

Abstract
Genome-wide association studies implicate multiple loci in risk for systemic lupus erythematosus (SLE), but few contain exonic variants, rendering systematic identification of non-coding variants essential to decoding SLE genetics. We utilized SNP-seq and bioinformatic enrichment to interrogate 2180 single-nucleotide polymorphisms (SNPs) from 87 SLE risk loci for potential binding of transcription factors and related proteins from B cells. 52 SNPs that passed initial screening were tested by electrophoretic mobility shift and luciferase reporter assays. To validate the approach, we studied rs2297550 in detail, finding that the risk allele enhanced binding to the transcription factor Ikaros (encoded by IKZF1), thereby modulating expression of IKBKE. Correspondingly, primary cells from genotyped healthy donors bearing the risk allele expressed higher levels of the interferon / NF-κB regulator IKKε. Together, these findings define a set of likely functional non-coding lupus risk variants and identify a regulatory pathway involving rs2297550, Ikaros, and IKKε implicated by human genetics in risk for SLE.
Similar content being viewed by others

Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease predominantly affecting women. Identical twin concordance rates range between 30–50%, and first-degree relatives exhibit an approximately 20-fold increased risk of SLE compared with the general population1,2,3. While monogenic SLE has proven extremely informative with respect to causative pathways, most SLE is polygenic, arising through a complex interplay of genetic and environmental factors4,5. Correspondingly, genome-wide association studies (GWAS) have defined dozens of loci that modulate disease risk6,7,8,9,10. Deciphering these different genetic contributors, each individually minor but collectively of major importance, will be central to defining SLE heterogeneity at a genetic level.
Unfortunately, translating GWAS data into pathogenic understanding has proven difficult. Few common disease-associated variants affect coding regions, indicating that the associated variants – predominantly single-nucleotide polymorphisms (SNPs)—likely exercise a regulatory role. Distinguishing functional non-coding SNPs from the many incidental SNPs that reside in close linkage disequilibrium (LD) remains a needle-in-the-haystack problem of great complexity. Strategies to address this problem in polygenic diseases include fine-mapping11, proteome-wide profiling of DNA-binding proteins over SNPs12, integration of expression quantitative trait loci (eQTL) with epigenetic and/or transcriptomic data13,14,15, massively parallel reporter assays16,17,18, and SNP-seq19,20.
Here, we pursue a combined experimental and bioinformatic approach to identify functional non-coding variants in SLE that act by regulating the binding of transcription factors and other DNA-binding proteins. We focus on B cells as a lineage implicated genetically as critical to SLE pathogenesis21. We used SNP-seq to screen for alleles that confer differential binding to B cell nuclear proteins, enriching targets further through epigenetic markers of open chromatin and predictors of regulatory function. Beginning with 2180 candidates, we identify 52 plausible regulatory SNPs, each interrogated experimentally to prioritize five high-likelihood functional variants. To test our approach, we examined rs2297550 in detail, finding that this variant modulates the binding of the transcription factor Ikaros to regulate the expression of the noncanonical IκB kinase IKKε. Thus, our strategy bridges the gap between GWAS and mechanism while defining a regulatory pathway implicated by population genetics in risk for SLE.
Results
Candidate functional non-coding variant identification via SNP-seq and bioinformatic screens
To identify functional non-coding variants associated with SLE, we began with GWAS hits with genome-wide significance (P value <5 × 10−8) from five studies7,9,10,22,23, finding 87 loci of interest, each defined by a tagging SNP (Supplementary Data 1). We identified all common variants in LD with these SNPs from 1000 Genomes Phase 3 (European population), filtering on minor allele frequency [MAF] >1%, LD R2 > 0.8, and location within 1 Mb of the tagging SNP (Fig. 1A). This strategy yielded 2180 candidate SNPs for downstream screening and validation (Supplementary Data 1).

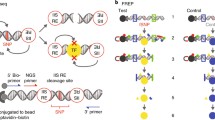
A Flow chart illustrating the identification of functional candidate SNPs from SLE GWAS data. B SNP-seq: To generate the SNP-seq construct (top), a 31 bp sequence centered on the SNP is positioned between two type IIS restriction enzyme (IIS RE) binding sites. SNPs that fail to bind regulatory proteins such as transcription factors (TF) are negatively selected (bottom), allowing enrichment of protected constructs by PCR. The whole construct can be amplified using primers as per Supplementary Data 9. Bio biotin. Figure 1B was created with BioRender.com released under a Creative Commons Attribution-NonCommerical-NoDerivs 4.0 International license. C The experimental procedure for SNP-seq; NE nuclear extract, Bio biotin. D Spearman’s correlation with a two-tailed test of SNP-seq allele counts normalized to control between PBMC and BL2 samples (ρ = 0.89, P < 1 \(\times\) 10−15). E We selected 248 SNPs that passed next-generation sequencing quality control (NGS-QC) and demonstrated progressive allele-specific protection (Suppl. Fig. 1C). Source data are provided as a Source Data file.
To narrow this pool, we applied SNP-seq19. In this method, a library of double-stranded DNA constructs is generated that includes each allele of each candidate SNP. A 31 bp sequence centered on each SNP is flanked by binding sites for the type IIS restriction enzyme BpmI that cleaves DNA in a sequence-independent manner 16 bp (5′)/14 bp (3′) adjacent to its binding site. The binding of transcription factors or other nuclear proteins to the SNP site protects the construct from enzymatic digestion (Fig. 1B), as recognized by subsequent amplification and DNA sequencing19,24. Here, our library contained 4363 dsDNA constructs reflecting each allele of the 2180 SNPs, including three with three alleles (Supplementary Data 2). The library was then incubated with nuclear extract from either human peripheral blood mononuclear cells (PBMCs) from healthy individuals or BL2 B cells, washed, and then digested with BpmI. As a control, the constructed pool was incubated without nuclear extract. The remaining sequences were amplified by PCR and used as the library for the next round of selection. The whole procedure was repeated for 5 cycles, with barcoding at cycles three and five to identify enriched SNPs by next-generation sequencing (NGS) (Fig. 1C). The entire procedure was performed in three independent biological replicates, finding high correlation (Spearman ρ = 0.89) between sample pairs (Supplementary Fig. 1A, B) and between PBMC and BL2 samples (Fig. 1D).
Two analytical approaches were employed to identify SNPs offering allele-specific protection, as established previously19. First, we chose SNPs that exhibited at least a 20% difference in protection between alleles in cycle 5 in samples treated with nuclear extract from both PBMC and BL2 cells; 452 SNPs exhibited such an allelic differential. Second, we sought SNPs demonstrating progressive allele-specific protection across cycles (cycles 0, 3, and 5) (Supplementary Fig. 1C); 496 SNPs exhibited this pattern. In the end, 248 SNPs met both criteria (Fig. 1E and Supplementary Data 3). A diagram of our analytical strategy is shown in Supplementary Fig. 2.
Next, we examined the same 2180 SNPs using two bioinformatic approaches. First, we employed informative chromatin marks to identify areas of open chromatin, calculating H3K4me3 chromatin scores from five chromatin immunoprecipitation (ChIP)-seq studies in human B cells in the Encyclopedia of DNA Elements (ENCODE) database, as per a described method25 (Supplementary Fig. 3). We selected for further study those SNPs that ranked in the top 100 from at least half of studies, yielding 91 variants (Supplementary Data 4). Second, we used combined annotation-dependent depletion (CADD), a composite measure that employs 60 genomic features to estimate the probability that a variant is functional26. We ranked all 2180 SNPs as per the CADD database (https://cadd.gs.washington.edu/), retaining the 25 SNPs that scored in the top 0.5% of all ~8.6 billion single-nucleotide variants (Supplementary Data 5).
To reduce the number of SNPs to a tractable number for experimental validation, we chose the top 20 SNPs identified uniquely by SNP-seq, the top eight SNPs identified uniquely by epigenetic and CADD analysis, and 18 SNPs identified in at least two of three analyses (six SNP-seq + H3K4me3, four SNP-seq + CADD, eight H3K4me3 + CADD), for a total of 52 plausible functional SNPs (Fig. 2 and Supplementary Table 1).

SNPs selected for experimental screening were the top 20 of 248 emerging form SNP-seq, the top eight emerging from H3K4me3 score, the top eight emerging from CADD, and 18 SNPs identified by at least two analyses: six SNP-seq+H3K4me3, four SNP-seq+CADD, and eight SNPs H3K4me3 + CADD.
Experimental validation of plausible functional SNPs in SLE
As experimental validation of the 52-SNP enriched variant pool, we first employed the electrophoretic mobility shift assay (EMSA). Biotinylated 31 bp DNA fragments centered on each allele of each the 52 candidate SNPs (111 alleles) were interrogated using nuclear extract from BL2 cells (triplicate biological repeats) followed by a validation EMSA using nuclear extract from human PBMCs. Overall, seven biallelic and two triallelic (rs2297550 and rs936394) SNPs exhibited allele-imbalanced binding for all nuclear extracts: rs2297550, rs906868, rs936394, rs7302634, rs276461, rs13213604, rs3768056, rs2814955, and rs9907966 (Fig. 3A, shift binding bands marked as red circles). All EMSA blots for every experiment and a summary table can be found in Supplementary Fig. 4 and Supplementary Data 6.

A Seven biallelic and two triallelic (rs2297550 and rs936394) SNPs showed consistent allele-imbalanced binding for nuclear by EMSA. Allele-specific gel shift/binding marked with red circles. Experiments were repeated three times using distinct nuclear extracts. Representative blots from PBMCs are shown here; BL2 data and negative SNPs are provided in Supplementary Fig. 4 and Supplementary Data 6. B Luciferase reporter assay between the reference (red) and alternative (blue) alleles of nine candidate regulatory SNPs from A in Daudi B cells (mean \(\pm\) s.d, n = 8 biological replicates, Mann–Whitney two-tailed U-test with two-stage step-up correction for multiple hypothesis testing: rs2297550 p = 0.00047, rs906868 p = 0.0013, rs936394 p = 0.00047, rs9907966 p = 0.0071, rs13213604 p = 0.0085). C Diagram displaying the position of 5 SNPs that showed consistent significant differences between alleles through EMSA and luciferase reporter assay; rs2297550 in promoter of IKBKE, rs906868 in intergenic region between YPEL5 and LBH, rs936394 in intron of WBP2, rs9907966 in intron of IKZF3, rs13213604 in intron of BLTP3A. The unit of chromosome position is in kilobases (Kb). A diagram of gene features including exon, intron, and UTR was generated using GSDS 2.0 (http://gsds.gao-lab.org/index.php). Source data are provided as a Source Data file.
Next, we assessed the allele-specific gene regulatory capacity of these 9 SNPs using a luciferase reporter assay; for rs2297550 and rs936394, we included only two alleles, since the third for each has a MAF <0.1% and is thus unlikely to mediate GWAS signal. We cloned the same risk or non-risk allele DNA fragments as for EMSA (but without biotinylation) into the pGL3 vector, and then transfected each construct into Daudi B cells, simpler to transfect than BL2, together with control vector pRL to normalize transfection efficiency. We found 5 SNPs to retain a significant difference between alleles and thus represent high-probability functional non-coding variants: rs2297550, rs906868, rs936394, rs9907966, and rs13213604 (Fig. 3B). These SNPs are diagrammed in Fig. 3C.
Identification of potential transcription factor binding
To provide independent validation of the identified SNPs, we sought to confirm the binding of transcription factors and/or other nuclear proteins in an allele-determined manner. Again, we approached this problem both experimentally and bioinformatically.
First, we employed pulldown and silver staining in SNPs that had exhibited allele-specific differential binding by EMSA, excluding rs2814955 because of its low luciferase activity, again with 31 bp oligonucleotides for each allele but now using nuclear extract from Daudi cells27. In two biological repeats, four high-probability functional variants again showed differential binding (red circles) (Supplementary Fig. 5). Each pulldown product for each allele of the five high-probability SNPs was evaluated by mass spectrometry to identify binding nuclear proteins, yielding a set of candidate proteins for each (Supplementary Data 7). Second, we employed a bioinformatic algorithm based on published ChIP-seq data to identify candidate transcription factors likely to recognize each of the 5 high-probability variants28,29. Bioinformatic predictions are shown in Supplementary Data 8. Evaluating these data together, we observed that the transcription factor Ikaros (IKZF1) was identified in the pulldown product for rs2297550 and was predicted by the algorithm, including preferential binding to the SLE-risk G allele as observed by EMSA. We, therefore, chose rs2297550 as a test case for our selection process.
rs2297550 regulates IKBKE/IKKε expression
As depicted in Fig. 3C, rs2297550 resides in a promoter/enhancer region, 47 bp upstream of IKBKE, encoding the kinase IKKε30,31,32. This SNP emerged from Chinese and European GWAS data as a leading candidate in SLE, with IKBKE as its likely target gene7,33. To assess the role of rs2297550 in the regulation of IKBKE, we used CRISPR homology-directed repair to generate base-edited Daudi cell clones with CC, CG, and GG genotypes. GG clones exhibited lower mRNA for IKBKE (Fig. 4A) and lower expression of IKKε protein (Fig. 4B), confirming a regulatory relationship between SNP and gene. Comparable findings were observed in LPS-stimulated Daudi cells (Supplementary Fig. 6).

A Using base-edited clones (4C/C, 3C/G, and 3G/G), mRNA levels of IKBKE and \(\beta\)-actin were measured by RT-qPCR, and B protein levels of IKK\(\epsilon\) (80 KDa) and GAPDH (37 KDa) were measured by Western blotting, performed once (mean \(\pm\) s.d, P values from one-way ANOVA corrected by Tukey’s multiple comparisons). Source data are provided as a Source Data file.
Next, we recruited healthy donors bearing different genotypes of rs2297550 to test the impact of genotype in primary PBMCs, using the Joint Biology Consortium (JBC) patient recruitment infrastructure and the Mass General Brigham Biobank (www.jbcwebportal.org). In resting cells, IKKε protein levels were comparable across genotypes in PBMCs collectively and in all immune subtypes tested (CD19+ B cells, CD3+ CD4+ T cells, CD3+CD8+ T cells, CD3-CD56+ NK cells, and CD14+ monocytes) (Fig. 5A–F). However, after 4 h of LPS stimulation, cells from GG donors exhibited differential protein levels in PBMCs generally as well as in B cells, CD8+ T cells, and NK cells; a heterozygote (allele dose) effect was observed in B cells and CD8+ T cells (Fig. 5G–L). Interestingly, unlike in Daudi cells, the effect of the risk allele was to increase rather than decrease IKKε expression.

IKK\(\epsilon\) expression was determined by flow cytometry for 35 healthy subjects with C/C (n = 12), C/G (n = 11), and G/G (n = 12) genotype at SNP rs2297550. The mean fluorescence intensity (MFI) of IKK\(\epsilon\) expression in (A–F) resting and (G–L) 4 h LPS-stimulated PBMCs, including CD19+ B cells, CD3+CD4+ T cells, CD3+CD8+ T cells, CD3-CD56+ NK cells, and CD14+ monocytes (mean \(\pm\) s.d, P values from one-way ANOVA corrected by Dunnett’s multiple comparisons test). Source data are provided as a Source Data file.
Ikaros binds to rs2297550 to regulate IKBKE
Given concordant findings from mass spectrometry and bioinformatic prediction, we pursued the possibility that Ikaros binds to rs2297550 to regulate IKBKE. We, therefore, performed an oligonucleotide pulldown assay using nuclear extract from Daudi cells, with or without an excess non-biotinylated competitor, followed by a western blot using anti-Ikaros antibody. Ikaros bound both C and G alleles but exhibited a preference for the G allele, as predicted, reflected in the disappearance of signal with excess non-biotinylated G competitor (Fig. 6A). To confirm this result, we employed EMSA-supershift, finding that anti-Ikaros weakened binding to rs2297550-G compared with isotype control or antibody (to a lesser degree) to another transcription factor candidate from the bioinformatic prediction, E2F transcription factor 4 (E2F4) (Fig. 6B). These studies establish that Ikaros binds rs2297550 and that this interaction is allele-specific.

A Oligonucleotide pulldown western blot assay testing Ikaros binding to rs2297550-G; binding is eliminated in the presence of a 30-fold excess of non-biotinylated rs2297550-C or G competitor; positive control, nuclear extract. Pulldown western blot was performed once. B EMSA supershift assay showed that anti-Ikaros caused fading of the shifted band (oligo rs2297550-G, Daudi nuclear extract), whereas isotype control did not; anti-E2F4 also exhibited specificity. EMSA supershift was repeated twice. All blots are provided in the Source Data. C ChIP-qPCR using anti-Ikaros confirms binding to rs2297550 (Daudi cells, mean \(\pm\) s.d, three biological replicates). D Allele discrimination ChIP-qPCR shows Ikaros preferentially binds to G allele over C allele of rs2297550 in CRISPR-HDR edited C/G Daudi cells (mean \(\pm\) s.d, three biological replicates). All studies were done in unstimulated cells. Statistical analysis for C and D: paired t-test with two tails, without correction. Source data are provided as a Source Data file.
To assess rs2297550-Ikaros binding in living cells, we performed chromatin immunoprecipitation (ChIP)-qPCR. In Daudi cells, the anti-Ikaros antibody precipitated rs2297550 significantly more than did the isotype control antibody (Fig. 6C). Further, in CRISPR-HDR edited CG Daudi cells, we observed preferential pulldown of the G allele over the C allele (Fig. 6D), again confirming its allelic preference.
To test whether the interaction of Ikaros with rs2297550 modulated IKKε expression, we generated three Ikaros knockout clones using CRISPR-Cas9. All clones showed Ikaros deficiency (Fig. 7A) and corresponding loss of the ability of anti-Ikaros to pull down rs2297550 (Fig. 7B). Consistent with the inhibitory effect of G allele carriage in these cells, loss of Ikaros increased IKBKE mRNA (Fig. 7C) and IKKε protein (Fig. 7D). These findings establish a rs2297550-Ikaros-IKKε regulatory axis implicated by human population genetics in risk for SLE.

CRISPR-Cas9 Ikaros knockout Daudi clones show A decreased Ikaros protein expression by western blotting, B loss of Ikaros binding to rs2297550 by ChIP-qPCR (mean \(\pm\) s.d, three biological replicates), and C, D increased IKBKE mRNA by RT-qPCR (mean \(\pm\) s.d, three biological replicates) and increased IKK\(\epsilon\) protein expression by western blotting. Statistical analysis panels B and C: one-way ANOVA corrected by Dunnett’s multiple comparisons test. Western blotting (A, D) was performed once. Source data are provided as a Source Data file.
Discussion
Genetic risk for SLE is borne by many susceptibility loci, most of which reflect common non-coding variants34. Despite the modest effect of particular variants on disease risk, each unambiguously implicates its associated pathway in the pathogenesis of human disease. Further, as the number of risk variants accumulates, the incidence of SLE increases and the age of onset declines, suggesting a cumulative hit model whereby variants operate synergistically to generate disease susceptibility9. Understanding which specific variants drive risk and how they do so offers a unique window into disease biology as well as into general mechanisms of immune control35,36.
Here, we conducted a systematic experimental and bioinformatic screen across 87 loci implicated by GWAS in SLE, identifying 52 plausible casual non-coding SNPs, of which five emerged through EMSA and luciferase as high-likelihood variants. Extrapolation from surrounding genes suggests that these 5 variants likely interface with functional pathways relevant to SLE biology, including the interferon pathway (IKBKE), pyroptosis (IKBKE), B cell differentiation (IKZF3), and WNT signaling (LBH) (Fig. 3C).
To test our approach, we studied one variant in detail: rs2297550, a triallelic SNP (frequency of the three alleles C 0.862, G 0.138, T < 0.001 per gnomAD) that resides 4462 bp upstream of the start codon for IKBKE, encoding the kinase IKKε. The minor allele G confers an increased risk of SLE with an odds ratio of 1.16 (95% CI: 1.11–1.21, p = 1.31 × 10−11) compared with the major C allele7. Base editing showed that allelic variation at rs2297550 changes the expression of IKBKE/IKKε. Variation in IKKε with genotype at rs2297550 was confirmed in healthy volunteer donors. As an orthogonal line of evidence, we showed that Ikaros exhibited an allele-dependent affinity for rs2297550 and that deletion of Ikaros altered IKBKE/IKKε expression. These results establish a previously undefined rs2297550-Ikaros-IKKε regulatory axis.
Some groups have found that the risk allele G at rs2297550 is associated with lower IKBKE mRNA in unstimulated PBMCs32,37. IKKε protein was not tested, raising the possibility that transcript and protein levels could be discordant. Indeed, we observed a trend toward lower levels of IKBKE mRNA in GG PBMC cells, though this effect did not reach significance (Supplementary Fig. 7). The basis for transcript-protein divergence is not clear but could relate to the relative half-lives of mRNA and protein, among other factors38,39.
Interestingly, the relationship between genotype and IKKε varied with context. In Daudi B lymphoblasts, a line derived from a patient with Burkitt lymphoma, the introduction of the risk GG genotype decreased IKKε expression and deletion of Ikaros enhanced it, establishing Ikaros as an inhibitor of IKBKE. This effect persisted after stimulation with LPS. By contrast, in healthy donors, circulating B cells showed an increase in IKKε with risk allele carriage, including a gene dose effect, but only after LPS stimulation. Similar regulation was observed in CD8 + T cells, NK cells, and monocytes. The dependence of the impact of genetic variants on cell type and developmental/activation state is well recognized40. Since Ikaros can toggle between activator and repressor depending on associated cofactors, it may be that the variation observed here reflects differential expression of these proteins, or on other genetic/epigenetic differences between the lineages studied41. Under conditions other than those tested here, it is possible that the risk allele at rs2297550 could suppress IKKε in primary cells, as it does in Daudi cells.
An important intracellular intermediate in innate immune responses, IKKε can activate IRF3, IRF7, and NF-κB pathways to drive gene transcription in response to extracellular and intracellular stimuli, resulting in the production of inflammatory mediators, including type I interferons (Fig. 8)42,43,44,45. The known role of interferons in SLE makes this pathway an appealing potential explanation for why rs2297550 is a risk SNP for SLE46. IKKε can also protect cells from TNF-induced apoptosis, plausibly thereby attenuating autoantigen production47,48. IKBKE mRNA expression has been reported to be either higher49,50 or lower32,51,52 in whole blood and/or PBMC of SLE patients compared with healthy donors. Our primary cell data suggest that the risk phenotype attributable to rs2297550 likely reflects enhanced rather than suppressed expression of IKKε, though further work will be required to define the mechanistic connection between IKKε and SLE.

The lupus risk SNP rs2297550 allelically modulates the binding of the transcription factor Ikaros (encoded by IKZF1) to regulate the expression of IKBKE, encoding IKKε, a protein that modulates multiple processes implicated in SLE pathogenesis. Note that Ikaros binding can either amplify or repress IKBKE depending on context; amplification, shown here, reflects the direction of effect observed in genotyped healthy donors. The role of mechanisms downstream of IKKε remains to be defined. Figure 8 was created with BioRender.com released under a Creative Commons Attribution-NonCommerical-NoDerivs 4.0 International license.
IKZF1 encodes Ikaros, an essential regulator of lymphocyte development41,53. Ikaros is implicated in SLE by multiple lines of evidence. A region including IKZF1 carries genetic risk for SLE33,54,55,56,57,58. Individuals bearing mutant IKZF1 can present with SLE-like autoimmune disease59,60. Clinical efficacy has been established in human SLE for iberdomide, an agent that accelerates degradation of Ikaros as well of the related transcription factor Aiolos61,62. Mechanistically, Ikaros is involved in B cell tolerance and plasmacytoid dendritic cell differentiation41,63,64,65. In B cells from SLE patients, iberdomide attenuated in vitro differentiation to plasmablasts and plasma cells, as well as antibody production66. Thus, our findings implicating Ikaros in SLE risk via rs2297550 are consistent with genetic, clinical, and mechanistic observations in SLE.
Our study establishes a connection between Ikaros, IKKε, and SLE, showing that the regulation of this intracellular signaling molecule is part of the portfolio of this transcription factor, and implicating both proteins in risk of incident SLE in humans. This finding has potential therapeutic relevance, since drug candidates supported by human genetic evidence are much more likely to succeed in clinical trials than those without such support67,68. However, our data illustrate the challenge of extrapolating from a genetic variant to a single mechanism. Depending on the cellular context, we find that the same variant has opposite effects on gene expression. Further, while we began our search in B cells, in human primary cells the rs2297550-IKKε effect spans multiple immune lineages, both adaptive and innate. Thus our data do not establish which lineages, and which pathways downstream of IKKε, contribute to the mechanisms through which rs2297550 drives SLE risk.
Other limitations arise from our methodology. Non-coding functional variants may operate in ways not detectable by SNP-seq or by our bioinformatic screens, for example, through chromosome conformation, microRNA, and mRNA splicing. We would not have detected variants that operate via DNA-binding proteins absent from the nuclear extract employed, for example, transcription factors specific for other B cell developmental/activation states or in different immune or non-immune lineages. For this reason, while the high-probability SNPs described here are likely functional, we expect that many genuinely functional non-coding SNPs will have been filtered out. Definitive validation of the remaining four high-probability SNPs will require a dedicated study of the kind devoted here to rs2297550. While our findings suggest the possibility of epistasis between IKBKE and IKZF1, inadequate resolution of the IKZF1 locus in the Immunochip rendered an attempt to identify such an interaction impossible, although we could confirm through colocalization analysis that B cells are likely to be a relevant lineage for SLE risk at IKZF1 (Supplementary Fig. 8)9. Thus, the existence of an epistatic interaction between these loci remains to be tested.
Our studies in genotyped donors employed healthy donors rather than SLE patients. This choice was deliberate. We derived our initial candidate SNPs from GWAS that compared people with SLE to people lacking SLE. This design identifies variants modulating the risk that a healthy person will develop disease, and therefore that must be operative before SLE appears—that is, in healthy individuals. Such variants may also modulate disease phenotype, severity, or response to therapy in patients with established SLE, but such effects remain conceptually distinct from how they predispose to incident disease, as studied here.
Our combined experimental and bioinformatic approach also has clear strengths. The number of SNPs that survived each individual filter was substantial, illustrating the difficulty of separating functional from incidental SNPs. Multiple orthogonal approaches are required to validate any individual SNP and remain highly labor-intensive. The value of such an effort arises from the fact that each functional variant points the way to a pathway relevant to human SLE. It is key to remember that the effect size of a non-coding variant reflects the impact of the polymorphism on disease risk, but not the importance of the gene itself in disease pathogenesis, since regulatory SNPs often alter gene expression only modestly, exactly as observed here. Further, non-coding variants can be expected to identify pathways that coding variants cannot, either because mutations that disrupt a gene are poorly tolerated and thus eliminated from the population or because they lead to a phenotype that is different from that induced by a more subtle alteration in gene expression. As a result, any effort to understand highly polygenic disease must seek to understand not only highly penetrant coding variants but also each non-coding variant.
In summary, we provide a combined experimental and bioinformatic approach to identify plausible regulatory non-coding variants in SLE, proceeding from our initial screen to establish a previously unrecognized pathway through which Ikaros regulates IKKε, as modified by common allelic variation at rs2297550. Further investigation is required to understand how IKKε participates in SLE, including which lineages and downstream pathways are essential to this role. Our approach is broadly applicable and will help to translate GWAS into the pathogenic understanding that is a prerequisite to the personalized treatment of polygenic diseases such as SLE.
Methods
Cells and culture
Human B cell line BL2 was from DSMZ (catalog number ACC 625) and human B cell line Daudi was kindly provided by Dr. Baglaenko Yuriy. BL2 and Daudi cells were cultured in RPMI1640 medium (Thermo Fisher Scientific, catalog number 11875093) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, catalog number 26140079) and 1% Penicillin-Streptomycin (Thermo Fisher Scientific, catalog number 15140122).
SNP-seq oligos and primers
All primers were purchased from IDT as listed in Supplementary Data 9. SNP-seq oligo pool (Supplementary Data 2) was purchased from TwistBiosciences and oligos for EMSA screening and luciferase reporter assay was purchased from EtonBiosciences.
SNP-seq
SNP-seq constructs were built according to Fig. 1B. The SNP sequence is 31 bp long DNA centered on the SNP of interest. The sequences of PCR amplification primers for the library oligonucleotides are listed in Supplementary Data 9 and the library itself is detailed in Supplementary Data 2. For oligo library amplification, 100 ng of pooled DNA was amplified with bioMagF-G5 and MagR-G3 by PCR for 25 cycles with AccuPrime Taq (Thermo Fisher Scientific, catalog number 12346086) at 94 °C for 60 s, 58 °C for 60 s, and 68 °C for 40 s. After gel purification, 10 ng of biotinylated DNA was attached to 4 μl streptavidin-Dynabeads (Invitrogen, catalog number 11205D) according to the manufacturer’s protocol. The DNA-bound beads were then incubated with 60 μg nuclear extract from BL2 cell line or PBMC or no nuclear extract for 1 h at room temperature in LightShift Chemiluminescent EMSA Kit reaction buffer (Thermo Fisher Scientific, catalog number 20148X). After washing and separation, the DNA-bound beads were digested with 2 μl BpmI (NEB, catalog number R0565L) for 30 min at 37 °C. After another wash and separation, the DNA was amplified again with bioMagF and MagR and reattached to the Dynabeads for the next SNP-seq cycle. Five cycles were performed in total. DNA from cycles 0, 3, and 5 were used to prepare a Next-Generation Sequencing (NGS) library by two consecutive PCRs (PCR with L1seq and R1 primers, 20 cycles of 98 °C 1:30 min, 60 °C 1:40 min, 72 °C 0:40 min; PCR with L2 and R2 primers, 25 cycles of 98 °C 1:30 min, 60 °C 1:40 min, 72 °C 0:40 min) for each independent sample using Herculase polymerase (Agilent, catalog number 600677). The primers used are also listed in Supplementary Data 9. PCR products were run in a 2% agarose gel and the correct band (around 200 bp) was purified with QiaQuick gel extraction Kit (QIAGEN, catalog number 28706) (at least two columns) following manufacturer´s instructions. The elution was sent for NGS at the Dana Farber Genomics Core, Harvard Medical School.
Human PBMC isolation
Human peripheral blood mononuclear cells (PBMCs) used in SNP-seq were extracted from apheresis leukoreduction collar blood via Ficoll gradient (Cytiva, catalog number GE17-1440-02). For studies in genotyped donors, whole blood was collected from genotyped healthy human subjects recruited from the Mass General Brigham Biobank through the Joint Biology Consortium (JBC) recruitment core (www.jbcwebportal.org). The genotype was confirmed in each donor by Sanger sequencing. PBMCs were isolated from whole blood collected in BD Vacutainer™ Plastic Blood Collection Tubes with K2EDTA (BD, catalog number 366643) via Ficoll gradient and stored in liquid nitrogen in 10% DMSO in complete FBS.
Human subjects research was approved under Brigham and Women’s Hospital IRB protocol 2019P003709 and the Boston Children’s Hospital IRB protocol P000043108. All participants provided informed consent.
Chromatin marks and CADD
Chromatin mark score calculation is based on published protocol25. In short, for a variant, the score is the ratio of the H3K4me3 epigenetic peak value from CHIP-seq data to the distance between the variant and its closest H3K4me3 peak. Five H3K4me3 CHIP-seq experiments for B cells from ENCODE are used for chromatin mark score calculation. The experimental ID and final sheet for all the scores are provided in Supplementary Data 4. Combined annotation-dependent depletion (CADD) scores (C-scores) for all SNPs investigated in this study were extracted from the available database69. The sheet for all the C-scores is in Supplementary Data 5.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific, catalog number 20148) according to manufacturer instructions. For the oligo probe, a 31 bp fragment centered upon the SNP was made by annealing two biotinylated oligonucleotides. Nuclear proteins were extracted from BL2 cells and PBMC using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, catalog number 78835) per manufacturer instructions. For gel supershift, 1 μg of Isotype antibody or 10 μl of specific antibodies were added to the 20 μl reaction after for additional 30 min incubation. Antibodies used are anti-Ikaros antibody (Cell Signaling, clone D10E5, catalog number 9034S, validated by the manufacturer in Jurkat, Ramos, and Raji cells), anti-E2F4 antibody (Cell signaling, clone E3G2G, catalog number 40291S, validated by the manufacturer in Jurkat, SEM, HCT15, HT1080, and COS-7 cells) and IgG isotype control antibody (Thermo Scientific, catalog number 02-6102). The oligonucleotide sequences used for the EMSA are listed in Supplementary Data 9.
Luciferase reporter assay
The luciferase reporter assay was performed exactly according to the manufacturer’s manual (Promega, catalog number E1751). The pGL3 expression vector (0.5 μg) with or without SNPs sequence was co-transfected with the control vector pRL by Nucleofection (Lonza 4D-nucleofector) into 5 × 105 Daudi cells. After 48 h incubation, luciferase activity was measured with the Dual-Glo luciferase assay system (Promega, catalog number E2920).
Pulldown assay and mass spectrometry
We conducted pulldown assays for eight SNPs (rs2297550, rs13213604, rs276461, rs9907966, rs7302634, rs906868, rs936394, and rs3768056) using oligonucleotides for each allele with nuclear extract form Daudi cells in two biological replicates. One pulldown product for each allele of five SNPs (rs2297550, rs13213604, rs276461, rs9907966, and rs7302634) was evaluated by mass spectrometry to identify binding nuclear proteins. Pulldown assay is modified from a published protocol for DNA Affinity Purification Assay (DAPA)27. Similar to SNP-seq, 500 ng of different alleles (31 bp, biotinylated) from SNPs were attached to 25 μl streptavidin-Dynabeads (Invitrogen) and then incubated with 100 μg of Daudi nuclear extract for 1 h with or without non-biotinylated competitor (the same alleles). The incubation mixture was then washed in PBST five times before eluting with elution buffer (25 mM Tris-Cl PH: 7.5, 1.5 M NaCl). The elutions were then precipitated using a precipitation kit (EMD Millipore, cat# 539180) and sent for mass spectrometry core at Harvard Medical School.
Samples were incubated with 200 ng/μl of modified sequencing-grade trypsin (Promega, catalog number V5117) in a 37 °C room overnight. The digested peptides were then resuspended in 50 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid) and desalted by STAGE tip. A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 µm C18 spherical silica beads into a fused silica capillary (100 µm inner diameter × ~30 cm length) with a flame-drawn tip. After equilibrating the column each sample was loaded via a Famos autosampler (LC Packings, San Francisco, CA) onto the column. A gradient was formed, and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid). As peptides eluted, they were subjected to electrospray ionization and then introduced into a Velos Orbitrap Elite ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences were identified by searching the acquired tandem mass spectra against human protein databases, including UniProt and RefSeq, using the database search program SEQUEST (Thermo Fisher Scientific, Waltham, MA). All databases include a reversed version of all the sequences, and the data were filtered to achieve a false discovery rate between one and two percent. The oligonucleotide sequences used for the pulldown are listed in Supplementary Data 9. All proteins identified are listed in Supplementary Data 7.
Western blots
Western blots for pulldown assay and knockout validation were performed using corresponding antibodies. All antibodies for western blot were diluted at a ratio of 1:1000. Anti-Ikaros antibody (Cell Signaling, clone D10E5, catalog number 9034S, validated by the manufacturer in Jurkat, Ramos, and Raji cells), anti-GAPDH antibody (Cell Signaling, clone 14C10, catalog number 2118S, validated by the manufacturer in NIH/3T3, C6, HUVEC, and L929 cells), anti-IKKε antibody (Cell Signaling, clone D20G4, catalog number 2905 S, validated by the manufacturer in Ramos, and RL cells), anti-vinculin antibody (Bio-Rad, clone V284, catalog number MCA465GA, validated by the manufacturer in Hela cells), goat anti-Rabbit IgG (H + L) cross-adsorbed secondary antibody HRP (Thermo Scientific, catalog number G-21234), and goat anti-mouse IgG-HRP (Santa Cruz, catalog number sc-525408).
Transcriptional factors binding prediction
Candidate TFs binding was analyzed using a published model to identify sequence-specific binding proteins28,29.
Flow cytometry
For IKKε intracellular staining in different subtype cells of PBMCs, cells were treated with or without LPS for four hours and then blocked with Fc Receptor Binding Inhibitor Polyclonal Antibody (eBioscience, catalog number 14-9161-71) and stained with live/dead-e506 stain (eBioscience, 65-0866-14), followed by staining with CD19-APC-e780 (eBioscience, clone HIB19, catalog number 47-0199-41, validated by the manufacturer in human PBMC); CD3-BV421 (Biolegend, clone UCHT1, catalog number 300433, validated by the manufacturer in human PBMC); CD4-PE-eFluor 610 (eBioscience, clone RPA-T4, catalog number 61-0049-41, validated by the manufacturer in human PBMC); CD8-BV786 (eBioscience, clone RPA-T8, catalog number 417-0088-42, validated by the manufacturer in human PBMC); CD56-PE-Cy7 (eBioscience, clone NCAM, catalog number 25-0567-41, validated by the manufacturer in human PBMC); CD14-APC (eBioscience, clone 61D3, catalog number 17-0149-41); anti-IKKε-PE for intracellular staining. IKKε antibody (Cell Signaling, clone D20G4, catalog number 2905S) was labeled with PE (Biotium, catalog number 92299). IKKε-PE for intracellular staining was validated in this study by comparing it to the background signal from an isotype control in human PBMCs. For intracellular staining, cells were fixed with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, catalog number 00-5523-00) after surface marker staining. Cells were analyzed on a BD LSRFortessa™ Cell Analyzer (BD Biosciences). All the above antibodies were used at 1:200 dilution.
CRISPR/Cas9 knockout
IKZF1 in Daudi cells was knocked out using CRISPR-Cas9 through nucleofection (4D-nucleofector from Lonza). About 1 µl of 40 µM sgRNAs (two sgRNAs to generate a deletion) for IKZF1 were mixed with 1 µl of 20 µM spCas9 and incubated at 37 degrees for 15 min to form ribonucleoprotein (RNP). The formed RNP was kept on ice before use. Daudi cells were then washed with PBS twice and resuspended in SF solution (Lonza, catalog number V4XC-2032), 300k cells per 20 µl solution. Next, RNP was added into each 20 µl solution with Daudi cells, and the whole mixture was transferred into nucleofection strip wells for nucleofection with program code CA137. After nucleofection, cells were transferred into a pre-warmed culture medium and incubated for 72 h before analyzing knockout efficiency. sgRNAs used are: 5′-AGGTGGTGGAGAGGTCCTCG-3′ and 5′-TGAGCCCATGCCGATCCCCG-3′.
CRISPR/Cas9 homology-directed repair (HDR)
CRISPR/Cas9 mediated HDR was applied for the generation of base-edited cell lines using sgRNA targeting DNA sequences near rs2297550 and asymmetrical single-stranded DNA donors as previously described70. 3 × 105 Daudi cells were nucleofected with 20 pmol of sgRNA-Cas9 complex and 100 pmol of DNA donor template using program CA137 of Amaxa 4D-Nucleofector and SF cell line kit S (Lonza, V4XC-2032). The edited single-cell clones were sorted into 96-well plates by BD Aria II sorter and expanded for 4 weeks. Genotype of individual clones were analyzed by Sanger sequencing to identify C/G wild-type clones (n = 3), C/C edited clones (n = 4), G/G edited clones (n = 3). sgRNA and single-stranded donor DNA for generating CC genotype are 5′-GAGAAAGAGAGAGCGAGAGACGG-3′ and 5′- TTGTGGCGGTGGGACACCAAGCTCAGCACTGAGTCCTGGCTCTCAGCTCTCTCGCACTTCCTGATTTCCGTCTCTCGCTCTCTCTTTCTCTCTTAAAGAGACAGAGGCA-3′; sgRNA and single-stranded donor DNA for generating GG genotype are 5′- TCTTTAAGAGAGAAAGAGAGAGG-3′ and 5′- TTGTGGCGGTGGGACACCAAGCTCAGCACTGAGTCCTGGCTCTCAGCTCTCTCGCACTTCCTGATTTCCGTCTCTCCCTCTCTCTTTCTCTCTTAAAGAGACAGAGGCA-3′.
RT-qPCR
Total RNA was isolated with Absolutely RNA Miniprep Kit (Agilent, catalog number 400800). cDNA was synthesized with AffinityScript QPCR cDNA Synthesis Kit (Agilent, catalog number 600559). All procedures were performed following the manufacturer’s protocols. RT-qPCR was done with an Agilent AriaMX qPCR machine according to the protocol for Brilliant II SYBR QPCR Low ROX Mstr Mx (Agilent, catalog number 600830). IKZF1 qPCR primers: forward, 5′- CGTGCTGGACAGACTAGCAA -3′, reverse, 5′- ATTTCGTTCTCCTTCTCGTAGC-3′. IKBKE qPCR primers: forward, 5′-CTTGTGAATCTGCTCCTCGTT-3’, reverse, 5’-GGTACATGAGGACAGAAGCATC-3′.
Chromatin immunoprecipitation (ChIP)-qPCR
ChIP was performed for Daudi cells using the CHIP-it kit (Active motif, catalog number 53042) according to the manufacturer’s protocols. Antibodies used are isotype control antibody and anti-Ikaros antibody (Cell Signaling, clone D10E5, catalog number 9034S, validated by the manufacturer in Jurkat cells) from EMSA. 10 μl of antibody was incorporated into 200 μl ChIP reaction. The primers used for Daudi cell CHIP-qPCR to detect rs2297550 are forward, 5′-CTCAGAGCATCCTCTTTCTCTTT-3′, reverse, 5′- CTCTCTCGCACTTCCTGATTT-3′.
Allele discrimination CHIP-qPCR
CHIP was performed using HDR clones with rs2297550 C/G genotype. Antibodies used are isotype control antibody and anti-Ikaros antibody from EMSA. Probes and primers used are included in the Custom TaqMan™ SNP Genotyping Assay, human for rs2297550 from Thermo Fisher.
Data analyses
SAS 9.4 was used for the analysis of SNP-seq data. All statistical analysis was done using GraphPad software (Prism 9) using one-way analysis of variance (ANOVA) for comparison of multiple groups, or unpaired t-test (non-parametric Mann–Whitney test) for two groups, with P values as indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. The Human reference genome utilized in this study was sourced from the genome assembly GRCh37 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/). The mass spectrometry proteomics data have been deposited to the ProteomeXchnage Consortium via the PRIDE71 partner repository (https://www.ebi.ac.uk/pride/archive/projects/PXD048367) with the dataset identifier PXD048367 and https://doi.org/10.6019/PXD048367. Source data are provided with this paper.
References
Deapen, D. et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 35, 311–318 (1992).
Block, S. R. et al. Studies of twins with systemic lupus erythematosus. A review of the literature and presentation of 12 additional sets. Am. J. Med. 59, 533–552 (1975).
Alarcon-Segovia, D. et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 52, 1138–1147 (2005).
Vinuesa, C. G., Shen, N. & Ware, T. Genetics of SLE: mechanistic insights from monogenic disease and disease-associated variants. Nat. Rev. Nephrol. 19, 558–572 (2023).
Caielli, S., Wan, Z. & Pascual, V. Systemic lupus erythematosus pathogenesis: interferon and beyond. Annu Rev. Immunol. 41, 533–560 (2023).
Zhang, H. et al. Meta-analysis of GWAS on both Chinese and European populations identifies GPR173 as a novel X chromosome susceptibility gene for SLE. Arthritis Res. Ther. 20, 92 (2018).
Morris, D. L. et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 48, 940–946 (2016).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015).
Langefeld, C. D. et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 8, 16021 (2017).
Gateva, V. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233 (2009).
Westra, H. J. et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 50, 1366–1374 (2018).
Butter, F. et al. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 8, e1002982 (2012).
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6, e1000888 (2010).
Perez, R. K. et al. Single-cell RNA-seq reveals cell type–specific molecular and genetic associations to lupus. Science 376, eabf1970 (2022).
Ulirsch, J. C. et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 165, 1530–1545 (2016).
Tewhey, R. et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165, 1519–1529 (2016).
Lu, X. et al. Global discovery of lupus genetic risk variant allelic enhancer activity. Nat. Commun. 12, 1611 (2021).
Li, G. et al. High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat. Gene. 50, 1180–1188 (2018).
Wang, Q. et al. Identification of a regulatory pathway governing TRAF1 via an arthritis-associated non-coding variant. Cell Genom. 3, 100420 (2023).
Karrar, S. & Cunninghame Graham, D. S. Abnormal B cell development in systemic lupus erythematosus: what the genetics tell us. Arthritis Rheumatol. 70, 496–507 (2018).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015).
Armstrong, D. L. et al. GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 15, 347–354 (2014).
Hardenbol, P. & Van Dyke, M. W. Sequence specificity of triplex DNA formation: Analysis by a combinatorial approach, restriction endonuclease protection selection and amplification. Proc. Natl Acad. Sci. USA 93, 2811–2816 (1996).
Trynka, G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 45, 124–130 (2013).
Rentzsch, P. et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–d894 (2019).
Miller, D. E. et al. Screening for functional non-coding genetic variants using electrophoretic mobility shift assay (EMSA) and DNA-affinity precipitation assay (DAPA). J. Vis. Exp. 54093 (2016).
Weirauch, M. T. et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126–134 (2013).
Weirauch, M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Dvir, S. et al. Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc. Natl Acad. Sci. USA 110, E2792–E2801 (2013).
Mignone, F. et al. Untranslated regions of mRNAs. Genome Biol. 3, reviews0004.1 (2002).
Zhu, T. et al. The decreased expression of IKBKE in systemic lupus erythematosus. Clin. Rheumatol. 39, 2611–2617 (2020).
Wang, Y. F. et al. Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat. Commun. 12, 772 (2021).
Kwon, Y. C. et al. Update on the genetics of systemic lupus erythematosus: genome-wide association studies and beyond. Cells 8, 1180 (2019).
Wang, Q. et al. Identification of a regulatory pathway governing TRAF1 via an arthritis-associated non-coding variant. Cell Genom. 3, 100420 (2022).
Kim, T. et al. Non-coding autoimmune risk variant defines role for ICOS in T peripheral helper cell development. Nat Commun 15, 2150 (2024).
Chen, L., Morris, D. L. & Vyse, T. J. Genetic advances in systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 29, 423–433 (2017).
Pott, J. et al. Genetically regulated gene expression and proteins revealed discordant effects. PLoS ONE 17, e0268815 (2022).
Robinson, J. W. et al. Evaluating the potential benefits and pitfalls of combining protein and expression quantitative trait loci in evidencing drug targets. Preprint at bioRxiv 2022.03.15.484248 (2022).
Gutierrez-Arcelus, M. et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet. 52, 247–253 (2020).
Cytlak, U. et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat. Commun. 9, 1239 (2018).
Clément, J. F., Meloche, S. & Servant, M. J. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 18, 889–899 (2008).
Fitzgerald, K. A. et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Tsukamoto, H. et al. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1–IKKϵ–IRF3 axis activation. J. Biol. Chem. 293, 10186–10201 (2018).
Peters, R. T., Liao, S. M. & Maniatis, T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 5, 513–522 (2000).
Psarras, A., Wittmann, M. & Vital, E. M. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat. Rev. Rheumatol. 18, 575–590 (2022).
Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Renner, F., Moreno, R. & Schmitz, M. L. SUMOylation-dependent localization of IKKepsilon in PML nuclear bodies is essential for protection against DNA-damage-triggered cell death. Mol. Cell 37, 503–515 (2010).
Kennedy, W. P. et al. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci. Med. 2, e000080 (2015).
Akita, K. et al. Interferon α enhances B cell activation associated with FOXM1 induction: potential novel therapeutic strategy for targeting the plasmablasts of systemic lupus erythematosus. Front. Immunol. 11, 498703 (2021).
Wu, C. et al. Bioinformatics analysis of differentially expressed gene profiles associated with systemic lupus erythematosus. Mol. Med. Rep. 17, 3591–3598 (2018).
Romano, M. et al. Expanded regulatory T cells induce alternatively activated monocytes with a reduced capacity to expand T helper-17 cells. Front. Immunol. 9, 1625 (2018).
Heizmann, B., Kastner, P. & Chan, S. The Ikaros family in lymphocyte development. Curr. Opin. Immunol. 51, 14–23 (2018).
Chen, L. et al. IKZF1 polymorphisms are associated with susceptibility, cytokine levels, and clinical features in systemic lupus erythematosus. Medicine 99, e22607 (2020).
Han, J. W. et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 41, 1234–1237 (2009).
Cunninghame Graham, D. S. et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 7, e1002341 (2011).
Wang, C. et al. Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. Eur. J. Hum. Genet. 21, 994–999 (2013).
Vyse, T. J. & Cunninghame Graham, D. S. Trans-ancestral fine-mapping and epigenetic annotation as tools to delineate functionally relevant risk alleles at IKZF1 and IKZF3 in systemic lupus erythematosus. Int. J. Mol. Sci. 21, 8383 (2020).
Hoshino, A. et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J. Allergy Clin. Immunol. 140, 223–231 (2017).
Van Nieuwenhove, E. et al. A kindred with mutant IKAROS and autoimmunity. J. Allergy Clin. Immunol. 142, 699–702.e12 (2018).
Merrill, J. T. et al. Phase 2 trial of iberdomide in systemic lupus erythematosus. N. Engl. J. Med. 386, 1034–1045 (2022).
Lipsky, P. E. et al. Biological impact of iberdomide in patients with active systemic lupus erythematosus. Ann. Rheum. Dis. 81, 1136–1142 (2022).
Wojcik, H. et al. Expression of a non-DNA-binding Ikaros isoform exclusively in B cells leads to autoimmunity but not leukemogenesis. Eur. J. Immunol. 37, 1022–1032 (2007).
Schwickert, T. A. et al. Ikaros prevents autoimmunity by controlling anergy and Toll-like receptor signaling in B cells. Nat. Immunol. 20, 1517–1529 (2019).
Allman, D. et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood 108, 4025–4034 (2006).
Rivellese, F. et al. Effects of targeting the transcription factors Ikaros and Aiolos on B cell activation and differentiation in systemic lupus erythematosus. Lupus Sci. Med. 8, e000445 (2021).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
King, E. A., Davis, J. W. & Degner, J. F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019).
Rentzsch, P. et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2018).
Richardson, C. D. et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
Q.W., T.K., and M.-M.B. were supported by Joint Biology Consortium microgrants off parent grant NIH/NIAMS P30AR070253. J.C. was supported by P30AR070253. J.A.S. was supported by NIH/NIAMS R01AR077607, P30AR070253, and P30AR072577, the R. Bruce and Joan M. Mickey Research Scholar Fund, and the Llura Gund Award for Rheumatoid Arthritis Research and Care. M.T.W. was supported by NIH grants R01NS099068, R01HG010730, U01AI130830, R01AI024717, and R01AR073228. M.-G.A. was supported by NIH/NIAMS P30AR069625, Gilead Sciences, a Global Team Science Award from the Lupus Research Alliance, and the Arthritis National Research Foundation Vic Braden Family Fellowship. P.A.N. was supported by a Global Team Science Award from the Lupus Research Alliance and NIH/NIAMS 2R01AR065538, R01AR075906, R01AR073201, and P30AR070253. We thank Dr. Kaur Alasoo for making available full summary statistics from the eQTL Catalog and for recommendations on colocalization analysis.
Author information
Authors and Affiliations
Contributions
Q.W. and P.A.N. conceived and designed the study. Q.W., T.K., M.M.-B., and S.S. performed experiments and data analysis. T.K. led revision experiments. V.A., J.C., and M.G.-A. performed data analysis. J.A.S. led the recruitment of genotyped human subjects. X.C. and M.T.W. identified candidate transcription factors. M.T. and B.W. performed experiments. M.C.M. and C.D.L. investigated epistasis between loci. Q.W., T.K., and P.A.N. drafted the manuscript and all authors edited and approved the manuscript. P.A.N. supervised the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sun Jung Kim, Betty Tsao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Kim, T., Martínez-Bonet, M. et al. High-throughput identification of functional regulatory SNPs in systemic lupus erythematosus. Nat Commun 15, 6804 (2024). https://doi.org/10.1038/s41467-024-50710-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50710-5
- Springer Nature Limited