residues R2234 and Y2181. In particular, Y2181 is positioned between the adenine and NAM moieties of NAD+, and therefore obstructs rotation of the adenine ring during the ADPR cyclization reaction (Fig. 3d).
We further evaluated the ADP-ribosyl cyclase activity of RDTND complexed with RID/CaM by using a modified HPLC-UV method31. Consistent with previously reported results26,28,29, the analyses showed that CD38 mostly hydrolyzes NAD+ to ADPR (99.28%) and generates a small amount of cADPR (0.74%) (Supplementary Fig. 6). RDTND complexed with RID/CaM hydrolyzed NAD+ to ADPR (99.69%) and cADPR (0.31%). The structural restriction on the movement of NAD+ moieties may lead to the RDTND-RID/CaM complex having lower ADP-ribosyl cyclase activity than that of CD38. Thus, the structural features together with the biochemical analysis suggest that RDTND is a NAD(P)+-hydrolyzing enzyme, although it cannot be ruled out that it has ADP-ribosyl cyclase activity.
Cryo-electron microscopic (EM) structure of the RDTND-RID complexed with CaM and Rac1
To clarify a mutual dependency of RDTND and RID during interaction with host targets, we conducted cryo‐EM studies. Size-exclusion chromatography assays clearly showed that RDTND-RIDC/A interacts with both CaM and the active form of Rac1 (i.e., Rac1Q61L), and forms a complex with them (Supplementary Fig. 7a). Consistent with previous results14, the duet did not interact with the inactive form of Rac1 (i.e., Rac1T17N) but bound with CaM, suggesting critical involvement of the Switch I region of Rac1 in the interaction with RID, and the independence of CaM and Rac1 during the interaction with the effector duet (Supplementary Fig. 7b). Subsequently, we obtained a complex comprising RDTND-RIDC/A, CaM, and Rac1Q61L (residues 1–177) through a sequence of purification procedures, including chemical cross-linking to stabilize the complex (Supplementary Fig. 8). Note that we deleted the C-terminal PBR of Rac1 to prevent non-specific cross-linking between lysine residues within the PBR. Finally, single particle analysis enables us to generate a reconstructed 3D map of the complex at an average resolution of 4.32 Å (Supplementary Figs. 8 and 9, and Supplementary Table 2). We initially tried to determine the cryo-EM structure of the non-cross-linked complex under various conditions including the use of grids and vitrification. However, single particle cryo-EM image analysis typically resulted in low-resolution 2D class averages, indicating flexible movement within the complex (Supplementary Fig. 10), caused possibly by low binding affinity between RID and Rac1. Cross-linking reduced flexible movement in the sample and improved the overall resolution of the cryo-EM map. The cryo-EM density map of the RDTND-RIDCBD complexed with CaM is observed at a contour of 8−12 σ, while the regions of RIDC/A complexed with Rac1Q61L are relatively weak in the cryo-EM density map contoured at ~4.5–6.0 σ (Supplementary Fig. 11); these data correlate with the binding affinity between MARTXVv RDTND-RIDC/A and CaM (Supplementary Fig. 1e, KD = 80 nM), and between MARTXVv RID and Rac1 (KD = 20 μM)14. Consequently, each protein structure in the complex fitted well into the cryo-EM density map contoured at 4.5 σ (Supplementary Fig. 11).
The overall shape and dimensions of the cryo-EM map show good agreement with the crystal structures of RDTND-RIDCBD complexed with CaM (this study), and RIDC/A (PDB ID, 5XN7)14 (Fig. 4a, b and Supplementary Fig. 11a–d). An extra density map was observed near to RIDCD, which fitted well with the crystal structure of the active form of Rac1 (PDB ID 3TH5) (Fig. 4a, b and Supplementary Fig. 11e, f). The cryo-EM map indicated that Switch I is critical for the interaction with RIDCD (Supplementary Fig. 11e). These structural conformations reveal that the PBR of Rac1 is accessible to the catalytic site of RID (Fig. 4b). The complex forms an L-shaped structure (Fig. 4a, b). Consistent with the biochemical analyses (Supplementary Fig. 7), these data suggest that CaM and Rac1 are associated independently with the effector duet within the complex.

Reconstructed 3D cryo-EM map (a) and the fitted model (cartoon representation) of RDTND-RIDC/A complexed with CaM and Rac1Q61L (b). Catalytic residue C2838 in RID, and the C-terminal polybasic (PBR, red dashed line) and Switch 1 regions in Rac1Q61L, are indicated. c Surface charge distribution of the RDTND-RIDC/A/CaM/Rac1Q61L complex. The negative and positive charges are shown as red and blue, respectively. The positively charged surface of RIDMLD_C and RIDCD is highlighted by a dashed line. d Subcellular localization of the RDTND-RID module and its truncates. The GFP, GFP-fused RDTNDE/Q-RIDC/A, or GFP-fused RDTND-RID truncates (RDTNDE/Q-RIDCBD/MLD_C, RDTNDE/Q-RIDCBD/MLD, and RDTND) were expressed in HeLa cells and analyzed. The HeLa cell plasma membrane and nucleus were stained with CF® 594-conjugated wheat germ agglutinin (WGA) and Hoechst 33342, respectively. Data are representative of three independent experiments, each with similar results. e Cartoon summarizing the re-localization of RDTND-RID to the membrane after CPD-mediated release from the MARTX toxin, as well as the concerted invasion strategy used by the effector duet at the membrane and near the membrane.
RID functions as an Nε-fatty acyltransferase that modifies lysine residues within the PBR of Rac1 at the plasma membrane14. The cryo-EM structure reveals a positively charged surface spanning the entire RID, except for RIDCBD (Fig. 4c). To elucidate the cellular localization of the effector module, we expressed the enzymatically inactive RDTNDE/Q-RIDC/A duet fused with the C-terminal green fluorescent protein (GFP), as well as its truncated proteins, ectopically in HeLa cells. A green fluorescence signal was emitted by the effector duet exclusively at the plasma membrane, suggesting that the duet re-localizes to the plasma membrane after CPD-mediated release from the MARTX toxin (Fig. 4d). A previous study used a protein-lipid overlay assay to reveal a specific interaction between the MARTXVv RID and PI(4,5)P214. That study suggested that a positively charged pocket in the RID N2 subdomain (corresponding to the RIDMLD_C in this study) may bind to PI(4,5)P2. In the present study, further domain analysis revealed that the membrane localization of the duet is dependent on the entire RID, including both RIDMLD_C and RIDCD (Fig. 4d). The truncated forms of RDTND-RIDCBD/MLD_C (without RIDCD) or RDTND-RIDCBD/MLD are insufficient for localization to the plasma membrane. The WT RDTND lacking RID localized exclusively to the cytoplasm and did not induce any morphological changes in cells, further indicating the RID-dependent function of RDTND (Fig. 4d).
Upon discharge from the MARTX toxin, the positively charged surface of RID mediates re-localization to the membrane, and this conformation localizes RDTND exclusively in the cytoplasm (Fig. 4e, upper). RIDCBD-dependent CaM binding transforms RDTND into a functional enzyme that seeks out NAD(P)+, located either in the cytoplasm or near to the membrane (Fig. 4e, lower). The structural conformation also enables RID to interact with Rac1 located at the plasma membrane via interaction between the lysine residues in the PBR of the substrate and the active site (Fig. 4e, lower). It should be mentioned however that the interaction between the MARTXVv RID and Rac1 (KD = 20 μM) is significantly weaker than that between the MARTXVc RID and Rac1 (KD = 4 μM)14. MARTXVv RID (residues 2264–2901, NCBI accession ID: WP_015728045.1) and MARTXVc RID (residues 2465–3098, NCBI accession ID: WP_108347803.1) share a high level of sequence identity (85.6%) and similarity (93.5%) (Supplementary Fig. 12a). We used AlphaFold 332 to generate a structural model of the MARTXVc RID using the template structure of MARTXVv RID14. We compared positively charged residues on the surfaces of MARTXVv RID, MARTXVv RID within the cryo-EM complex structure, and the modeled MARTXVc RID (Supplementary Fig. 12b). Positive residues are distributed similarly on the surfaces of the RID proteins (Supplementary Fig. 12c). More positive residues occur on the surface of MARTXVc RID than on the surface of MARTXVv RID, particularly in the membrane localization domain (MLD)-containing domain (i.e., MLD_C), suggesting that membrane binding of MARTXVc RID may be stronger than that of MARTXVv RID. The weaker membrane binding of MARTXVv RID may be due the structural flexibility needed to transform RDTND into a NAD(P)+-hydrolyzing enzyme via hijacking of CaM, which may be more associated with the weaker interaction between MARTXVv RID and Rac1 than the interaction between MARTXVc RID and Rac1. The binding affinities of MARTXVc RID to Rac1 and MARTXVv RID to Rac1 were calculated in solution14. Since RID proteins are localized to the membrane and RID-catalyzed Rac1 modification is achieved in the membrane14, the binding affinities may be higher between both RID proteins and Rac1 in the membrane. Thus, the architectural organization of the duet in complex with host targets provides important insights into the concerted invasion strategy mediated by the MARTX toxin.
RDTND-RID silences the ROS/ERK/JNK/NF-κB axis critically required for first line immune responses in the host
Upon bacterial infection, host immune cells such as macrophages and neutrophils rapidly generate ROS to clear bacteria through NOX family proteins sited either at the plasma membrane or within mitochondria33,34,35,36,37,38. ROS activate MAPK signaling, which in turn triggers pro-inflammatory immune responses39,40. Therefore, eliminating ROS allows bacteria to survive and colonize the host41,42. Since depletion of NAD+ from immune cells causes a failure of cellular redox homeostasis, which is critical for ROS production via NOX family proteins43, and small GTPase Rac1 or Rac2 acts as a regulatory subunit during activation of various NOX family members15, we evaluated the significance of the effector duet in bone marrow-derived macrophages (BMDMs) infected with a series of engineered V. vulnificus strains that secrete MARTX toxins carrying effector-free (EF), effector duet (EF::RDTND-RID), or mutant duet (EF::RDTND-RIDC/A, EF::RDTNDE/Q-RIDC/A, or EF::RDTND4mt-RIDC/A) (Fig. 5a).

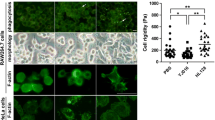
a Schematic diagram showing MARTX toxins from engineered V. vulnificus mutant strains. Substituted residues in RDTND-RID within the MARTX toxin from each mutant strain are indicated. Intracellular NAD+ (b) and NADP+ (c) levels in BMDMs infected for 2 h with the indicated V. vulnificus strains (MOI = 5, 5 × 106 CFU). NAD(P)+ levels relative to PBS are presented (n = 3 per group). ROS level in RAW 264.7 cells infected for 2 h with the indicated V. vulnificus strains (MOI = 5, 5 × 106 CFU). ROS production was measured by flow cytometry (d), and fold changes in mean fluorescence intensity (MFI) compared with the PBS control are shown (n = 3 per group) (e). f Immunoprecipitation assay. Lysates of BMDMs infected for 2 h with the indicated V. vulnificus strains were immunoprecipitated by an anti-gp91phox antibody, followed by immunoblot analysis with anti-Rac1, anti-p47phox, anti-p67phox, and anti-gp91phox antibodies. g Immunoblot analysis of MAPK signaling molecules and transcription factors in BMDMs infected for 2 h with the indicated V. vulnificus strains. EF, effector-free; WT-WT, EF::RDTND-RID; WT-C/A, EF::RDTND-RIDC/A; E/Q-C/A, EF::RDTNDE/Q-RIDC/A; 4mt-C/A, EF::RDTND4mt-RIDC/A. ELISA of pro-inflammatory cytokines IL-6 (h), TNF-α (i), and MCP-1 (j) in supernatants from BMDMs infected for 2 h with the indicated V. vulnificus strains (n = 6 per group). Data shown are representative of three independent experiments, each with similar results (mean ± SEM). P-values were calculated using one-way ANOVA with multiple comparisons. ns, not significant. Source data are provided as a Source Data file.
First, we evaluated intracellular NAD+ and NADP+ levels, both of which were significantly decreased in BMDMs infected with V. vulnificus strains secreting MARTX toxins containing EF::RDTND-RID or EF::RDTND-RIDC/A, but not in BMDMs infected by other strains containing inactive RDTND (EF::RDTNDE/Q-RIDC/A) or an effector duet defective in CaM binding (EF::RDTND4mt-RIDC/A). These data further demonstrate the function of RDTND as an NAD(P)+-hydrolyzing enzyme at the strain level (Fig. 5b, c).
Next, we used flow cytometry to analyze intracellular ROS levels in RAW 264.7 macrophage cells infected with different strains (Fig. 5d, e, and Supplementary Fig. 13). The intracellular ROS level increased upon infection with EF or inactive RDTND-containing toxin strains (EF::RDTNDE/Q-RIDC/A) due to LPS-mediated ROS generation33,44; however, infection by the EF::RDTND-RIDC/A strain reduced ROS production by macrophages significantly. We were unable to calculate ROS production induced by the V. vulnificus strain secreting a MARTX toxin carrying EF::RDTND-RID due to severe cytotoxic effects on immune cells. Nevertheless, a previous study showed that MARTXVc RID inhibits ROS production by mouse macrophage cells14. We then examined involvement of Rac1 in formation of NOX2 complexes to defend against infection. Immunoprecipitation (IP) assays showed that infection by the EF, EF::RDTND-RIDC/A, EF::RDTNDE/Q-RIDC/A, or EF::RDTND4mt-RIDC/A led to a marked increase in the NOX2/gp91phox interaction with Rac1, p47phox, and p67phox in macrophages (Fig. 5f). However, infection by the EF::RDTND-RID strain infection led to a significant reduction in the association between the key component Rac1 and these subunits (Fig. 5f), resulting in NOX2 inactivation and inhibition of ROS generation. The cryo-EM structure reveals that RID interacts with active Rac1 via the Switch I region and allows Rac1 to access the PBR for fatty acylation modification (Fig. 4b and Supplementary Fig. 11e). A previous structural and interaction study using purified proteins expressed in Escherichia coli showed that active Rac1 interacts with p67phox (KD = 1.7 μM) via the Switch I region45. Rac1 contains two potent membrane-localizing elements (the PBR and a geranylgeranyl modification) at its C-terminus46. The transmembrane gp91phox catalytic subunit (i.e., NOX2) is activated by the assembly of four cytosolic subunits (p47phox, p67phox, Rac1 or Rac2, and p40phox), which translocate to the membrane to form the active NOX2 complex. In preparation for membrane translocation, p67phox interacts with p47phox prior to the interaction with the GTP-bound Rac protein47. Although the binding affinity between MARTXVv RID and Rac1 (KD = 20 μM)14 is weaker than that between p67phox and Rac1, the IP results (Fig. 5f) suggest that the association between modified Rac1 and the p67phox and p47phox complex may reduce the binding affinity, and that RID competitively hijacks and fatty acylates Rac1 to block its association with the NOX2 complex in macrophages infected by the EF::RDTND-RID strain. Collectively, these data suggest that both NAD(P)+ depletion and Rho GTPase modification by the RDTND-RID duet are critically linked to reduced production of ROS.
During early responses, ROS act as secondary messengers capable of regulating expression of pro-inflammatory genes by altering kinase cascades and activating transcription factors48,49,50,51,52. Therefore, we examined MAPK cascades in BMDMs infected with V. vulnificus strains at different time points. We found no significant difference in cellular levels of phosphorylated extracellular signal-regulated kinase (ERK) or c-Jun NH2-terminal kinase (JNK) between BMDMs infected with EF, EF::RDTND-RIDC/A, or EF::RDTNDE/Q-RIDC/A up until 1 h post-infection. However, there was a significant reduction in phosphorylation of ERK and JNK in immune cells infected with strain EF::RDTND-RIDC/A, but not with strain EF or EF::RDTNDE/Q-RIDC/A, at 2 h post-infection (Supplementary Fig. 14a). Thus, we further analyzed the signaling pathways at 2 h post-infection, and observed that activation of ERK and JNK was reduced markedly in cells infected with EF::RDTND-RID or EF::RDTND-RIDC/A (Fig. 5g). However, activation of p38 was unchanged in cells infected with these two strains when compared with strains EF, EF::RDTNDE/Q-RIDC/A, or EF::RDTND4mt-RIDC/A (Fig. 5g).
Mitogen- and stress-activated kinases (MSK) are activated by either ERK or p38 kinases in response to stress, including bacterial infection; activated ERK or p38 kinases subsequently activate pro-inflammatory mediators such as NF-κB in monocyte/macrophage cells44,53,54. The silenced phosphorylation of ERK and JNK mediated by RDTND-RID resulted in reduced phosphorylation of MSK1/2, which was linked to a significant reduction in phosphorylation of CREB (S133) and NF-κB (S276) (Fig. 5g). In addition to phosphorylation of the NF-κB p65 subunit at S276, acetylation at K310 is required for NF-κB-mediated transactivation of pro-inflammatory genes. We found that effector duet-mediated suppression of MAPK signaling also silenced acetylation of NF-κB p65 at K310 significantly (Fig. 5g). By contrast, reduced phosphorylation of JNK inhibited activation of c-Jun, a critical regulator of pro-inflammatory cytokine expression in macrophages (Fig. 5g).
Next, we evaluated the expression levels of pro-inflammatory cytokines in BMDMs infected with V. vulnificus strains. While BMDMs infected with EF or inactive RDTND-RID duet-containing MARTX toxin-secreting V. vulnificus strains produced significant levels of IL-6, TNF-α, and MCP-1, cells infected with the enzymatically active effector duet-containing toxin-secreting strains (i.e., EF::RDTND-RID strain harboring both NAD(P)ase and Nε-fatty acyl-transferase functions and EF::RDTND-RIDC/A strain harboring only NAD(P)ase function) showed a clear reduction in pro-inflammatory cytokine production (Fig. 5h–j). We further validated the function of RDTND alone in pro-inflammatory cytokine suppression using NOX2-silenced macrophage cells infected with V. vulnificus strains. Overall cytokine levels in the NOX2-silenced macrophage cells infected with the strains were significantly lower than those in WT macrophage cells. Nevertheless, the results showed that only NAD(P)ase activity (i.e., EF::RDTND-RIDC/A) significantly suppressed pro-inflammatory cytokine production in the NOX2-silenced macrophage cells (Supplementary Fig. 14b–e), indicating that depletion of NAD(P)+ by RDTND is critically involved in pro-inflammatory cytokine suppression via ROS reduction, independent of RID-mediated inhibition of NOX2 complex formation. Thus, these data demonstrated that MARTX toxin-secreting pathogenic bacteria uses the RDTND-RID duet-mediated strategy to suppress efficiently ROS generation in immune cells, which silences the ERK/JNK/NF-κB signaling required for the evasion of the pro-inflammatory immune responses critical for the first line of defense against infection.
RDTND-RID promotes bacterial dissemination and sepsis in mice
To demonstrate the pathogenicity of the effector duet in vivo, mice were subcutaneously challenged with a series of V. vulnificus strains. First, we assessed NAD+ levels in peripheral blood mononuclear cells (PBMCs) isolated from the blood of mice at 7 h post-infection with the indicated V. vulnificus strains. The NAD+ levels in PBMCs from mice infected with EF::RDTND-RID or EF::RDTND-RIDC/A strains were significantly lower than those in mice infected with the EF::RDTNDE/Q-RIDC/A or EF::RDTND4mt-RIDC/A strains (Fig. 6a). Next, we measured bacterial colonization of the blood, spleen, and liver in mice. The largest numbers of bacteria were found in the blood, spleen, and liver of the mice challenged with the EF::RDTND-RID strain, with slightly lower numbers in those challenged with the EF::RDTND-RIDC/A strain (Fig. 6b–d). However, mice infected with the EF::RDTNDE/Q-RIDC/A or EF::RDTND4mt-RIDC/A strains had significantly lower bacterial populations in the blood, spleen, and liver (Fig. 6b–d).

a Intracellular NAD+ levels in PBMCs isolated from 5-week-old female ICR mice 7 h post-infection with the indicated V. vulnificus strains. NAD+ levels relative to PBS are presented (n = 8 for PBS; n = 12 for EF::RDTND-RID, EF::RDTND-RIDC/A, EF::RDTNDE/Q-RIDC/A, and EF::RDTND4mt-RIDC/A). Single bacterial colonies in blood (b), spleen (c), and liver (d) collected from 5-week-old female ICR mice at 10 h post-infection with the indicated V. vulnificus strains; normalized data are shown (n = 14 per group). ELISA of pro-inflammatory cytokines of IL-6 (e) and MCP-1 (f) in serum collected from 5-week-old female ICR mice infected with the indicated V. vulnificus strains at the designated time (n = 2 for 0 h, and n = 6 for 3 h, 5 h, and 7 h post-infection per group). P-values were calculated versus the PBS control at each time point, and are indicated above each graph. ICR mice were challenged with indicated V. vulnificus strains (n = 10, 5 × 106 CFU per group), and the ventral surface temperature (g) and survival rates (h) of the infected mice are shown. h.p.i., hours post-infection. The ventral surface temperature at 14 h was analyzed, and P-values compared with those from PBS-treated mice. Data shown in a–d are pooled from three independent experiments (mean ± SEM). Data shown in e and f are representative of three independent experiments, each with similar results (mean ± SEM). Data shown in g and h are pooled from n = 5 per group from n = 2 independent experiments. P-values were calculated using one-way ANOVA with multiple comparisons (a, g), a non-parametric Mann-Whitney U-test (two-tailed) (b–d), or a two-way ANOVA with multiple comparisons (e, f) or log-rank test (h). ns, not significant. Source data are provided as a Source Data file.
Consistent with the colonization results showing bacterial dissemination into different tissues in mice, we found that IL-6 and MCP-1 levels in sera were also considerably higher (i.e., a cytokine storm), and body temperature was substantially lower, in mice infected with the EF::RDTND-RID or EF::RDTND-RIDC/A strain (particularly at 5 h and 7 h post-infection); these are typical signs and symptoms of bacterial sepsis (Fig. 6e–g). Furthermore, significant infiltration of the liver by immune cells was observed in mice infected with the EF::RDTND-RID or EF::RDTND-RIDC/A strains at 10 h post-infection (Supplementary Fig. 14f).
Consequently, all mice challenged with the parental WT or EF::RDTND-RID strains succumbed to sepsis within 20 h post-infection (Fig. 6h). Mice challenged with the EF::RDTND-RIDC/A strain showed a 60% survival rate, while all mice challenged with EF::RDTNDE/Q-RIDC/A or EF::RDTND4mt-RIDC/A strain survived until the end of the experiment (Fig. 6h).
A previous study using mice infected with V. vulnificus secreting MARTX toxin showed that the bacterial population increased time-dependently at the dorsally located subcutaneously infected site55. The tissue surrounding the infected area revealed disintegrated myofibrils and necrotic adipocytes. Proliferation and dissemination of the bacteria to internal organs through the bloodstream was observed at 9 h post-infection55. V. vulnificus lacking the MARTX toxin was defective in conferring cytotoxicity to immune cells and was sensitive to phagocytosis by immune cells, as evidenced by the subsequent reduction in colonization and dissemination of the bacteria to internal organs55. Consistent with a previous study55, our study also found that pro-inflammatory cytokine levels were up-regulated time-dependently (Fig. 6e, f), and that bacterial loads appeared in the blood, spleen, and liver at 10 h post-infection (Fig. 6b–d), suggesting that the subcutaneously infected V. vulnificus RDTND-RID strain or RDTND-RIDC/A strain may colonize and subsequently spread to distal organs through the bloodstream. Taken together, these data demonstrate that MARTX toxin-secreting pathogenic bacteria utilize the RDTND-RID duet, which acts cooperatively to evade ROS-mediated pro-inflammatory immune responses and to promote systemic dissemination, ultimately resulting in bacterial sepsis.
Discussion
Due to its essential role in maintaining cellular homeostasis, NAD+ is a key target of pathogens43,56,57,58,59,60,61. Pathogenic bacteria secrete NAD+‐utilizing toxins capable of damaging hosts either by modulating the activity of host proteins through post-translational modification (i.e., ADP-ribosylation)62 or by depleting NAD+ 63,64,65,66,67. Deadly human pathogens such as Streptococcus pyogenes and Mycobacterium tuberculosis produce endogenous NADase, together with cognate immunity factors that act as an antitoxin by forming complexes with NADase to inactivate it and prevent “self-poisoning” (NAD+ is also essential for metabolic and energy homeostasis in pathogens)65,68,69,70. Upon infection of macrophages, the pathogen-secreted NADase translocates to the host cytosol where it hydrolyzes NAD+, thereby exerting cytotoxic effects65,71,72,73.
Here, we report that, unlike NADase-secreting pathogens, MARTX toxin-secreting pathogens have evolved distinct strategies to attack their hosts. The MARTX toxin RDTND-RID effector duet takes advantage of host cell machinery to turn itself into a critical weapon that paralyzes first line immune defense mechanisms by disrupting host cell NAD+ homeostasis and disabling Rac1. As suggested by its original name, DUF1 (i.e., domain of unknown function in the first position), the function of RDTND was never thought to be that of a NAD+-hydrolyzing enzyme. Its structural transformation into an NADase resembling host CD38 in infected host cells, a process assisted by its closest neighboring effector (i.e., RID), is sublimely unaware of evolutionary strategy of pathogens.
Although the mechanisms underlying CD38-mediated immune responses remain poorly understood and controversial, we do know that CD38 expression is induced robustly in macrophages and monocytes in response to infection, after which it regulates inflammatory responses during the early innate immune response74,75,76,77,78,79. CD38, in addition to NAD+-hydrolyzing enzyme, is one of the main mammalian ADP-ribosyl cyclases. CD38 generates Ca2+-mobilizing second messengers ADPR and cADPR from NAD+, and nicotinic acid adenosine dinucleotide phosphate (NAADP) from NADP+ 80. A recent study shows that CD38-mediated production of cADPR and NAADP, but not that of ADPR, induces expression of tristetraprolin (TTP), which downregulates expression of CD38 expression, leading to an increase in NAD+ levels (peaking 12 h after LPS treatment) and Sirtuin 1 activation in LPS-challenged macrophages79. The Sirtuin 1-dependently activated TTP then suppresses acute inflammatory responses and promotes autophagic bacterial clearance during the late phase of innate immunity, highlighting the importance of timely expression and activity of CD38 for resolution of inflammation and prevention of sepsis79. Here, we show that in mice, V. vulnificus secreting a MARTX toxin harboring the RDTND-RID effector duet significantly depletes NAD(P)+ levels and suppresses production of pro-inflammatory cytokines (including TNF-α) by downregulating ROS generation within 2 h post-infection (Fig. 5). This promotes dissemination of the bacteria and induces typical signs and symptoms of bacterial sepsis within 7–10 h post-infection (Fig. 6b–g). Taken together, these data suggest that the marked depletion of NAD(P)+ by RDTND-RID inhibits production of Ca2+ mobilizing second messengers during the early phase of infection, thereby paralyzing early innate immune defense mechanisms that normally promote resolution of late phase inflammation and prevent onset of sepsis.
A study reports that, unlike the MARTXVc RID MLD, the corresponding domain in MARTXVv RID (which is 93% similar) shows only weak membrane localization81. Our analysis confirmed that the entire MARTXVv RID, except the RIDCBD, is required for plasma membrane targeting (Fig. 4d). These features may correlate with the evolutionary conservation of V. vulnificus strains that secrete MARTX toxins carrying the RDTND and the RID, rather than the RDTND alone (Fig. 1b, c); this allows concurrent targeting of two important host proteins: CaM in the cytoplasm and Rac1 at the membrane (Fig. 4e). In addition to NAD+ depletion, RDTND located in close proximity to the cell membrane via RID may allow efficient depletion of NADP+ (the by-product of NOX2), a process required for NADPH production (the substrate of NOX2)43, further contributing to blockade of ROS generation.
The present study revealed that V. cholerae strains mainly secrete RID-containing MARTX toxins, a significant portion of which are RDTND-RID-containing toxins (Fig. 1a, b). Remarkably, nearly all clinical strains of V. cholerae secrete RID-carrying MARTX toxins (Fig. 1c). These results suggest that MARTXVc may have evolved to robustly target Rac1. Note that MARTXVc RID binds to Rac1 with an affinity that is five-fold higher than that of MARTXVv RID binding to Rac114. In general, the MARTX toxins secreted by V. cholerae strains deliver RID together with ACD (rather than with RDTND) for effective actin cytoskeleton collapse via the suppression of Rac1-mediated inflammatory responses to cytoskeletal damage, which promotes a non-inflammatory diarrheal disease (i.e., cholera)13. Since MARTXVc RID interacts with CaM, it remains to be clarified whether CaM is involved in V. cholerae pathogenesis.
It is evident that NAD+ homeostasis is a key determinant of core kidney function82. Interestingly, renal failure is a common sequelae of V. vulnificus infection83; indeed, a few cases reported in different countries highlight acute kidney injury in patients with cholera84,85,86. Our study proposes that Vibrio-related kidney problems may be interrelated with NAD+-depletion by the RDTND-RID duet.
In summary, pathogenic bacteria constantly evolve a wide range of strategies to invade and colonize their hosts. Our findings provide mechanistic insight into how pathogenic bacteria-generated effectors can act in a mutually-dependent manner, and may prove useful for the future design of tools or strategies to combat MARTX toxin-related human diseases.
Methods
Ethical statement
All animal experiments were carried out according to a protocol approved by the Institutional Animal Use and Care Committee of the Korea Research Institute of Bioscience and Biotechnology (KRIBB-AEC-18186). We have complied with all relevant ethical regulations for animal use.
Bacterial strains, culture media, cell culture, and mice
The bacterial strains, culture media, cell lines, and mice used in this study are listed in Supplementary Data 1. Escherichia coli strains were grown at 37 °C in Luria-Bertani medium (LB) supplemented with appropriate antibiotics. As previously reported1,87,88, Vibrio vulnificus MO6-24/O strains were grown at 30 °C in LB medium supplemented with 2% (w/v) sodium chloride (LBS). HEK293T, HeLa, and RAW 264.7 cells were obtained from the American Type Culture Collection. HEK293T, HeLa, RAW 264.7, and murine bone marrow-derived macrophage (BMDM) cells were cultured at 37 °C/5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Corning) containing 10% (v/v) fetal bovine serum (Gibco) and antibiotic-antimycotic solution (Cytiva). All mice (specific pathogen-free; 5 or 7-week-old female CrlOri:CD1 (ICR) mice) used in this study were purchased from Orientbio and acclimatized in the institutional facility for at least 24 h before the infection experiments (12 h light/dark cycle at 22 ± 1 °C temperature with 50 ± 5% humidity).
DNA and plasmids
Genomic DNA extracted from V. vulnificus MO6-24/O was used as a template for amplification of DNA fragments encoding MARTX toxin proteins. Amplified DNA fragments corresponding to the DUF1-RID effector module [referred to hereafter as RDTND-RID (residues 1959–2901)] and its C-terminally-deleted proteins [RDTND-RIDND/MLD (residues 1959–2468); RDTND-RIDND (residues 1959–2374); and RDTND (residues 1959–2252)] were subcloned individually into the pProEX HTa vector expressing a 6×His-TEV cleavage site at the N-terminus (Invitrogen). The primers used are listed in Supplementary Data 2. If necessary, the DNA fragments mentioned above were subcloned individually into the pHis-Parallel1 vector89 to express non-tagged recombinant proteins. Human calmodulin2 (CaM) DNA was purchased from the Korea Human Gene Bank and subcloned into the pHisGST-Parallel1 vector89 to express the protein with an N-terminal 6×His/GST tag. For expression in mammalian cells, DNA fragments encoding RDTND-RID variants [RDTND-RID; RDNTD; RID (residues 2263–2901); RDTND-RIDC/A (mutation of C2838 to alanine); RDTNDE/Q-RIDC/A (mutations of E2186 and C2838 to glutamine and alanine, respectively); and RDTNDE/Q-RIDCBD/MLD_C (residues 1959–2581); RDTNDE/Q-RIDCBD/MLD (residues 1959–2468); RIDC/A] were subcloned individually into the pAcGFP_N1 vector (Clontech), which generates proteins carrying GFP at the C-terminus. To identify interacting partners of RDTND-RID or RID in mammalian cells, DNA encoding RDTNDE/Q-RIDC/A or RIDC/A was subcloned individually into pEXPR-IBA 103 (IBA Lifesciences) to express proteins carrying a strep tag at the C-terminus. DNA encoding human Ras-related C3 botulinum toxin substrate 1 (Rac1) was purchased from the Korea Human Gene Bank. DNA fragments encoding Rac1 (residues 1–192) or C-terminally-deleted Rac1 (residues 1–177) were subcloned individually into the pProEX HTa vector to express proteins carrying a 6×His-TEV cleavage site at the N-terminus. Plasmids expressing RDTND-RID mutants and Rac1 mutants (Rac1Q61L, and Rac1T17N) were generated by site-directed mutagenesis using the primers described in Supplementary Data 2, Pfu DNA polymerase (NanoHelix), and the DpnI restriction enzyme (NEB).
Sequence analysis of MARTX toxins
MARTX toxins containing the RDTND-RID module and its variants were subjected to sequence analysis using the amino acid sequence of RDTND-RID (residues 1959–2901) of V. vulnificus MO6-24/O strain as a template. The full sequences of 1858 MARTX toxins were downloaded from the NCBI database and compared with that of RDTND-RID and its variants using NCBI Blast+ (version 2.14.0 + ), with the BLASTp option. Each sequence that exhibited a similarity of at least 60% (and a coverage of at least 90%) with the RDTND-RID module sequence was deemed identical. To detect the presence of a MARTX toxin containing the RDTND-RID module in clinical isolates of V. cholerae and V. vulnificus, a total of 5979 and 1373 genomes, respectively, that had been assembled at least to the scaffold-level or higher were downloaded from the NCBI Genome database. Only genomes originating from Homo sapiens were downloaded from the NCBI BioSample database (which lists the origin of strains) for use in this study. The representative RDTND-RID module sequence from V. vulnificus MO6-24/O was aligned to each genome using NCBI Blast+ (version 2.14.0 + ), with the BLASTn option. Genome sequences exhibiting at least 90% coverage of the RDTND-RID module or RID were considered as matches, and subsequently classified.
Expression and purification of recombinant proteins
E. coli BL21-CodonPlus (DE3) RIPL strain (Agilent) was transformed with plasmids containing the 6×His-tagged RDTND-RID module or its variants [RDTND-RIDC/A; RDTNDE/Q-RIDC/A; RDTND4mt-RIDC/A (mutations: W2183, R2195, E2285, and R2328 to leucine for RDTND4mt); RDTND-RIDCBD; and RDTND]. E. coli strains were grown at 37 °C in LB broth containing 100 μg mL−1 ampicillin until the optical density at 600 nm (OD600) reached 0.5–0.6. Then, expression of recombinant proteins was induced at 18 °C by addition of 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG; LPS Solution) for 18 h. The cells were harvested by centrifugation at 4000 × g for 10 min, resuspended in buffer A [50 mM Tris-HCl (pH 7.5), 300 mM NaCl, and 5% (v/v) glycerol] supplemented with 10 mM imidazole, and then lysed using a high pressure homogenizer (Nano DeBee). After centrifugation at 14,000 × g for 1 h at 4 °C, the supernatants were subjected to immobilized metal affinity chromatography (IMAC) using a nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) pre-equilibrated with buffer A. The column was washed with buffer A supplemented with 25 mM imidazole, and 6×His-tagged recombinant proteins bound to the resin were eluted with buffer A supplemented with 250 mM imidazole. When necessary, the N-terminal 6×His-tag was removed from proteins by treatment with TEV protease (Sigma Aldrich) for 18 h at 4 °C during dialysis into buffer B [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 5% (v/v) glycerol]. Proteins not digested by TEV proteases were removed by a second affinity chromatography step using Ni-NTA resin, and eluted proteins were further purified by size-exclusion chromatography using a HiLoad 16/60 SuperdexTM 200 prep grade column (GE Healthcare) pre-equilibrated with buffer B. To purify 6×His/GST-fused CaM in the presence of CaCl2 (6×His/GST-CaM/Ca2+), harvested E. coli cells expressing 6×His/GST-fused CaM were lysed in buffer A supplemented with 5 mM CaCl2 and 10 mM imidazole, and further purified using the procedures described above. To purify 6×His/GST-fused CaM in the absence of CaCl2 (6×His/GST-CaM), harvested E. coli cells were lysed in buffer C [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM EDTA, and 10 mM EGTA] using a high pressure homogenizer (Nano DeBee). After centrifugation at 14,000 × g at 4 °C for 1 h, the supernatant was subjected to immobilized affinity chromatography using Glutathione SepharoseTM 4 Fast Flow resin (Cytiva) pre-equilibrated with buffer C. The column was washed with buffer C, and 6×His/GST-fused recombinant proteins bound to the resin were eluted with buffer C supplemented with 30 mM reduced glutathione. The 6×His/GST tags were cleaved from the recombinant proteins by TEV protease, and non-digested proteins and 6×His/GST were removed by a second affinity chromatography step using Ni-NTA resins. The eluted proteins were further purified by size-exclusion chromatography using a HiLoad 16/60 SuperdexTM 75 prep grade column (GE Healthcare) pre-equilibrated with buffer B. To prepare Ca2+-bound CaM in complex with RDTND-RIDC/A or RDTND-RIDCBD, harvested cells expressing 6×His/GST-CaM and harvested cells expressing RDTND-RID variants without a tag were mixed together and lyzed in buffer A supplemented with 5 mM CaCl2 and 10 mM imidazole. Proteins were purified as described for 6×His/GST-CaM/Ca2+. To purify CaM complexed with RDTND-RIDC/A or RDTND-RIDCBD, harvested cells expressing 6×His/GST-CaM or RDTND-RID variants without a tag were mixed together, lyzed in buffer C, and purified as described for 6×His/GST-CaM. To prepare selenomethionine (SeMet)-labeled proteins (RDTND-RIDCBD, RDTND-RIDCBD/CaM, and RDTND-RIDCBD/CaM/Ca2+), E. coli B834 (DE3) strain (Novagen) was transformed with the corresponding expression plasmids, and grown in minimal M9 medium supplemented with 50 mg mL−1 of SeMet and 100 μg mL−1 of ampicillin until OD600 = 0.8. Expression of SeMet-labeled proteins was induced at 25 °C by addition of 0.5 mM IPTG until OD600 = 1.5 (at least). SeMet-labeled proteins were purified as described above. All purified proteins were concentrated to 15–20 mg mL−1 using an Amicon Ultra-15 30 K device (EMD Millipore) prior to crystallization and kept at −80 °C until use. To purify Rac1 proteins, harvested E. coli cells expressing N-terminal 6×His-tagged Rac1 proteins (Q61L mutant for GTP-bound form and T17N mutant for GDP-bound form) were lyzed in buffer A supplemented with 1 mM 1,4-dithiothreitol (DTT; LPS Solution) and 10 mM imidazole, and purified as described for the 6×His-tagged RDTND-RID module. To purify the RDTND-RIDC/A complexed with Ca2+-bound CaM and Rac1Q61L, E. coli cells expressing RDTND-RIDC/A and 6×His/GST-CaM were mixed together and lyzed in buffer A supplemented with 5 mM CaCl2 and 10 mM imidazole. Meanwhile, E. coli cells expressing 6×His-tagged Rac1Q61L (residues 1–177) were lyzed in buffer A supplemented with 1 mM DTT and 10 mM imidazole. Then, each sample was purified as described above. All purified proteins were pooled in a beaker and incubated with gentle stirring for 3 h at 4 °C. After incubation, the RDTND-RIDC/A/CaM/Rac1Q61L complex was further purified using a SuperdexTM 200 10/300 GL column (GE Healthcare) pre-equilibrated with buffer D [50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, and 1% (v/v) glycerol] prior to cryo-EM.
In vitro pull-down assay
To investigate the interaction between RDTND-RID variants and CaM, 0.5 mg of each purified protein (RDTND-RID, RDTND-RIDND/MLD, RDTND-RIDND, or RDTND), and 0.25 mg of purified 6×His/GST-CaM, were used. The protein mixtures were reacted at 4 °C for 1 h in TBS buffer [50 mM Tris-HCl (pH 7.4) and 137 mM NaCl] containing 10 mM EDTA and 10 mM EGTA. Then, the mixtures were loaded onto 0.1 mL of Glutathione SepharoseTM 4 Fast Flow affinity resins. The resins were washed with a 150-fold resin volume of TBS buffer, and samples were eluted with TBS buffer supplemented with 50 mM reduced glutathione. Eluates were analyzed by SDS-PAGE and visualized using Coomassie Brilliant Blue staining. To validate the Ca2+-dependency of the interaction between RDTND-RID and CaM, 0.5 mg of RDTND-RID module protein was incubated with 0.25 mg of 6×His/GST-CaM or 6×His/GST-CaM/Ca2+ in TBS buffer containing 10 mM EDTA and 10 mM EGTA, or in TBS buffer containing 10 mM CaCl2, respectively, at 4 °C for 1 h.
Isothermal titration calorimetry
To measure the binding affinity of CaM or CaM/Ca2+ for the RDTND-RID module, purified CaM, CaM/Ca2+, or RDTND-RIDC/A proteins were dialyzed in buffer B at 4 °C for 18 h. All purified proteins were degassed at 25 °C for 20 min using a ThermoVac (Microcal). CaM or CaM/Ca2+ (450 μM) was loaded into the syringe and titrated against 30 μM RDTND-RIDC/A in the reaction cell. Isothermal titration calorimetry measurements were performed at 25 °C, with an injection volume of 6 μL, 35 injections with an interval of 240 s between each, and a stirring speed of 300 rpm. Heat data derived from injection of CaM or CaM/Ca2+ into buffer B were subtracted from those derived from injection of CaM or CaM/Ca2+ into RDTND-RIDC/A. Raw data were processed, fitted, and plotted using the Origin software (version 7) supplied with the VP-ITC Microcalorimeter (Microcal).
Crystallization
Purified SeMet-labeled RDTND-RIDCBD proteins were screened initially at 20 °C prior to crystallization using the sitting-drop vapor-diffusion method conducted in 96-well MRC crystallization plates (Swissci); proteins were crystallized in a reservoir solution containing 8% (v/v) Tacsimate (pH 6.0) and 20% (w/v) polyethylene glycol (PEG) 3350 from the PEG/Ion screening kit (Hampton Research). Optimized SeMet-labeled RDTND-RIDCBD crystals were grown at 20 °C in 2 μL droplets containing equal volumes of protein and reservoir solution [4% (v/v) Tacsimate (pH 6.0) and 22% (w/v) PEG 3350]. For data collection, SeMet-labeled RDTND-RIDCBD crystals were cryoprotected by transferring them into reservoir solution containing an additional 10% (v/v) ethylene glycol and flash-frozen in liquid nitrogen. After crystallization of complex proteins using the sitting-drop vapor-diffusion method in 96-well MRC crystallization plates (Swissci), RDTND-RIDCBD/CaM/Ca2+ and SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ microcrystals were generated at 20 °C after 3 days in reservoir solution containing 20% (w/v) PEG 3350 and 0.2 M magnesium acetate from the MCSG1 screening kit (Anatrace). Optimized RDTND-RIDCBD/CaM/Ca2+ and SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ crystals were grown at 20 °C in 2 μL droplets containing equal volumes of protein and reservoir solution [17% (w/v) PEG 3350 and 0.18 M magnesium acetate, or 20% (w/v) PEG 3350 and 0.17 M magnesium acetate, respectively]. For data collection, RDTND-RIDCBD/CaM/Ca2+ and SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ crystals were cryoprotected by transferring them into a reservoir solution containing an additional 10% (v/v) ethylene glycol and flash-frozen in liquid nitrogen. SeMet-labeled RDTND-RIDCBD/CaM crystals were produced at 20 °C after 1 day in reservoir solution containing 25% (w/v) polyethylene glycol (PEG) 3350, 0.1 M HEPES-NaOH (pH 7.5), and 0.2 M MgCl2 from the MCSG1 screening kit (Anatrace). Optimized RDTND-RIDCBD/CaM crystals were generated at 20 °C in 2 μL droplets containing equal volumes of protein and reservoir solution [20% (w/v) PEG 3350, 0.1 M HEPES-NaOH (pH 7.5), 0.28 M MgCl2, and 6 mM MnCl2]. For data collection, crystals were flash-frozen in liquid nitrogen. To determine the structure of the NAD+-bound RDTNDE2186Q-RIDCBD/CaM/Ca2+ complexes, proteins were screened initially for crystallization using the sitting-drop vapor-diffusion method in 96-well MRC crystallization plates. RDTNDE2186Q-RIDCBD/CaM/Ca2+ microcrystals were produced at 20 °C after 2 days in reservoir solution [0.1 M Tris-HCl (pH 8.5), 30% (w/v) PEG 4000, and 0.2 M sodium acetate] from the SG1TM Screen screening kit (Molecular Dimensions). Optimized RDTNDE2186Q-RIDCBD/CaM/Ca2+ crystals were grown at 20 °C in 2 μL droplets containing equal volumes of protein and reservoir solution [0.1 M Tris-HCl (pH 8.5), 32.5% (w/v) PEG 4000, and 0.15 M sodium acetate]. RDTNDE2186Q-RIDCBD/CaM/Ca2+ was soaked for 2 h in a soaking solution containing an additional 10 mM NAD+ (Sigma Aldrich). For data collection, NAD+-bound RDTNDE2186Q-RIDCBD/CaM/Ca2+ crystals were mounted directly on the goniometer.
Collection of X-ray diffraction data and structure determination
SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ crystals were diffracted at a resolution of 3.05 Å, and single wavelength anomalous dispersion (SAD) data were collected at beamline 5 C at the Pohang Acceleratory Laboratory (PAL). All data were indexed, processed, and scaled using the HKL2000 software package90. The initial phases for SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ were obtained by the molecular replacement (MR)-SAD method using the Phenix program91, with the structure of human CaM (PDB ID 1CLL) as the MR template. Crystals of SeMet-labeled RDTND-RIDCBD were diffracted at a resolution of 3.38 Å, and SAD data were collected at beamline 5 C at PAL. The initial phases of SeMet-labeled RDTND-RIDCBD were determined by the MR-SAD method using Phenix91, with the structure of RDTND-RIDCBD within the SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ complex used as the MR template. Crystals of the NAD+-bound RDTNDE2186Q-RIDCBD/CaM/Ca2+ were diffracted at beamline 5 C at PAL at a resolution of 2.35 Å. The structure of NAD+-bound RDTNDE2186Q-RIDCBD/CaM/Ca2+ was determined by MR using Molrep in the CCP4i suite using the RDTND-RIDCBD/CaM/Ca2+ structure as the MR template. Crystals of SeMet-labeled RDTND-RIDCBD/CaM were diffracted at beamline 11 C of PAL at a resolution of 2.82 Å, and SAD data were collected at beamline 11 C at PAL. All diffraction data were indexed, processed, and scaled using the HKL2000 software package and the XDS program package. The initial phases of SeMet-labeled RDTND-RIDCBD/CaM were obtained by the MR method using Molrep in the CCP4i suite, with the SeMet-labeled RDTND-RIDCBD/CaM/Ca2+ used as the MR template. Model building was carried out using Coot92, and refinement was performed by Refmac593. Ramachandran statistics were analyzed by MolProbity94. All data collection and refinement statistics are summarized in Supplementary Table 1. Superimposition of structures was carried out using PyMOL align (Schrödinger) and Coot SSM superpose. The Dali server was used to identify proteins with structures similar to the RDTND-RIDCBD of RDTND-RIDCBD/CaM/Ca2+. All molecular graphics were generated using PyMOL version 1.5.0.4 (Schrödinger) and ChimeraX version 1.595.
Preparation of samples for cryo-EM and data acquisition
For the cryo-EM studies of native samples, the purified RDTND-RIDC/A/CaM/Rac1Q61L complex (0.75 mg mL−1) was applied to a glow-discharged holey-carbon copper grid (Quantifoil, R1.2/1.3, 200-mesh; EMS). The grids were blotted for 3.0 s and plunge-frozen in ethane cooled with liquid nitrogen using Vitrobot Mark IV (FEI), with the environmental chamber set at a humidity of 100% and a temperature of 4 °C. Automatic cryo-EM data were collected on a Glacios (FEI) at an acceleration voltage of 200 kV using EPU software (FEI), and images were recorded using a Falcon IV direct electron detector (FEI). A total of 12,472 micrographs were recorded at a defocus range of −1.4 to −2.2 μm, with a nominal magnification of 120,000, resulting in a physical pixel size of 0.87 Å. Each image was dose-fractionated into 50 movie frames, with a total exposure time of 6.98 s, resulting in a total dose of 55 electrons per A2.
For the cryo-EM studies of cross-linked samples, the purified RDTND-RIDC/A/CaM/Rac1Q61L complex proteins were incubated for 2.5 h at 4 °C in buffer D containing 0.5% (v/v) glutaraldehyde. Then, the cross-linked proteins were loaded to a 5–20% glycerol gradient and ultracentrifuged at 160,000 × g at 4 °C for 18 h using a swinging bucket rotor (Eppendorf Himac Technologies). After ultracentrifugation, the samples were fractionated with a Piston Gradient FractionatorTM (Biocomp Instruments), and the fraction containing homogenous cross-linked proteins was further purified by size-exclusion chromatography using a SuperdexTM 200 10/300 GL column (GE Healthcare). The procedures for purification are shown in Supplementary Fig. 8. The prepared sample (0.75 mg mL−1) was applied to a glow-discharged holey-carbon copper grid (Quantifoil, R1.2/1.3, 300-mesh; EMS). The grids were blotted for 3.0 s and plunge-frozen in ethane cooled with liquid nitrogen using Vitrobot Mark IV (FEI), with the environmental chamber set at a humidity of 100% and a temperature of 4 °C. Automatic cryo-EM data were collected on a Titan Krios G4 (FEI) at an acceleration voltage of 300 kV using EPU software (FEI), and images were recorded using a BioQuantum K3 direct electron detector (Gatan). A total of 11,236 micrographs were recorded at a defocus range of −1.4 to −2.2 μm, with a nominal magnification of 130,000, resulting in a physical pixel size of 0.66 Å. Each image was dose-fractionated into 50 movie frames, with a total exposure time of 2.8 s, resulting in a total dose of 50 electrons per A2 (Supplementary Table 2).
Processing of cryo-EM data
The procedures used for data processing of cross-linked samples are shown in Supplementary Fig. 9. All data processing was done in cryoSPARC version 4.1.296. Motion correction and contrast transfer function estimation were done in cryoSPARC version 4.1.2. The initial 4,482,761 particles were picked automatically from 11,236 micrographs using Blob picker. Next, 179,094 particles were selected by Extract and repetitive 2D Classification, and classified to generate 2D templates. For further automated particle picking from the 179,094 particles, TOPAZ Train & Extract was used, with a box size of 300 pixels, and 3,357,262 particles were picked from 11,236 micrographs. The auto-picked particles were 2D classified over multiple rounds, and 437,183 particles were separated from all particles in bad 2D classes. The particles were re-extracted with a box size of 500 pixels and 2D classified again through multiple rounds. The selected 247,635 particles were then used for ab initio 3D reconstruction and subjected to heterogeneous 3D refinement. There was no significant difference between the four 3D reconstruction maps obtained from the heterogeneous 3D refinement. All 247,635 particles was subjected to several rounds of Non-uniform 3D Refinement, yielding a final reconstruction of the RDTND-RIDC/A/CaM/Rac1Q61L complex at 4.32 Å. The resolution was measured based on the gold-standard Fourier shell correlation cut-off criterion of 0.143. The procedures used for data processing of native samples are shown in Supplementary Fig. 10. All data processing was done in cryoSPARC version 4.3.196. Motion correction and contrast transfer function estimation were done in cryoSPARC version 4.3.1. The initial 1,869,419 particles were picked automatically from 11,558 micrographs using Blob picker. Next, 47,009 particles were selected by Extract and repetitive 2D Classification, and classified to generate 2D templates. For further automated particle picking from the 47,009 particles, TOPAZ Train & Extract was used with a box size of 300 pixels, and 1,437,704 particles were picked from 11,558 micrographs. The auto-picked particles were 2D classified over multiple rounds, and 181,398 particles were separated from all particles in bad 2D classes. The selected 2D classified particles showed low resolution due to high flexible movement.
Fitting of models into cryo-EM map
The L-shaped overall feature of the RDTND-RIDC/A/CaM/Rac1Q61L complex was observed in a cryo-EM map contoured at 4.5 σ. Individual X-ray structures of RDTND-RIDCBD complexed with CaM (this study), RIDMLD_C/CD (PDB ID: 5XN7), and active Rac1 (PDB ID: 3TH5) were well docked in the cryo-EM map using ChimeraX version 1.595. In detail, the structure of RDTND-RIDCBD complexed with CaM was docked in the cryo-EM density map contoured at ~8–12 σ. The structures of RIDMLD_C/CD and the active mimic Rac1Q61L (residues, 1–177) were fitted in the cryo-EM density map contoured at ~4.5–6 σ (Supplementary Fig. 11). The Cα-backbones were fitted in the cryo-EM map; however, the RID flexible region (residues, 2702–2764) and the side chains of the amino acids could not be modeled due to a weak cryo-EM map. The structural regions of RIDMLD_C/CD and Rac1Q61L were further refined using GalaxyRefineComplex on the GalaxyWEB server97, based on repeated structure perturbations and subsequent overall structural relaxation by short molecular dynamic simulation. The overall structure of RDTND-RIDC/A/CaM/Rac1Q61L was finally fitted into the cryo-EM map by WinCoot92 and phenix.cryo_fit, using Phenix version 1.20.1–1448791,98. The structural model of the complex fitted into the cryo-EM map is provided in Supplementary Data 3.
Confocal microscopy
To assess the cytotoxicity of RDTND-RID variants, plasmids expressing GFP or C-terminally GFP-fused RDTND-RID, RDTND-RIDC/A, RDTND, RID, or RIDC/A were transiently transfected into 1 × 105 of HEK293T cells in an 8-well chamber slide (Ibidi) using a LipofectamineTM 3000 transfection reagent (Thermo Fisher Scientific). After 18 h, cells were washed twice in Dulbecco’s phosphate-buffered saline (DPBS; Cytiva) and fixed for 20 min at room temperature using 4% (v/v) formaldehyde (Thermo Fisher Scientific) in DPBS. Next, 50% methanol in DPBS was added, followed by permeabilization in 100% methanol at −20 °C for 20 min. To analyze the subcellular localization of the RDTND-RID variants, 2 × 104 HeLa cells in an 8-well chamber slide (Ibidi) were transfected with plasmids expressing GFP, or C-terminally GFP-fused RDTNDE/Q-RIDC/A, RDTNDE/Q-RIDMLD_C, RDTNDE/Q-RIDMLD, or RDTNDE/Q, using X-tremeGENETM HP DNA transfection reagent (Roche). After 24 h, cells were washed twice with DPBS. Prepared cells were stained with corresponding reagents (Hoechst 33342 for nuclei, Thermo Fisher Scientific; Rhodamine phalloidin for F-actin, Thermo Fisher Scientific; CF® 594-conjugated wheat germ agglutinin (WGA) for the plasma membrane; Biotium). Images were obtained under a C2plus laser scanning confocal microscope (Nikon) and analyzed using NIS-Elements software (Nikon).
Nuclear magnetic resonance (NMR) analysis
Purified RDTND-RID complexed with CaM (66 nM) was incubated with 1 mM NAD+ in NMR reaction buffer [20 mM NaH2PO4/Na2HPO4 (pH 7.4), and 80 mM NaCl] in a final volume of 500 μL at room temperature for 30 min. The 1H NMR spectra of the reaction products were acquired on an Avance 700 MHz spectrometer (Bruker) equipped with a cryogenic triple resonance probe, operating at a proton frequency of 700 MHz at 298 K. The acquisition was carried out using Topspin software version 3.5 (Bruker) and a zgesgp pulse program (Bruker). Each spectrum for each time point was obtained with 4 scans. The spectral width was 9803.922 Hz, and the acquisition time was 835.584 ms. The spectra were processed with a window function of a 90°-shifted sine bell using Topspin software version 3.5.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
To identify RID-interacting proteins in mammalian cells, individual HEK293T cells were transfected with either empty vector (pEXPR_IBA103) or plasmid expressing C-terminally strep-tagged RIDC/A, and then incubated for a further 48 h to allow protein expression. Then, the cells were harvested and lysed with RIPA lysis buffer [50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1% (v/v) NP-40, 1 mM EDTA, 0.25% (w/v) Na-deoxycholate, and protease and phosphatase inhibitor cocktail (GenDepot)]. Cell lysates were centrifuged at 16,000 × g at 4 °C for 20 min, and the supernatants were incubated at 4 °C for 3 h with 50 μL of Strep-Tactin® Sepharose® resin (IBA Lifesciences), with gentle agitation. The supernatant was removed from the resin, and the resin was washed with a 200-fold resin volume (10 mL) of Strep wash buffer [100 mM HEPES-NaOH (pH 8.0), 150 mM NaCl, and 1 mM EDTA]. The proteins bound to the resin were eluted with Strep elution buffer [100 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 2.5 mM desthiobiotin]. Samples (n = 1 for empty vector and RIDC/A-Strep) were digested using Straps micro (PROTIFI) following the manufacturer’s protocol version 4.7, which includes the use of tris(2-carboxyethyl)phosphine (TCEP) and iodoacetamide (IAA) for reduction and alkylation. One μg of trypsin/lysC (Pierce) was added to each sample and incubated overnight at 37 °C. The eluted samples were dried with a speed-vac. The peptides were then resuspended in a 2% acetonitrile/0.1% formic acid solution and injected into a U3000 RSLCnano/Orbitrap Exploris 240 system (Thermo Fisher Scientific). The trap column used was an AcclaimTM PepMapTM 100 C18 (75 μm × 2 cm, 3 μm; Thermo Fisher Scientific), and the analytical column was a Double nanoViperTM PepMapTM Neo (75 μm × 50 cm, 2 μm; Thermo Fisher Scientific). Both columns were maintained at 60 °C. The mobile phases were 0.1% formic acid in HPLC-grade water (buffer X) and 0.1% formic acid in acetonitrile (buffer Y). The following gradient was used at a flow rate of 300 nL min−1: 2% to 22% buffer Y over 100 min, then 22% to 35% buffer Y over 20 min. The survey scan settings were as follows: Resolution = 120,000, Max IT = 50 ms, AGC target = 300%, and mass range = 350–1200 Th. The selected precursor ions were fragmented by HCD and analyzed by the Orbitrap. Additional parameters for the MS/MS scan were as follows: Top15 data-dependent acquisition, resolution = 30,000, Max IT = 200 ms, AGC target = standard, intensity threshold = 1 × 105, normalized collision energy = 30%, isolation width = 2, dynamic exclusion after 1 occurrence, exclusion duration = 20 sec, and mass tolerance window = ± 10 ppm. LC-MS/MS raw data were analyzed using MaxQuant version 2.4.0.099 and Perseus version 2.0.6.0100. The MaxQuant parameters included using the Uniprot Homo sapiens database, setting the enzyme to Trypsin/P, and allowing for variable modifications such as Oxidation (M) and Acetylation (protein N-term). Carbamidomethylation (C) was set as a fixed modification, and the match between runs feature was enabled. After reading the data into Perseus, protein groups were filtered out based on the following criteria: reverse sequences, proteins identified only by site, contaminants, and those with fewer than two razor + unique peptides. The LC-MS/MS results for the RIDC/A-interacting candidates are listed in Supplementary Data 4. To analyze the product of NAD(P)+ hydrolyzed by RDTND-RID module, 1 mM NAD(P)+ was incubated with 1 μM RDTND-RIDC/A/CaM protein in TBS buffer at 37 °C for 1 h. The reaction mixture was loaded onto an Amicon Ultra-0.5 3 K device (EMD Millipore) to remove proteins and then centrifuged at 14,000 × g at 4 °C for 20 min. The flow-through was diluted (1:5 ratio) with distilled water and analyzed by an Acquity UPLC/Synapt HDMS (Waters) system fitted with an Acquity UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm; Waters). The mobile phases were buffer X and buffer Y, with the column temperature set to 35 °C. The gradient elution was performed at a flow rate of 0.2 mL min−1 with the following program: 0% buffer Y for 5 min, 0% to 10% buffer Y over 1 min, and 10% to 40% buffer Y over 1 min. The survey scan settings included a mass range of 300–2000 Th, positive ion mode, and a scan time of 1 sec.
High-performance liquid chromatography (HPLC)
To analyze the by-products of NAD+ hydrolysis by CD38 or RDTND-RID, HPLC experiments were performed using a modified method from previous study31. One mM of NAD+ was incubated at 37 °C for 30 min with CD38 (10 nM; Sigma Aldrich) or RDTND-RID/CaM complex (80 nM) in TBS buffer. The reaction was placed in an Amicon Ultra-0.5 3 K device and centrifuged at 14,000 × g at 4 °C for 20 min. Then, 40 μL of the flow-through fraction was injected into a SUPELCOSILTM LC-18 HPLC column (25 cm × 4.6 mm, 5 μm; Supelco) in a HPLC system equipped with a diode array detector. The elution conditions were as follows: starting with 9 min at 100% buffer 1 (0.1 M potassium phosphate buffer, pH 6.0), followed by a gradual increase over 6 min to 12% buffer 2 (buffer 1 with 10% methanol). Then, the gradient increased to 45% buffer 2 over 2.5 min, and further to 100% buffer 2 over the next 2.5 min. The column was then held at 100% buffer 2 for 5 min. The flow rate was maintained at 1.0 mL min−1, and the assay was performed at room temperature. The absorbances of NAD+, ADPR, and cADPR were evaluated at a wavelength of 254 nm.
Engineering of V. vulnificus
To clarify the function of the RDTND-RID module within the MARTX toxins, four exotoxin gene-deleted (vvhA encoding hemolysin, vvpE encoding elastase, plpA encoding phospholipase A2, and vvpM encoding secretory protease) strains were used as the parental strains from which other V. vulnificus mutant strains were engineered as described previously1. The plasmid pDS_EF-MARTX, which contains an EF rtxA1 gene (EF-rtxA1)1, was used to engineer plasmids pDS_RDTND-RID, pDS_RDTND-RIDC/A, pDS_RDTNDE/Q-RIDC/A, and pDS_RDTND4mt-RIDC/A. Amplified DNA fragments of RDTND-RID, RDTND-RIDC/A, RDTNDE/Q-RIDC/A, or RDTND4mt-RIDC/A were subcloned into the XhoI restriction enzyme site of pDS_EF-MARTX. Consequently, plasmids pDS_RDTND-RID, pDS_RDTND-RIDC/A, pDS_RDTNDE/Q-RIDC/A, and pDS_RDTND4mt-RIDC/A were constructed. These constructs were delivered to the parental strain to exchange the rtxA1 locus for RDTND-RID, RDTND-RIDC/A, RDTNDE/Q-RIDC/A, or RDTND4mt-RIDC/A by in-frame double homologous recombination, as previously described1, thereby generating the EF::RDTND-RID, EF::RDTND-RIDC/A, EF::RDTNDE/Q-RIDC/A, and EF::RDTND4mt-RIDC/A strains, respectively (Fig. 5a).
Isolation of bone marrow-derived macrophages (BMDMs)
BMDMs were isolated from the thigh bones of 7-week-old female ICR mice and filtered through a cell strainer (40 μm; Falcon). The filtrate was treated for 10 min with ACK lysis buffer (Gibco) to remove red blood cells and centrifuged at 300 × g for 5 min at room temperature. Then, the isolated BMDMs were washed with PBS, cultured in DMEM containing 10% (v/v) FBS and 50 ng mL−1 GM-CSF (R&D Systems), and allowed to differentiate for 6 days in fresh complete medium, which was replaced every other day. The differentiated BMDMs were plated at 1 × 106 /well in 6-well plates and used for bacterial infection experiments.
V. vulnificus infection
V. vulnificus variants were grown in LBS until OD600 = 0.5. Bacteria were harvested by centrifugation at 2000 × g at room temperature. The cell pellet was washed and diluted in PBS to 1 × 108 colony-forming units (CFU) mL−1. Prior to infection of BMDMs, the culture medium was replaced with DMEM without FBS and antibiotic-antimycotic solution. Then, the prepared bacterial resuspension was added to the BMDMs in 6-well plates at the multiplicity of infection (MOI) of 5 and centrifuged in a swing rotor at 125 × g for 3 min at room temperature. BMDMs treated with V. vulnificus were incubated for 2 h (or other designated time) at 37 °C/5% CO2. For the mouse survival tests, 50 μL of bacterial suspension was subcutaneously injected into the region beneath the skin over the backs of the necks of 5-week-old female ICR mice (5 × 106 CFU; n = 10 per group), which were under slight isoflurane anesthesia. Infected mice were observed every 2 h until 48 h post-infection. If necessary, ventral surface temperature was measured every 2 h using a non-contact infrared thermometer (Daihan Scientific)101.
Isolation of peripheral blood mononuclear cells (PBMCs)
To isolate PBMCs from murine blood, 5-week-old female ICR mice infected by V. vulnificus (n = 8 for PBS; n = 12 for EF::RDTND-RID, EF::RDTND-RIDC/A, EF::RDTNDE/Q-RIDC/A, and EF::RDTND4mt-RIDC/A) were sacrificed at 7 h post-infection, and blood was collected from the infraorbital vein using heparin-coated capillary tubes (Kimble). Blood samples were diluted with PBS supplemented with 2% (v/v) FBS (up to 2 mL) and transferred to SepMateTM-15 in vitro diagnostic tubes (StemCell Technologies) filled with 4.5 mL Ficoll-PaqueTM PREMIUM 1.084 (Cytiva) for isolation of PBMCs. Tubes were centrifuged at 1200 × g for 10 min and plasma containing PBMCs was transferred to a new 15 mL tube. The plasma was then washed by adding 5 mL of 1 × PBS supplemented with 2% (v/v) FBS. Tubes were further centrifuged at 300 × g for 8 min, and the pellet cell was used for subsequent experiments.
NAD(P)+-hydrolyzing assay
To measure the NAD(P)+-hydrolyzing activity of RDTND-RID proteins, 1 μM of NAD+ or NADP+ (Sigma Aldrich) was incubated with 10 nM of protein in TBS buffer for 30 min at 37 °C. After completing the reaction, the remaining NAD+ was measured using EnzyChromTM NAD+/NADH assay kit (Bioassay Systems) or the EnzyChromTM NADP+/NADPH assay kit (Bioassay Systems). To measure the cellular NAD+ or NADP+ level in BMDMs, V. vulnificus was added to 1 × 106 of BMDMs in a 6-well plate at an MOI of 5, and then incubated at 37 °C for 2 h. The medium was exchanged for DMEM supplemented with 100 μg mL−1 of gentamycin to eliminate V. vulnificus. After further incubation for 4 h, the medium was removed and the BMDMs were washed with DPBS. Then, the intracellular NAD+ or NADP+ level was measured using an NAD+/NADH assay kit or NADP+/NADPH assay kit, respectively. Briefly, to measure intracellular NAD+ or NADP+ levels, BMDMs infected with V. vulnificus were homogenized in 100 μL of NAD(P)+ extraction buffer and incubated at 60 °C for 5 min. Then, the samples were neutralized by adding 20 μL of Assay buffer and 100 μL of NAD(P)H extraction buffer, and centrifuged at 16,000 × g for 5 min to remove cell debris. After centrifugation, the supernatant was used for the NAD+/NADH or NADP+/NADPH assay. To measure intracellular NAD+ levels in PBMCs, the number of isolated PBMCs from V. vulnificus-infected mice as described above was counted using a Luna-IITM automated cell counter (Logos Biosystems) after trypan blue staining. Then, the intracellular NAD+ level in PBMCs was measured using an EnzyChromTM NAD+/NADH assay kit and normalized to the number of PBMCs.
Enzyme-linked immunosorbent assay (ELISA)
To analyze pro-inflammatory cytokines, supernatants from in vitro-cultured BMDMs infected by V. vulnificus for 2 h as described above were obtained. To analyze pro-inflammatory cytokines in serum, murine blood samples collected from V. vulnificus-infected 5-week-old female ICR mice at different time points (0, 3, 5, and 7 h post-infection; n = 20 per group in total) were kept at room temperature for 4 h. The supernatant was transferred to a new 1.5 mL tube and centrifuged at 1000 × g at room temperature for 20 min to remove red blood cells. Then, cytokines (mouse IL-6, mouse TNF, and mouse MCP-1) in the medium or murine serum were measured using an ELISA kit (BD Biosciences).
Reactive oxygen species (ROS) detection assay
To detect intracellular ROS, RAW 264.7 cells were treated for 24 h with phorbol 12-myristate 13-acetate (2 μg mL−1). Then, the medium was exchanged for DMEM, and the cells were infected with V. vulnificus variants as described above (MOI = 5, 5 × 106 CFU). After incubation for 1.5 h, 20 μM 2’,7’-dichlorofluorescin diacetate (DCFDA; Sigma Aldrich) was added and further incubated for 30 min at 37 °C. The medium was removed and the cells were washed twice using FACS buffer [2% (v/v) FBS and 0.05% NaN3 in PBS]. Then, the cells were resuspended in fixation buffer [0.2% (v/v) formaldehyde in FACS buffer], and intracellular ROS were analyzed using a BD FACSCalibur® cytometer (BD Biosciences). The obtained data were analyzed by FCS ExpressTM software (version 7; De Novo Software).
Immunoblot analysis
BMDMs treated with V. vulnificus were lysed in RIPA lysis buffer and centrifuged at 16,000 × g for 30 min at 4 °C. The supernatant was mixed with 5 × SDS-PAGE sample buffer and subjected to denaturing gel electrophoresis (BoltTM 4–12% Bis-Tris Plus; Invitrogen). After electrophoresis, proteins were transferred to a PVDF membrane, which was then washed three times with TBST buffer [0.1% (v/v) Tween-20 in TBS]. The PVDF membrane was incubated for 1 h at room temperature in blocking buffer [4% (w/v) bovine serum albumin in TBST buffer], and further incubated at 4 °C overnight with blocking buffer containing a primary antibody. The membrane was then washed three times with TBST buffer and incubated again for 1 h at room temperature in blocking buffer containing the secondary antibody. Then, chemiluminescence was visualized using ECLTM Prime western blotting detection reagent (Cytiva) and a Davinch-ChemiTM chemiluminescence imaging system (Davinch-K).
Antibodies
The following antibodies were used for immunoblotting: anti-NOXA2/p67phox (1:1000; Abcam, ab109366), anti-NOX2/gp91phox (1:1000; Abcam, ab129068), anti-p-MSK2 Thr568 (1:1000; Assay Biotech, A8149), anti-MSK1 (1:1000; R&D Systems, AF2518), anti-p47phox (1:1000; Santa Cruz Biotechnology, sc-17845), anti-MSK2 (1:1000; Santa Cruz Biotechnology, sc-377151), anti-p-NF-kB p65 Ser276 (1:1000; Thermo Fisher Scientific, PA5-37718). The following antibodies were from Cell Signaling Technology: anti-Rac1/Cdc42 (1:1000, #4651), anti-p-ERK1/2 T202/Y204 (1:1000, #9101), anti-ERK1/2 (1:1000, #4695), anti-p-SAPK/JNK T183/Y185 (1:1000, #4668), anti-SAPK/JNK (1:1000, #9252), anti-p-p38 MAPK T180/Y182 (1:1000, #4511), anti-p38 MAPK (1:1000, #8690), anti-p-MSK1 T581 (1:1000, #9595), anti-p-CREB S133 (1:1000, #9198), anti-CREB (1:1000, #9197), anti-p-c-Jun S37 (1:1000, #3270), anti-c-Jun (1:1000, #9165), anti-ac-NF-kB p65 K310 (1:1000, #3045), anti-NF-kB p65 (1:1000, #8242), anti-β-Actin (1:5000, #12620). Details are provided in the Supplementary Data 1.
Immunoprecipitation assay
To analyze the assembly of the NOX2 complex, BMDMs were infected with the indicated V. vulnificus strains for 2 h as described above. Then, the medium was removed and the cells were lysed using RIPA lysis buffer containing a protease and phosphatase inhibitor cocktail (GenDepot). The cell lysates were incubated for 1 h at 4 °C with protein A/G agarose (Santa Cruz Biotechnology) to remove non-specifically bound proteins. Then, supernatants were incubated at 4 °C overnight with an anti-gp91phox antibody, followed by incubation with protein A/G agarose for 4 h at 4 °C. The immunoprecipitates were washed five times in RIPA lysis buffer, and subsequently eluted by adding 5 × SDS-PAGE sample buffer and boiling at 95 °C for 10 min. Finally, the samples were analyzed by SDS-PAGE followed by immunoblotting with anti-Rac1, anti-p47phox, anti-p67phox, and anti-gp91phox antibodies.
RNAi silencing of gp91phox
RAW 264.7 cells (1 × 106) in serum-free media were transfected with siRNA duplex (Supplementary Data 1) using TransIT-TKO transfection reagent (Mirus Bio). After incubation for 6 h, DMEM containing 20% FBS was added at a 1:1 ratio, and further incubated until 24 h for gene silencing. Non-targeting siRNA was used as a control, and the expression level of gp91phox was validated using immunoblot analysis.
Quantification of bacterial load
To measure the microbial populations in different tissues, liver, spleen, and blood were collected from 5-week-old female ICR mice (n = 14 per group) infected by V. vulnificus variants at 10 h post-infection. The weight of collected tissues was measured and lysed using TissueLyser II (30 Hz for 2 min; Qiagen) in 1 mL DPBS. Next, 50 μL of lysed tissue or blood was serially diluted in DPBS, plated on LBS agar supplemented with polymyxin B (100 units mL−1), and incubated overnight at 30 °C. Then, the number of bacterial colonies was counted, and normalized to grams (for liver and spleen) or liters (for blood), respectively.
Histological analysis of murine tissues
Mice were sacrificed at 10 h post-infection with V. vulnificus, and liver tissues were harvested and fixed overnight at room temperature with 10% formalin. The fixed tissues were washed in tap water for 8 h and embedded in paraffin blocks on a Tissue Embedding Center module (Sakura Finetek). The paraffin blocks were sectioned to a thickness of 7 μm using a microtome (Sakura Finetek) and deparaffinized in xylene. Liver tissues were further rehydrated in ethanol, and stained with hematoxylin and eosin (H&E). The stained tissue sections were imaged with an optical microscope (Olympus).
Statistical analysis
Statistical analyses were performed using Prism software (version 6; GraphPad), and P-values were calculated using one-way ANOVA with multiple comparisons, the non-parametric Mann-Whitney U-test (two-tailed), two-way ANOVA with multiple comparisons, and the log-rank test. All results are presented as the mean ± SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic models and structure factors have been deposited in the Protein Data Bank under accession codes 8KA2 (RDTND-RIDCBD), 8KA1 (RDTND-RIDCBD/CaM), 8K9Z (RDTND-RIDCBD/CaM/Ca2+), and 8KA0 (RDTNDE/Q-RIDCBD/CaM/Ca2+/NAD+). Crystal structures 1CFD (CaM), 1CLL (CaM/Ca2+), NAD+-bound or NAD+-unbound CD38 from human (2I65; 6VUA, 1YH3), 3ZWM (NAD+-bound ADP-ribosyl cyclase from A. californica), 1R12 (NAD+-unbound ADP-ribosyl cyclase from A. californica), 5XN7 (RIDVv), and 3TH5 (active Rac1) referred to in the manuscript can be found on the Protein Data Bank. The cryo-EM density map has been deposited in the Electron Microscopy Data Bank under accession code EMD-37593 (RDTND-RIDC/A/CaM/Rac1Q/L). The mass spectrometry proteomics data generated in this study have been deposited to the ProteomeXchange Consortium via the PRIDE database, with the dataset identifier PXD047927. The amino acid sequences of MARTXVv RID (NCBI accession ID: WP_015728045.1 [https://www.ncbi.nlm.nih.gov/protein/WP_015728045.1]) and MARTXVc RID (NCBI accession ID: WP_108347803.1 [https://www.ncbi.nlm.nih.gov/protein/WP_108347803.1]) were downloaded from the NCBI database for the sequence alignment in this study. Source data are provided with this paper.
References
Lee, Y. et al. Makes caterpillars floppy-like effector-containing MARTX toxins require host ADP-ribosylation factor (ARF) proteins for systemic pathogenicity. Proc. Natl Acad. Sci. USA 116, 18031–18040 (2019).
Choi, S., Kim, B. S., Hwang, J. & Kim, M. H. Reduced virulence of the MARTX toxin increases the persistence of outbreak-associated Vibrio vulnificus in host reservoirs. J. Biol. Chem. 296, 100777 (2021).
Kim, B. S. et al. MARTX Toxin-Stimulated Interplay between Human Cells and Vibrio vulnificus. mSphere 5, https://doi.org/10.1128/mSphere.00659-20 (2020).
Gavin, H. E. & Satchell, K. J. F. RRSP and RID Effector Domains Dominate the Virulence Impact of Vibrio vulnificus MARTX Toxin. J. Infect. Dis. 219, 889–897 (2019).
Gavin, H. E. & Satchell, K. J. MARTX toxins as effector delivery platforms. Pathog. Dis. 73, ftv092 (2015).
Kim, B. S. The Modes of Action of MARTX Toxin Effector Domains. Toxins (Basel) 10, https://doi.org/10.3390/toxins10120507 (2018).
Prochazkova, K. et al. Structural and molecular mechanism for autoprocessing of MARTX toxin of Vibrio cholerae at multiple sites. J. Biol. Chem. 284, 26557–26568 (2009).
Lupardus, P. J., Shen, A., Bogyo, M. & Garcia, K. C. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–268 (2008).
Jones, M. K. & Oliver, J. D. Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 77, 1723–1733 (2009).
Kim, B. S., Gavin, H. E. & Satchell, K. J. F. Variable Virulence of Biotype 3 Vibrio vulnificus due to MARTX Toxin Effector Domain Composition. mSphere 2, https://doi.org/10.1128/mSphereDirect.00272-17 (2017).
Kwak, J. S., Jeong, H. G. & Satchell, K. J. Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc. Natl Acad. Sci. USA 108, 1645–1650 (2011).
Agarwal, S. et al. Autophagy and endosomal trafficking inhibition by Vibrio cholerae MARTX toxin phosphatidylinositol-3-phosphate-specific phospholipase A1 activity. Nat. Commun. 6, 8745 (2015).
Woida, P. J. & Satchell, K. J. F. The Vibrio cholerae MARTX toxin silences the inflammatory response to cytoskeletal damage before inducing actin cytoskeleton collapse. Sci Signal 13, https://doi.org/10.1126/scisignal.aaw9447 (2020).
Zhou, Y. et al. N(epsilon)-Fatty acylation of Rho GTPases by a MARTX toxin effector. Science 358, 528–531 (2017).
Hordijk, P. L. Regulation of NADPH oxidases: the role of Rac proteins. Circ. Res. 98, 453–462 (2006).
Nisimoto, Y., Freeman, J. L., Motalebi, S. A., Hirshberg, M. & Lambeth, J. D. Rac binding to p67(phox). Structural basis for interactions of the Rac1 effector region and insert region with components of the respiratory burst oxidase. J. Biol. Chem. 272, 18834–18841 (1997).
Freeman, J. L., Abo, A. & Lambeth, J. D. Rac “insert region” is a novel effector region that is implicated in the activation of NADPH oxidase, but not PAK65. J. Biol. Chem. 271, 19794–19801 (1996).
Freeman, J. L., Kreck, M. L., Uhlinger, D. J. & Lambeth, J. D. Ras effector-homologue region on Rac regulates protein associations in the neutrophil respiratory burst oxidase complex. Biochemistry 33, 13431–13435 (1994).
Abo, A. et al. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353, 668–670 (1991).
Knaus, U. G., Heyworth, P. G., Evans, T., Curnutte, J. T. & Bokoch, G. M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 254, 1512–1515 (1991).
Glogauer, M. et al. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 170, 5652–5657 (2003).
Kim, C. & Dinauer, M. C. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J. Immunol. 166, 1223–1232 (2001).
Shen, A. et al. Mechanistic and structural insights into the proteolytic activation of Vibrio cholerae MARTX toxin. Nat. Chem. Biol. 5, 469–478 (2009).
Kuboniwa, H. et al. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2, 768–776 (1995).
Aarhus, R., Graeff, R. M., Dickey, D. M., Walseth, T. F. & Lee, H. C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 270, 30327–30333 (1995).
Howard, M. et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262, 1056–1059 (1993).
Lee, H. C. et al. Production and hydrolysis of cyclic ADP-ribose at the outer surface of human erythrocytes. Biochem Biophys. Res Commun. 191, 639–645 (1993).
Takasawa, S. et al. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 268, 26052–26054 (1993).
Shrimp, J. H. et al. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 136, 5656–5663 (2014).
Angeletti, C. et al. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 25, https://doi.org/10.1016/j.isci.2022.103812 (2022).
Balducci, E. et al. Assay methods for nicotinamide mononucleotide adenylyltransferase of wide applicability. Anal. Biochem. 228, 64–68 (1995).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Hsu, H. Y. & Wen, M. H. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J. Biol. Chem. 277, 22131–22139 (2002).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Panday, A., Sahoo, M. K., Osorio, D. & Batra, S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 12, 5–23 (2015).
Scaturro, P. & Pichlmair, A. Oxeiptosis: a discreet way to respond to radicals. Curr. Opin. Immunol. 56, 37–43 (2019).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Collins, Y. et al. Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806 (2012).
Son, Y. et al. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 792639 (2011).
Torres, M. & Forman, H. J. Redox signaling and the MAP kinase pathways. Biofactors 17, 287–296 (2003).
Laroux, F. S., Romero, X., Wetzler, L., Engel, P. & Terhorst, C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J. Immunol. 175, 5596–5600 (2005).
Nguyen, G. T., Green, E. R. & Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect. Microbiol. 7, 373 (2017).
Xie, N. et al. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target Ther. 5, 227 (2020).
Park, H. S. et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 173, 3589–3593 (2004).
Lapouge, K. et al. Structure of the TPR domain of p67phox in complex with Rac.GTP. Mol. Cell 6, 899–907 (2000).
Michaelson, D. et al. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152, 111–126 (2001).
Cipriano, A. et al. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J. Med Chem. 66, 11632–11655 (2023).
Chen, L. F., Mu, Y. & Greene, W. C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 21, 6539–6548 (2002).
Wan, F. & Lenardo, M. J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 1, a000067 (2009).
Morgan, M. J. & Liu, Z. G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21, 103–115 (2011).
Canton, M. et al. Reactive Oxygen Species in Macrophages: Sources and Targets. Front Immunol. 12, 734229 (2021).
Fang, F. C. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832 (2004).
Lim, S. G., Kim, J. K., Suk, K. & Lee, W. H. Crosstalk between signals initiated from TLR4 and cell surface BAFF results in synergistic induction of proinflammatory mediators in THP-1 cells. Sci. Rep. 7, 45826 (2017).
Vermeulen, L., Vanden Berghe, W., Beck, I. M., De Bosscher, K. & Haegeman, G. The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci. 34, 311–318 (2009).
Lo, H.-R. et al. RTX Toxin Enhances the Survival of Vibrio vulnificus During Infection by Protecting the Organism From Phagocytosis. J. Infect. Dis. 203, 1866–1874 (2011).
Singhal, A. & Cheng, C. Y. Host NAD+ metabolism and infections: therapeutic implications. Int Immunol. 31, 59–67 (2019).
Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 (2015).
Canto, C., Menzies, K. J. & Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 22, 31–53 (2015).
Adriouch, S., Haag, F., Boyer, O., Seman, M. & Koch-Nolte, F. Extracellular NAD(+): a danger signal hindering regulatory T cells. Microbes Infect. 14, 1284–1292 (2012).
Al-Shabany, A. J., Moody, A. J., Foey, A. D. & Billington, R. A. Intracellular NAD+ levels are associated with LPS-induced TNF-alpha release in pro-inflammatory macrophages. Biosci. Rep. 36, e00301 (2016).
Roussin, M. & Salcedo, S. P. NAD+-targeting by bacteria: an emerging weapon in pathogenesis. FEMS Microbiol. Rev. 45, https://doi.org/10.1093/femsre/fuab037 (2021).
Simon, N. C., Aktories, K. & Barbieri, J. T. Novel bacterial ADP-ribosylating toxins: structure and function. Nat. Rev. Microbiol 12, 599–611 (2014).
Ghosh, J., Anderson, P. J., Chandrasekaran, S. & Caparon, M. G. Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J. Biol. Chem. 285, 5683–5694 (2010).
Smith, C. L. et al. Structural basis of Streptococcus pyogenes immunity to its NAD+ glycohydrolase toxin. Structure 19, 192–202 (2011).
Sun, J. et al. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678 (2015).
Whitney, J. C. et al. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163, 607–619 (2015).
Skjerning, R. B., Senissar, M., Winther, K. S., Gerdes, K. & Brodersen, D. E. The RES domain toxins of RES-Xre toxin-antitoxin modules induce cell stasis by degrading NAD+. Mol. Microbiol 111, 221–236 (2019).
Stevens, D. L., Salmi, D. B., McIndoo, E. R. & Bryant, A. E. Molecular epidemiology of nga and NAD glycohydrolase/ADP-ribosyltransferase activity among Streptococcus pyogenes causing streptococcal toxic shock syndrome. J. Infect. Dis. 182, 1117–1128 (2000).
Meehl, M. A., Pinkner, J. S., Anderson, P. J., Hultgren, S. J. & Caparon, M. G. A novel endogenous inhibitor of the secreted streptococcal NAD-glycohydrolase. PLoS Pathog. 1, e35 (2005).
Danilchanka, O. et al. An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc. Natl Acad. Sci. USA 111, 6750–6755 (2014).
Timmer, A. M. et al. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J. Biol. Chem. 284, 862–871 (2009).
Bastiat-Sempe, B., Love, J. F., Lomayesva, N. & Wessels, M. R. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio. 5, e01690–01614 (2014).
Madden, J. C., Ruiz, N. & Caparon, M. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell 104, 143–152 (2001).
Amici, S. A. et al. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front Immunol. 9, 1593 (2018).
Partida-Sanchez, S. et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med 7, 1209–1216 (2001).
Cynamon, M. H., Sorg, T. B. & Patapow, A. Utilization and metabolism of NAD by Haemophilus parainfluenzae. J. Gen. Microbiol 134, 2789–2799 (1988).
Hogan, K. A., Chini, C. C. S. & Chini, E. N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front Immunol. 10, 1187 (2019).
Zeidler, J. D. et al. The CD38 glycohydrolase and the NAD sink: implications for pathological conditions. Am. J. Physiol. Cell Physiol. 322, C521–C545 (2022).
Joe, Y. et al. Cross-talk between CD38 and TTP Is Essential for Resolution of Inflammation during Microbial Sepsis. Cell Rep. 30, 1063–1076 e1065 (2020).
Lee, H. C. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev. Pharm. Toxicol. 41, 317–345 (2001).
Geissler, B., Tungekar, R. & Satchell, K. J. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proc. Natl Acad. Sci. USA 107, 5581–5586 (2010).
Ralto, K. M., Rhee, E. P. & Parikh, S. M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 16, 99–111 (2020).
Lerstloompleephunt, N., Tantawichien, T. & Sitprija, V. Renal failure in vibrio vulnificus infection. Ren. Fail 22, 337–343 (2000).
Qasem, A. & Rabbani, S. A. Acute Kidney Injury Associated With Cholera. Cureus 15, e34101 (2023).
Reyes-Corcho, A. et al. Cholera gravis associated with acute renal failure in a traveler from Haiti to the United States. Travel Med Infect. Dis. 10, 236–239 (2012).
Malaiyan, J. et al. Novel gas producing Vibrio cholerae: a case report of gastroenteritis with acute kidney injury. Access Microbiol. 1, e000005 (2019).
Hwang, J. et al. Structural insights into the regulation of sialic acid catabolism by the Vibrio vulnificus transcriptional repressor NanR. Proc. Natl Acad. Sci. USA 110, E2829–E2837 (2013).
Strom, M. S. & Paranjpye, R. N. Epidemiology and pathogenesis of Vibrio vulnificus. Microbes Infect. 2, 177–188 (2000).
Sheffield, P., Garrard, S. & Derewenda, Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39 (1999).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D. Biol. Crystallogr 67, 355–367 (2011).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40, W294–W297 (2012).
Kim, D. N. et al. Cryo_fit: Democratization of flexible fitting for cryo-EM. J. Struct. Biol. 208, 1–6 (2019).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Gavin, H. E. & Satchell, K. J. F. Surface hypothermia predicts murine mortality in the intragastric Vibrio vulnificus infection model. BMC Microbiol. 17, 136 (2017).
Acknowledgements
We thank the beamline staff at Pohang Accelerator Laboratory (BL-5C and 11 C) and the cryo-EM facility staff at the Institute of Membrane Proteins (Pohang) and KRIBB, Korea, for their kind help with data collection. Computational analysis of cryo-EM data was performed on the High Performance Computing Resources in the IBS Research Solution Center, Daejeon, Korea. This work was supported by the National Research Foundation of Korea (NRF-2021M3A9I4022934 to M.H.K.) funded by MSIT and by the KRIBB Research Initiative Program (KGM1382413 and KGM9942421 to M.H.K).
Author information
Authors and Affiliations
Contributions
M.H.K. conceived and supervised the work; S.C. designed and carried out crystallization and cryo-EM sample preparation, cell biological, immunological, and animal experiments, with the help of Y.L., S.P., S.-M.K., T.-H.K., and J.H.; Y.L. designed and performed biochemical and structural biological experiments, with the help of S.C., S.Y.J., and D.W.O.; J.P. analyzed amino acid sequences and domains of the MARTX toxins; G.S.L. and J.H.M. performed the LC/MS experiments for interactome analysis; G.S.L., D.L., E.-H.K., H.-K.C., and J.H.M. analyzed by-products of NAD(P)+ using LC/MS and NMR, with the help of S.C. and J.H.; S.C., Y.L., C.C., B.S.K., J.-J.S., J.H., and M.H.K. analyzed and interpreted the data; S.C., Y.L., and J.H. drafted the manuscript, and M.H.K. edited the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
: Nature Communications thanks Takashige Kashimoto and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, S., Lee, Y., Park, S. et al. Dissemination of pathogenic bacteria is reinforced by a MARTX toxin effector duet. Nat Commun 15, 6218 (2024). https://doi.org/10.1038/s41467-024-50650-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50650-0
- Springer Nature Limited