

Abstract
The arginyl-transferase ATE1 is a tRNA-dependent enzyme that covalently attaches an arginine molecule to a protein substrate. Conserved from yeast to humans, ATE1 deficiency in mice correlates with defects in cardiovascular development and angiogenesis and results in embryonic lethality, while conditional knockouts exhibit reproductive, developmental, and neurological deficiencies. Despite the recent revelation of the tRNA binding mechanism and the catalytic cycle of yeast ATE1, the structure-function relationship of ATE1 in higher organisms is not well understood. In this study, we present the three-dimensional structure of human ATE1 in an apo-state and in complex with its tRNA cofactor and a peptide substrate. In contrast to its yeast counterpart, human ATE1 forms a symmetric homodimer, which dissociates upon binding of a substrate. Furthermore, human ATE1 includes a unique and extended loop that wraps around tRNAArg, creating extensive contacts with the T-arm of the tRNA cofactor. Substituting key residues identified in the substrate binding site of ATE1 abolishes enzymatic activity and results in the accumulation of ATE1 substrates in cells.
Similar content being viewed by others


Introduction
Conserved from yeast to plants and mammals, arginyl-tRNA-protein transferase 1 (ATE1) is an enzyme that covalently transfers an arginine (arginylation) from tRNAArg to its protein substrates and has been implicated in a multitude of biological processes, including, prominently, in protein degradation1,2,3,4. Unlike other degron systems, the Arg/N-degron branch is unique as its action requires ATE1-mediated protein arginylation in a tRNA-dependent manner. Specifically, ATE1 transfers the nutrient arginine from high-energy arginyl-tRNAArg to substrate proteins containing an exposed N-terminal aspartate/glutamate residue (Fig. 1a). Arginyl-tRNA synthetase (ArgRS) then recharges tRNAArg released from the reaction. Therefore, this mechanism inherently links fundamental cellular processes, including protein synthesis, nutrient availability and metabolism, and protein turnover.

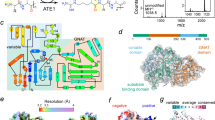
a Overview of protein arginylation and protein synthesis. b In vitro arginylation of the indicated substrate peptide using recombinantly purified ATE1 monitored by liquid chromatography–mass spectrometry (LC-MS) analysis. c Intact protein analysis of recombinantly purified human ATE1 after cleaving GST-tag by LC-MS. d Overview of the in-bacteria arginylation assay. The protein of interest (POI) fused to an N-terminal SUMO tag is co-expressed with Ulp1 protease and ATE1, producing recombinantly expressed arginylated proteins. e In E. coli arginylation of indicated substrate peptide co-expressed with ATE1 was monitored by intact protein analysis. f Superimposed 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the substrate DDIAA-Ub co-expressed with ATE1 (black) with recombinantly purified corresponding Nt-arginylated product RDDIAA-Ub (red) or unmodified DDIAA-Ub (blue) in the absence of ATE1. g Representative 2D class averages of ATE1 particles. Scale bar is 100 Å. h The cryo-EM structure of ATE1 (2.8 Å). Ribbon representation of ATE1 with the color-coded domain organization on a linear sequence: GNAT domain (orange), substrate binding domain (purple in chain A, green in chain B), and unstructured loop (gray). i, j Ribbon representation of ATE1 dimeric model (i) and volume of the map (j) colored by chain. a, d Created with BioRender.com, released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
Accumulating evidence positions ATE1 as a crucial player in normal development and in the progression of diseases1,3,5,6. Although not essential for yeast viability, deletion of both copies of Ate1 in mice results in embryonic lethality, characterized by abnormal cardiovascular development and angiogenesis1,2. Similarly, conditional ATE1 knockout in vertebrates yields a spectrum of issues, including reproductive defects, delayed growth, behavioral abnormalities, and brain irregularities7,8,9,10. Given that ATE1 is an enzyme regulating its substrate turnover, it is widely accepted that ATE1’s broad functions stem from controlling the half-life of its diverse substrates. For instance, regulators of G protein signaling (RGS) proteins, which are negative modulators of cardiovascular-specific Gq and Gi pathways, have been identified as ATE1 substrates and, once arginylated, are consequently degraded via the ubiquitin-proteosome pathway in vivo11. Likewise, under stress conditions, endoplasmic reticulum (ER) chaperone GRP78/Bip is arginylated to facilitate the clearance of misfolded proteins through SQSTM1/p62 mediated autophagy12,13,14.
While the prominent role of ATE1 has been attributed to regulating substrate degradation, other functions have also been linked with protein arginylation. For example, actin arginylation alters cell migration and autonomous cardiac myocyte functions, whereas α-synuclein arginylation augments neuroprotection functions15,16,17,18,19,20. Additionally, arginylation increases the half-life of calreticulin and its association with stress granules in the cytoplasm21,22. Other non-degradative roles of ATE1 involve transcriptional regulation of mitochondrial respiratory complexes23.
Given ATE1’s distinct enzymatic role and its substantial impact on physiological functions, we initiated a detailed study of its structure-function relationships. Our recent findings revealed that Saccharomyces cerevisiae ATE1 (scATE1) adopts a compact fold and primarily interacts with the major groove of the tRNA acceptor arm via its conserved GCN5-related N-acetyltransferases (GNAT) domain24. The N- and C-termini of scATE1 converge to form a putative substrate-binding domain, featuring a zinc-finger motif that is conserved in Kluyveromyces lactis ATE125. Human ATE1 shares only 32% sequence identity with scATE1, and the two enzymes share few common protein substrates, resulting in a significant gap in our understanding of its catalytic function, substrate selection, or the molecular interactions that regulate these important events.
In this report, we present structures of human ATE1 in an apo-state and as a ternary complex bound with substrate peptide and tRNAArg. Surprisingly, we discovered that human ATE1 exists as a symmetric dimer in its apo-state, a form that dissociates upon substrate binding. A long, unstructured loop in human ATE1 envelops tRNAArg, creating a unique interaction with the D- and T- arms of tRNA that is lacking in the yeast enzyme. Concurrently, the substrate peptide binds in a narrow groove that is situated near the interface of ATE1 homodimers leading to its dissociation in the ternary complex. We further identified crucial residues in both the tRNA and substrate binding sites and demonstrated that mutations in these residues inactivate ATE1, which leads to reduced turnover rates and substrate accumulation in human cells.
Results
Recombinant human ATE1 robustly arginylates the N-terminus of substrates
In addition to the well-documented N-terminal arginylation, mouse and human ATE1s are suggested to arginylate side chain carboxylates18,26,27,28. To investigate whether human ATE1 isoform 2 (hereafter referred to simply as hATE1) can facilitate midchain arginylation, we conducted liquid chromatography-mass spectrometry (LC-MS) analysis using a reconstituted arginylation reaction mixture or with purified recombinant ATE1. Similar to findings with yeast ATE124, the incubation of human ATE1 alongside in vitro-transcribed human tRNAArg, ArgRS, a model peptide substrate derived from the actin sequence (D1DIAALVVDNGSGMG16SGGS-Lys-biotin), and ATP led to the detection of the anticipated arginylated product with a 156 Da mass increase (Fig. 1b). Intriguingly, intact protein analysis of purified ATE1 (GST-tag removed) unveiled a secondary, albeit minor, peak with a mass of 59,214 Da—approximately 156 Da greater than its calculated mass of 59,056 Da—corresponding to a mono-arginylated ATE1 (Fig. 1c). As the recombinant ATE1 was produced as a GST-fusion protein without an exposed N-terminus, this finding suggests that arginylation occurred at an internal residue of the peptide substrate, consistent with prior observations for mouse ATE128.
Considering the challenges in enriching the small arginylated fraction of ATE1 for further analysis ( < 3% of total), we created an in-bacteria arginylation assay (Fig. 1d). Briefly, a 6xHis-tagged protein of interest (POI) is fused to plant SUMO and co-expressed with human ATE1 and Ulp1 protease in Escherichia coli. Following protease cleavage, the POI exposes a selectable N-terminal residue that is amenable to arginylation. This design facilitates targeted arginylation to enrich for modified product. To validate the concept, we designed a substrate containing a D1DIAALVVDNGSGMG16 sequence fused to ubiquitin (referred to as DDIAA-Ub hereafter). The predominant peak in the purified DDIAA-Ub sample registered at 11,321 Da (Fig. 1e), aligning with the anticipated mono-arginylated form. Both the unmodified form (11,165 Da) and a double-arginylated variant (11,477 Da) were identified. Moreover, we observed a leucinylated form (11,434 Da) of the Nt-arginylated protein, which is attributed to the presence of endogenous Leucyl/phenylalanyl-tRNA protein transferase (LFTR) in E. coli, which targets proteins with an exposed Nt-arginine for leucinylation and subsequent ClpS-mediated degradation29.
To pinpoint whether arginylation occurs internally or at the N-terminus, we produced 15N-labeled DDIAA-Ub and its Nt-arginylated analog, RDDIAA-Ub in the presence and absence of ATE1 protein. Each construct was subjected to 1H, 15N heteronuclear single quantum coherence (HSQC) spectral analysis. The HSQC spectrum of DDIAA-Ub closely aligns with that of RDDIAA-Ub only when co-expressed with ATE1 (Fig. 1f). This finding supports the notion that human ATE1 primarily arginylates the N-terminal aspartate/glutamate residues of its substrate, whereas the midchain arginiylated products are in lesser quantity and undetectable by NMR, consistent with in vitro arginylation assays on peptides using mouse ATE126.
Overall structure of human ATE1
Given the significant differences in human and yeast ATE1 sequences (Supplementary Fig. 1), we sought to determine the structure of human ATE1 to help understand this evolutionary divergence. Using single-particle cryo-electron microscopy (cryoEM), we achieved an average resolution of 2.8 Å for the full-length apo-state structure of hATE1 in dimeric form (Fig. 1g–j and Supplementary Table 1). Like scATE1, the N- and C-termini of hATE1 converge to create the substrate-binding domain, which lies adjacent to the GNAT domain. hATE1 also contains the conserved ‘CX2CXnCC’ zinc-finger motif within the substrate-binding domain (Fig. 2a, b). Intriguingly, in contrast to scATE1, hATE1 forms a near-symmetric homodimer (C2 symmetry), with an extensive interface of about 1183 Å2. The dimeric interface is located on the opposite side of the GNAT domains which coordinate tRNA binding (Fig. 1h, see discussion). Notably, hATE1 lacks the variable domain observed in yeast ATE1 but includes an additional unstructured loop (residues 105-268 of ATE1) residing between helix αC and β6 in the GNAT domain, although it is not accounted for in the cryoEM map.

a Ribbon diagram with a rainbow coloring from blue to red indicated N- to C-terminal position of ATE1 protomer. b 2D topological diagram of ATE1 with α-helices indicated by cylinders and β-strands by arrows. c The AlphaFold2 computational model of ATE1 (AF-O95260) color by RMSD calculated at each Cα compared with chain A of apo-ATE1 (this study). Residues that are missing in the cryoEM structure are colored red. The color bar indicates the spectrum representing distances between aligned Cα atom pairs, with blue and yellow specifying 0.1 and 3 Å, respectively. d The ribbon diagram of the ATE1 computational model colored based on the sequence conservation using ConSurf server. For visualization of conservation, the most variable positions were colored red, and the most conserved positions were colored blue per Consurf coloring scheme. e Web logo analysis of ATE1 (a.a. 191-220) based on multiple sequence alignment of ATE1 sequences with 35%-95% identity in all species generated during ConSurf analysis. f Representative EMSA assay of ATE1 binding to synthesized human tRNAArg A + T arms and acceptor (A) arm alone. Three independent experiments were performed with similar results. g Representative EMSA assay of ATE1 mutant lacking helix αC’ (Δ191-220) binding to synthesized human tRNAArg A + T arms and acceptor arm alone. Three independent experiments were performed with similar results. Source data are provided as a Source Data file.
To explore the characteristics of the unmodeled loop, we superimposed our cryoEM structure of hATE1 onto the computational model that is predicted by AlphaFold (AF-O95260-F1). The aligned structured regions displayed a mean root-mean-square deviation (RMSD) of around 1.2 Å for Cα atoms (Fig. 2c). Despite the medium-to-low confidence of this region in the AF2 model, the extended loop is predicted to contain three long and three short α-helices, which would be expected to interact with both the substrate-binding and GNAT domains of ATE1. Conservation mapping of the amino acid sequence among species that possess this loop revealed that, except for one conserved helix, it is predicted to be highly variable (Fig. 2d). Further sequence analysis of the conserved helix in this loop (residues 191-220 of ATE1) identifies a notable presence of positively charged lysine and arginine residues (Fig. 2e). Interestingly, this positively charged region clusters to the same side of helix αC on the GNAT domain, a conserved tRNA binding element in scATE1 and other tRNA-dependent aminoacyl-transferases24. We thus suspect this helix (denoted as αCloop hereafter) may facilitate tRNA recognition in hATE1. To test this hypothesis, we conducted EMSA experiments to determine the binding of wild-type (WT) or mutant ATE1 lacking helix αCloop (∆191-220) and defined their respective binding affinities for synthetic RNA segments that represent either the acceptor arm alone or the A- and T-arms (referred to as A + T arms) of tRNA (Fig. 2f, g). In alignment with scATE1, WT hATE1 displayed stronger binding affinity to the A + T arms than the acceptor arm alone, as indicated by Kd values of 0.4 µM and 1.6 µM, respectively (Fig. 3a). Conversely, the ∆191-220 mutant showed a 2-fold and 4-fold decrease in binding affinity to the acceptor arm (Kd = 3.4 µM) and A + T arms (Kd = 1.6 µM), respectively. These findings suggest that helix αCloop, which is absent in the yeast homolog, plays a direct role in tRNA binding in hATE1.

a Kd values of ATE1-RNA interaction in EMSA assays. Error represents SEM of at least three separate experiments. b Representative 2D class averages of ATE1/tRNA/substrate particles. Scale bar is 100 Å. c cryo-EM structure of ATE1 in complex with tRNA and substrate. d ATE1/tRNA/substrate complex. The tRNA molecule is colored based on specific regions: acceptor arm (reddish purple), T-arm (bluish green), variable loop (yellow), D-arm (orange), anticodon arm (blue). The substrate peptide is colored magenta. e Overlay of the structure of ATE1 in apo (blue) and tRNA-bound (orange) states. Structural regions unique to apo- and bound states are colored sea green and orange red, respectively. f A schematic illustration and formulae for proteasomal flux (P.F.) index. g Immunoblotting analysis of exogenously expressed C-RGS4-FLAG in WT or ATE1-KO HeLa cells ectopically expressing indicated ATE1 constructs with MG132 treatment (10 μM, 6 h). The proteasomal flux (P.F.) index was calculated based on the ratio of RGS4 levels in the presence or absence of MG132. The samples derive from the same experiment and the blots were processed in parallel. h Immunostaining analysis of recombinant RGS4-Myc/His with K88E/K92E mutant and WT hATE1 in ATE1 deficient HeLa cells. Scale bar, 10 µm. i Quantification of (h) based on normalized GFP signal intensity (n = 20 cells). Within each box, horizontal black lines denote median values; boxes extend from the 25th to the 75th percentile of each group’s distribution of values; vertical extending lines denote adjacent values. **p < 0.01 using the paired t-test. Source data are provided as a Source Data file.
The human ATE1·tRNAArg·substrate complex structure reveals tRNA contact sites that are absent in yeast
To decipher the molecular underpinnings of tRNA and substrate binding in hATE1, we reconstituted the ATE1·tRNAArg·substrate complex using hATE1, in vitro transcribed human tRNAArg, and the model substrate DDIAA-biotin peptide. Structural characterization by cryo-EM achieved an average resolution of 5.7 Å for the ATE1/tRNA/substrate complex (Fig. 3b, c and Supplementary Table 1). Quantification of computationally selected particles indicated that the inclusion of tRNA and substrate resulted in a substantial decrease in the ATE1 dimer population, dropping from 74% in the apo-state to 21% in the presence of tRNA and substrate peptide. This finding suggests that the ATE1 dimer dissociates upon binding of tRNA and/or substrate. Human ATE1 engages tRNA through an extensive interface measuring ~1474 Å2 (Fig. 3c). Similar to scATE1, the αC helix of hATE1 interacts with the acceptor arm of tRNA. Additionally, hATE1 establishes a secondary contact site with the D- and T-arms of tRNA (Fig. 3c, d). This interaction appears to stabilize part of the divergent long loop containing the helix αCloop in hATE1, as evidenced by features in the cryoEM map that account for residues 203-268 of this loop in the ternary complex, which was completely absent in the map of the apo form (Fig. 3e). Additionally, substrate engagement seems to induce an outward repositioning of the zinc-finger, likely facilitating the accommodation of substrate to the correct position in the active site. Concurrently, the N- and C-termini of ATE1 (residue 1-19 and 448-518) show notable destabilization in the ternary complex, which coincided with a loss of features for β1, αj-l, and β14-16 in the cryoEM map (Fig. 3e).
To evaluate the contribution of individual sites to tRNA-binding, we engineered an ATE1 K88E/K92E variant featuring charge reversal mutations in the αC helix, which interfaces with the major groove of the tRNA acceptor arm. We compared its RNA-binding affinity and enzymatic activity to the ∆191-220 ATE1 mutant. The K88E/K92E mutant showed about a 2-fold reduction in binding affinity to the A + T arms compared to the ∆191-220 variant (Fig. 3a). This observation supports the idea that interactions with the major groove of the acceptor arm are crucial for tRNA binding to ATE1, aligning with the conserved sequence of the αC helix in yeast and other tRNA-dependent aminoacyl-transferases.
To determine whether hATE1 specifically targets substrates in the N-degron pathway, we measured the proteasomal flux (P.F.) of a known substrate, RGS4, in WT and exon-5-deleted ATE1-KO HeLa cells. P.F. is assessed by RGS4 accumulation following proteasome inhibition via MG132 (Fig. 3f). ATE1-KO cells exhibited no RGS4 accumulation post-MG132 treatment, whereas WT ATE1 cells showed significant RGS4 accumulation with a normalized P.F. index (fold change) of ~29 (Fig. 3g). Transient re-expression of WT ATE1 restored the P.F. index, while neither the empty vector nor the RNA-binding-deficient mutant K88E/K92E could rescue the PF index. In line with this observation, immunostaining of FLAG-RGS4 indicated elevated RGS4 levels in ATE1-KO cells transfected with either the empty vector or the K88E/K92E mutant, but not in those transfected with WT ATE1 (Fig. 3h, i). These results affirm that the electrostatic interactions between the αC helix and bound tRNA are essential for human ATE1 function.
Protein substrate binding site
Upon initial analysis of the cryoEM map and model of the hATE1·tRNAArg·substrate complex, we noted that the substrate peptide could be accommodated in the cryoEM map. Since the resolution of the cryoEM map of the ternary complex does not allow for side-chain positioning, we integrated modeling to improve our prediction of the molecular interactions that are critical to ATE1 function. To better understand peptide substrate recognition by ATE1, we employed all-atom molecular dynamics flexible fitting (MDFF) using an explicit solvent model (see “methods”) to generate structural models of the ternary complex. For structural validation, we extended MD simulations for an additional 200 ns with MDFF and without MDFF. RMSF analysis of the ensemble with and without MDFF demonstrates a strong linear relationship (Supplementary Fig. 3a), indicating minimal overfitting of the model. The structural model predicts that the A76 of tRNAArg is situated close to the arginine-binding pocket at the opposite side of E387 in hATE1 (Fig. 4a). The space between the ribose O3’ atom of A76 of bound tRNA and the carboxyl group of E387 in ATE1 is ~13 Å, which is sufficient to accommodate the arginine residue that would exist on arginyl-tRNA. ATE1 residues surrounding the pocket around the first two residues in the substrate peptide display a volume increase of 258 Å2. This volume expansion aligns with the observed outward shift of the zinc finger and the β6-β8 strands of ATE1 that we observed in our structural analysis of the tRNA-substrate-bound complex.

a Zoom in view of the peptide and tRNA binding sites in ATE1/substrate/tRNA complex. Surfaces of the peptide substrate and ATE1 were shown in pink and white, while the last three nucleotides of tRNA were shown in yellow. The arginine binding pocket was shown as an orange oval. b–c Binding curves used to determine the Kd values of hATE1-peptide interaction in a direct binding assay by MST in the absence of RNA (b) or with 50 µM A + T arms (c). Data are presented as mean values +/− SD. Error bar represents s.d. in triplicate measurements. d–e Immunoblotting analysis of exogenously expressed C-RGS4-FLAG in ATE1 deficient HeLa cells with MG132 treatment (10 μM, 6 h). The protein levels of RGS4 with WT, R56E, H303A, E387A, and K419A mutants of hATE1 were compared. Proteasomal flux (P.F.) indices were calculated based on the ratio of RGS4 levels in the presence or absence of MG132. The samples were derived from the same experiment and the blots were processed in parallel. f Immunocytochemistry analysis of recombinant RGS4-Myc/His with R56E, K415E, K419A, and WT hATE1 in WT or ATE1 deficient HeLa cells. Scale bar, 10 µm. g–i Quantification of RGS4 level in WT or ATE1 deficient HeLa cells expressing indicated mutants based on normalized GFP signal intensity (n = 50 cells). Within each box, horizontal black lines denote median values; boxes extend from the 25th to the 75th percentile of each group’s distribution of values; vertical extending lines denote adjacent values. *p < 0.05, **p < 0.01 using the paired t-test. See also Supplementary Fig. 5b, e. Source data are provided as a Source Data file.
In the refined model, the substrate peptide (D1DIAALVVDNGS12) is largely buried within hATE1, covering an area of about 785 Å2—nearly 56% of its total surface area. Residues near the substrate mainly consist of the ZnF and residues including Y21-Y25, S58, K60, Y298-Q299, I302-H303, D305, E314, R317, F318, Y367, Y369, F408, I410, K415, and K419 of ATE1. These residues are in close contact with both D1-D2 regions of the substrate peptide and C75-A76 of the tRNA, hinting at their likely involvement in catalysis and/or substrate recognition. We further assessed residues R56 and R57 of hATE1, previously proposed to recognize the carboxylate group of the N-terminal Asp/Glu on the substrate protein25,30. The guanidine groups of both R56 and R57 reside more than 10 Å away from the N-terminus of the substrate and are also involved in intra-ATE1 contacts, making them unlikely contributors to catalysis. Consistently, an R56E mutant caused a shift in the size-exclusion profile of the recombinantly purified hATE1 protein (Supplementary Fig. 4), indicating a disruption in protein folding.
Building upon the structural information, we utilized differential scanning fluorimetry (DSF) to explore the stabilizing effects of specific molecules—free arginine, glutamine amide, and arginine amide—on ATE1. Our data reveal that the melting temperature of hATE1 decreased from 43.4 °C to 40.7 °C in the presence of 20 mM arginine amide, while no change was observed with free arginine or glutamine amide (Supplementary Fig. 5a). We interpret this result as a preferential binding of ATE1 to its product (arginine amide) over its substrate (glutamine amide) in the absence of tRNA, leading to dissociation of the dimer and exhibiting decreased thermal stability. Alternatively, arginine amide may induce local conformational changes that affect stability. Additionally, microscale thermophoresis (MST) assays demonstrated that, in the absence of tRNA, the binding affinity of hATE1 for the arginylated peptide product is greater than for its unmodified peptide substrate (Kd values: 160 and ~530 µM, respectively) (Fig. 4b). The presence of tRNA-mimetic (A + T arms) greatly enhanced the binding affinity for product and substrate peptides by 15- and 5-fold (Kd values: 11 and 99 µM, respectively) (Fig. 4c). We note that the A + T arms mimic the uncharged tRNA-Arg, therefore, representing the end state of the reaction which displays higher affinity toward the product peptide. We would expect that the presence of the charged, arginylated tRNA cofactor would contribute to the selection and binding of unmodified substrates/peptides.
To further interrogate the functional roles of the identified substrate binding sites, we carried out targeted mutagenesis of key residues. These mutations also led to a stabilization of RGS4 levels, corroborated by immunostaining (Fig. 4d–h, Supplementary Fig. 5). Additional mutations at E387A and K419A (arginine pocket), as well as R56E (Nt-GLU/ASP pocket), yielded similar outcomes. Furthermore, consistent with our previous finding that the zinc-finger domain is important for scATE1 stability and substrate binding, mutating the zinc-coordinating residue C26 of hATE1 to an alanine led to RGS4 stabilization (Fig. 4i, Supplementary Fig. 5d–e). Disruption of metal coordination by EDTA treatment also resulted in a thermal stability decrease of ~7 °C, reaffirming the critical role of the zinc-finger domain (Supplementary Fig. 5f). These results affirm the importance of these identified pockets in the binding of arginine and Nt-GLU/ASP, though higher resolution structures are still required for a more complete understanding of the precise molecular interactions that govern substrate binding to hATE1.
Dimerization of Human ATE1 is Critical for Substrate Arginylation
In human ATE1, dimer formation is a unique attribute compared to ATE1 from yeast, or in related tRNA-dependent aminoacyl-transferases. Our analysis of the dimeric interface identifies an array of interacting residues: G7-P9, V11, Y21, W38, H40, K60-Y61, S278-E280, K285, E305, D308-E309, G311-E314, R317-F318, F474, K476-R477, A478-I479, L514, and F516-N518. Residues W38 and H40 are located near the center, while residues D308-E309 and K476-R477 are positioned at the periphery, of the dimeric interface (Fig. 5a). While these residues are more distal to the substrate peptide binding site of their respective protomers, it is likely that their contribution to stabilizing the homodimer is important for substrate binding and/or catalysis. The dissociation constant measured by MST for dimerization of hATE1 was found to be 8.5 µM in vitro (Fig. 5b).

(a) Zoom-in view of the dimeric interface of human ATE1 in apo-state. The surfaces of the peptide substrate and tRNA were colored in purple and yellow, respectively. (b) Binding curves used to determine the Kd value of ATE1 dimerization by MST. Error bar represents s.d. in triplicate measurements. (c) Tm values derived from DSF assay for the WT and indicative mutant ATE1 proteins (n = 4). Data are presented as mean values +/− SD. Error bars represent SD determined from four independent experiments. d Immunoblotting of recombinant WT and W38F/H40E/K476E/R477E mutant of hATE1 in ATE1−/− HeLa cells using native gel and reducing SDS-PAGE gel. The samples were derived from the same experiment and the blots were processed in parallel. e–f Immunoblotting analysis of transiently expressed C-RGS4-FLAG in WT or ATE1-deficient HeLa cells transfected with WT, W38F/H40E (e), and K476E/R477E (f) mutants of hATE1 with MG132 treatment (10 μM, 6 h). The samples were derived from the same experiment and the blots were processed in parallel. g Co-IP assay in transiently co-transfected HEK293T cells with RGS4-FLAG and WT or W38F/H40E/K476E mutant hATE1 plasmids with MG132 treatment (10 µM, 6 h). The samples were derived from the same experiment and the blots were processed in parallel. h Immunostaining analysis of recombinant RGS4-Myc/His with indicative hATE1 mutants in WT or ATE1-deficient HeLa cells treated with MG132 (10 μM, 6 h). Scale bar, 10 µm. i Quantification of (h) based on normalized GFP signal intensity (n = 50 cells). Within each box, horizontal black lines denote median values; boxes extend from the 25th to the 75th percentile of each group’s distribution of values; vertical extending lines denote adjacent values. *p < 0.05, **p < 0.01 using the paired t-test. (j) PLA assay of recombinant C-RGS4-FLAG (PLUS) and ATE1 mutants (MINUS) in MG132 (10 μM, 6 h) treated ATE1−/− HeLa cells. Scale bar, 10 µm. k Quantification of (j) based on normalized PLA signal intensity (n = 50 cells). Within each box, horizontal black lines denote median values; boxes extend from the 25th to the 75th percentile of each group’s distribution of values; vertical extending lines denote adjacent values. **p < 0.01 using the paired t-test. See also Supplementary Fig. 6. Source data are provided as a Source Data file.
To evaluate the functional significance of dimerization, we selected two sets of mutations to disrupt the dimeric interface of ATE1: W38F and H40E, informed by sequence alignment with yeast ATE1, and K476E and R477E, selected for their likely electrostatic interactions with D308 and E309 of the opposite protomer. We hypothesized that these mutations would expose large surface areas of ATE1 to solvent, thus likely compromising protein stability. In line with this prediction, both pairs of mutation led to a reduced melting temperature of ~40 °C, while the quadruple mutant (W38F/H40E/K476E/R477E) displayed a similar decrease in melting temperature (Fig. 5c). Native gel electrophoresis using ATE1−/− HeLa cells substantiated this finding, showing that the quadruple mutant failed to form dimers, which contrasted with the WT human ATE1 (Fig. 5d).
To further explore the functional impact of mutations at the dimeric interface of ATE1, we examined the proteasomal flux of RGS4 in ATE1−/− cells carrying various mutations. Partial inhibition of RGS4 degradation was evident in cells expressing the W38F/H40E mutant. In contrast, cells with the K476E/R477E and the quadruple mutant showed significant inhibition of RGS4 proteasomal flux (Fig. 5e, f, Supplementary Fig. 6a). Immunocytochemical analysis of RGS4 levels further corroborated these findings (Fig. 5h, i). We thus reasoned that ATE1 dimerization might be a pivotal step in maintaining protein stability prior to substrate recognition and interaction in the Arg/N-degron pathway. In support of this notion, co-immunoprecipitation assays highlighted a substantial difference in RGS4 interaction with the triple mutant W38F/H40E/K476E compared to the WT hATE1 (Fig. 5g). Notably, this triple mutant alone was sufficient to halt proteasomal flux, mirroring the effects of the quadruple mutant (Supplementary Fig. 6b).
To further validate these observations, we performed a proximity ligation assay (PLA) using recombinant RGS4 and hATE1 constructs. The PLA technique enables the detection of protein interactions that occur within ~40 nm of each other. Consistent with prior data, mutations at the dimeric interface disrupted RGS4-ATE1 complex formation (Fig. 5j, k and Supplementary Fig. 6c). Overall, our comprehensive dataset confirms that the dimeric interface of ATE1 is essential for its catalytic activity, presumably by stabilizing the ATE1 protein to facilitate substrate binding.
Discussion
In this study, we unveil the 3D structures of human ATE1 with and without a tRNA cofactor and peptide substrate. Notably, human ATE1 forms a symmetric homodimer in its apo state (Fig. 6a), which cannot be confidently predicted by the AlphaFold2 algorithm, likely due to a lack of homologs and pre-existing structural patterns. Compared to its yeast counterpart, hATE1 lacks the variable domain, which packs onto the substrate binding domain and, thus, likely contributes to increased stability of scATE1 (Tm ~ 46 °C)24. Consequently, hATE1 has evolved to assemble as a homodimer as a means of stabilization; a notion that is supported by homodimeric interface disrupting mutations resulting in decreased stability of the free enzyme (Fig. 5c). Importantly, residues on the dimeric interface are conserved in mice and humans, but not in lower organisms such as fly or Arabidopsis (Supplementary Fig. 1, blue dash boxes). Many residues that reside on the dimeric interface, including S278-E280, K285, E305, D308-E309, and G311-E314, are in exon 7b of human ATE1-2 (also denoted as isoform 1b7b). Although the residues on the dimeric interface are mostly conserved or highly similar between exons 7b and 7a, we cannot rule out a potential disruption of the dimeric interface in isoforms containing exon 7a. Future studies of ATE1 in other organisms and isoforms will be needed to determine whether the dimerization of hATE1 is unique to mammals and shared among other isoforms.

a Comparison of the overall structures of ATE1 from S. cerevisiae (left), human (middle), and AF2 computational model (right) in apo-state. The variable, substrate-binding, and GNAT domains were colored in deep green, purple, and coral, respectively. b Comparison of the overall structures of ATE1 from S. cerevisiae (left) and human (right) in complex with tRNA. The peptide substrate in the human ATE1 complex was colored cyan.
In cells, disruption of the dimeric interface of hATE1 led to loss of activity and substrate binding, resulting in slower turnover and, thus, accumulation of RGS4 (Fig. 5e–k). Our data suggest that the activity and protein levels of ATE1 are very likely regulated by its oligomeric state. Specifically, our data implies that the dimerization of hATE1 partially occludes the substrate peptide binding site, thus likely inhibiting ATE1 activity. However, given the variety of ATE1 substrates, we cannot exclude the possibility that other substrates may accommodate the binding pocket without dissociating the dimeric structure. Furthermore, while the two substrate binding sites on an hATE1 dimer are 50 Å away from each other, having both cofactors bound simultaneously would increase the local concentration, which may help to promote the binding of substrates.
Another distinct feature of human ATE1 is its extensive interactions with tRNAArg that extend beyond those interactions with the acceptor arm that were observed in yeast ATE1 (Fig. 6b). We show that hATE1 contains a highly conserved helix within its unstructured loop contacting the D- and T-arms and the variable loop on tRNA. Interestingly, this region is largely conserved from fly to human, but is lacking in Arabidopsis, amoeba, and yeast, suggesting a unique evolutionary selection of additional tRNA contacts in higher organisms (Supplementary Fig. 1, green dash boxes). S. cerevisiae and humans contain a total of 275 and 501 tRNAs, respectively, among which 19 and 28 are tRNAArg. It is possible that ATE1s in higher organisms evolved to have additional tRNA contacts to account for the expansion of tRNA species. Due to the existence of additional tRNA contacting sites in human ATE1, we expect arginyl-tRNAArg fragments containing both the A- and T-arms will be most efficient for arginylation, even though the acceptor arm alone of arginyl-tRNAArg is sufficient for arginine transfer using mouse ATE131.
Collectively, our study provides insights into the structure-function relationship of human ATE1, which is essential for understanding the mechanism and regulation of its activity in diverse cellular processes. Critical amino acids within the substrate binding regions of human ATE1 were identified and mutations that disrupt the homodimer assembly render the enzyme inactive, and results in reduced turnover rates and accumulation of ATE1 substrates within cells. Our study builds a foundation enabling future research to elucidate the specificity and selectivity of protein substrates and provides a starting point for the rational design of small molecules attenuating ATE1 activity in humans.
Methods
Protein expression and purification
The hATE1 was cloned into a pGEX 6p-1 vector with an N-terminal GST-tag and PreScission cleavage site and expressed in BL21(DE3)-RIL cells (Agilent). This RIL strain contains a rare codon plasmid encoding extra copies of argU, ileY, and leuW and overexpresses tRNAArg. Protein production was induced with 0.4 mM IPTG overnight at 16 °C in Luria broth (LB). The GST-tagged hATE1 proteins were purified on glutathione Sepharose 4B beads (GE Healthcare) in 50 mM Tris (pH 7.5), 0.5 M NaCl, and 5 mM DTT. The GST tag was cleaved with PreScission protease overnight at 4 °C. The pET21a ΔNHsArgRS plasmid with an N-terminal His-tag is a generous gift from Aaron Smith's lab at the University of Maryland. The His-tagged proteins were purified on Ni-NTA beads (Qiagen) in 50 mM Tris-HCl (pH 7.5) buffer, supplemented with 0.5 M NaCl, 5 mM β-ME, and 10 mM Imidazole. The His-tag was cleaved overnight at 4 °C with TEV protease. The model substrate containing a D1DIAALVVDNGSGMG16 sequence fused to ubiquitin (DDIAA-Ub) was cloned into a pET-28a vector with a C-terminal His-tag. Proteins were further purified by size exclusion chromatography and concentrated in Millipore concentrators. All mutants were generated by site-directed mutagenesis using the Q5 polymerase mutagenesis protocol, grown, and purified as WT proteins. The sequences of oligonucleotides used were included in Supplementary Data 1.
Intact Protein Mass Spectroscopy Analysis
For intact protein analyzes, proteins were directly injected onto a 2.1 × 5 mm Acquity UPLC BEH300 C4, 1.7 µm VanGuard Pre-Column (Waters), using a Waters Acquity classic UPLC. Protein was loaded and washed at 0.2 mL/minute for 3 min, then eluted with a gradient from 3% to 85% ACN in 3 min then to 95% ACN in 0.5 min and detected using a Synapt G2 Q-Tof mass spectrometer (Waters). Precursor mass spectra (MS1) were acquired in positive ES resolution mode from 200 to 2500 m/z with 3.0 kV capillary voltage and a source temperature of 80 °C. Intact protein masses were deconvoluted using Mass Lynx v4.2 Maximum Entropy. Full scan spectra and deconvoluted intact mass spectra were included in the Source Data file.
NMR experiments
NMR experiments were carried out at 298 K on Bruker 800 MHz spectrometers equipped with a cryoprobe. Uniformly labeled model substrates DDIAA-Ub were expressed in the presence or absence of co-expressing human ATE1. Uniformly labeled corresponding model product RDDIAA-Ub was expressed without ATE1. NMR samples contained 0.1 mM uniformly 15N-labeled DDIAA-Ub or RDDIAA-Ub in 20 mM Tris-HCl (pH 7.5) buffer supplemented with 150 mM NaCl, 5 mM DTT, and 8% D2O. 1H,15N HSQC spectra were collected using Topspin. NMR data processing and spectra analysis was performed using NMRbox32.
Fluorescent microscale thermophoresis (MST) binding assay
The MST experiments were performed using a Monolith NT.115 instrument (Nanotemper) as described previously33. All experiments were performed with the purified hATE1 in a buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 5 mM DTT, and 0.08% Tween-20. The final concentration of the hATE1 labeled with red Tris-NTA dye was 5 nM. Dissociation constants for the interaction of labeled hATE1 with unlabeled hATE1 were determined using a direct binding assay in which increasing amounts of unlabeled hATE1 were added stepwise. The measurements were performed at 40% excitation and medium MST power with 20 s MST on time and 1 s off time. For all measurements, samples were loaded into standard capillaries and 10000–20000 counts were obtained for the fluorescence intensity. The Kd values were determined using the MO. Affinity Analysis software from manufacture. All measurements were done in triplicates.
Differential scanning fluorimetry
Spectra were recorded on a CFX96 Touch real-time PCR detection system (Bio-Rad). The reaction volume was 30 μL containing 5 ~ 10 μM hATE1 in 20 mM Tris (pH 7.5), 150 mM NaCl, 5 mM DTT, and SYPRO orange (Thermo Fisher 5000X) to a final concentration of 2.5 ~ 5X with or without possible ligands in variable concentrations. The thermal gradient was conducted between 25 and 90 °C with 0.2 °C/min intervals.
Gel shift assays (EMSA)
Increasing amounts of hATE1 were incubated with 1 μM tRNAArg acceptor arms, or 0.25 μM tRNAArg A + T arms in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM DTT for 30 min on ice. The reaction mixtures were loaded on 4–20% non-denaturing gradient gels (GenScript) and electrophoresis was performed in 1x TB buffer at 100 V for 3 h at 4 °C. Gels were stained with SYBR green and visualized with Bio-Rad Gel Doc XR Imaging System.
Cryo-electron microscopy data collection and image processing
The apo hATE1 protein was polished by a size-exclusion chromatography step and diluted with the peak fraction to a concentration of 0.6 mg/mL. For electron microscopy, the purified protein was deposited onto an UltraAuFoil R1.2/1.3 grid (300 mesh) that had been freshly glow-discharged. Subsequently, the grid was plunge-frozen using a Vitrobot Mark IV (Thermo Fisher) with parameters set to a blot force of 0, a blot time of 2.5 s, 100% humidity, and a temperature of 4 °C. Data acquisition was performed at our in-house Cryo-electron Microscopy Core Facility. The movie stacks were generated using an FEI Titan Krios G3i transmission electron microscope, operating at 300 keV. The microscope was outfitted with a Gatan K3 direct electron detector and a Gatan BioQuantum image filter. The filter was operated in zero-loss mode, with a slit width established at 20 eV. The automated data collection process was facilitated by SerialEM software34. It was conducted at a nominal magnification of 64,000x, translating to a physical pixel size of 1.34 Å/pixel (or 0.67 Å/pixel at super-resolution). The set nominal dose was 40 e/Å2, achieved with a dose rate of 20 electrons/pixel/second. During this process, 3.5-sec exposure times were fractioned across 70 frames.
For the hATE1·tRNAArg·substrate complex, the peak fraction of purified hsATE from SP column was incubated with 1.5-fold excess in vitro transcribed tRNAArg and 2-fold excess substrate peptide with a sequence of NH2-DDIAALVVDNGSGMGSGGS-Lys-biotin (synthesized by SynPeptide). 3.5 µL of 10 µM assembled complex was deposited onto an UltraAuFoil R1.2/1.3 grid (300 mesh) that had been freshly glow-discharged. Subsequently, the grid was plunge-frozen using a Vitrobot Mark IV (Thermo Fisher) with parameters set to a blot force of 0, a blot time of 2.5 s, 100% humidity, and a temperature of 4 °C. Data acquisition was performed at our in-house Cryo-electron Microscopy Core Facility similar to the apo hATE1 sample. The specified imaging condition includes a nominal magnification of 165,000x, translating to a physical pixel size of 0.52 Å/pixel (or 0.26 Å/pixel at super-resolution). The set nominal dose was 100 e/Å2, achieved with a dose rate of 20 electrons/pixel/second. During this process, 1.3-sec exposure times were fractioned across 27 frames. The utilized defocus range was set between -0.5 and -2.0 µm. A comprehensive list detailing the number of movies per dataset can be found in Supplementary Table 1.
Image processing was performed using CryoSPARC version 4.3.0. The particles were automatically picked using the blob picker with an 100 Å diameter. Reference-free 2D classification was performed in streaming with 200 classes and limited maximum resolution to 18 Å. 2D class averages with expected size and shape were subjected to another round of 2D classification with 200 classes with a maximum resolution set to 6 Å. After the second round of 2D classification, particles from classes with resolution better than 8 Å and ECA less than 2 were selected for 2D rebalance. The rebalanced particle set was then used for ab-initio reconstruction to generate the initial volume allowing the use of a maximum resolution of 6 Å. 3D refinement was first performed using non-uniform 3D refinement with an initial lowpass resolution of 8 Å to preserve map features reconstituted in the ab initio 3D volume. Local refinement was subsequently performed with the following options turned on: (1) Use pose/shift Gaussian prior during alignment, (2) Re-center rotation each iteration, (3) Re-center shifts each iteration, (4) Force re-do GS split and (5) FSC Noise-Substitution. The gold-standard Fourier shell correlation (FSC) of 0.143 criterion was used to report the resolution and the FSC curves were corrected for the effects of a soft mask using high-resolution noise substitution. Comprehensive image processing workflows can be found in Supplementary Figs. 7 and 8.
Model building and refinement
For apo hATE1 model building, AlphaFold II predicted hATE1 structure was used as the initial model35. A zinc atom was placed to coordinate cysteine residues 23, 26, 71, and 72 in Coot (version 0.9.6)36, and metal coordination restrains were generated using phenix.metal_coordinate37. The model was refined into the final cryoEM map using phenix.real_space_refine37. All statistics for structural models were reported in Supplementary Table 1. Figure panels depicting cryoEM maps or atomic models were generated using ChimeraX version 1.638. Maps colored by local resolution were generated using RELION 4.039.
Molecular dynamic flexible fitting (MDFF) of ATE1·tRNAArg·substrate complex
The structure of human tRNAArg was modeled using PDB entry 7UXA40 as the initial model. Both the tRNA model and apo-form of hATE1 structure were docked into cryoEM map as a rigid body, and the substrate peptide was manually built in Coot into the additional density in the substrate binding pocket. The final assembly was energy minimized with the final cryoEM map using phenix.real_space_refine37. Then all-atom MDFF simulations41 with explicit solvent model were performed starting from this starting structure. The system was built with amber ff19sb42 for protein, RNA.OL3 for RNA43, ZAFF for zinc42, and ions with the TIP3P water model44. The system was placed in a rectangle water box with 32 Å padding and charge-neutralizing sodium ions with additional salt (Na+, Cl−) around the solute resulting in 50 mM salt condition using tleap45. Input files for MDFF were prepared using the mdff package in VMD 1.9.646 with the scaling factor (gscale=0.3) coupling to backbone atoms and sampled for 100 ns using NAMD347 on two NV-link NVidia A100 GPU cards. The variable SCALING_1_4 was set to 0.8333333 to be compatible with amber force field. Collective variable calculations (colvars) module in NAMD47 was used to constrain the distance between the N atom of the first Asp residue in the substrate peptide and O3′ of A76 in tRNA with a distance of 3 Å with a force constant of 10 kcal/mol/Å2. Cross-correlation values for snapshots from the MD trajectory with cryoEM map were calculated using Situs package48. A frame from the last 10 ns of the trajectory with a cc value of 0.81 was selected (Supplementary Fig. 3b) and subsequently minimized using Geometry Minimization (Supplementary Fig. 3c) in phenix37. Using this model as the starting structure, an additional 200 ns MD simulations were performed with MDFF (gscale = 0.3) and without MDFF (gscale = 0). Root-mean-square-fluctuation (RMSF) of individual residues was used for comparing the ensemble with and without MDFF49.
Cell culture
HEK293T was purchased from ATCC (American Type Culture Collection, CRL-3216). Wild-type and ATE1-deficient HeLa cell lines were obtained from KRIBB (Korea Research Institute of Bioscience and Biotechnology). ATE−/− HeLa cells were constructed by deleting exon 5 of ATE1. These cells were cultured in DMEM (Gibco, 11995-062) supplemented with 10% FBS (Gibco, 11570636) and incubated in a 5% CO2 incubator.
Plasmids and other reagents
Mutant plasmids of human ATE1 were constructed by conducting site-directed mutagenesis from recombinant pcDNA3.1-ATE1-Myc/His plasmid. The sequences of oligonucleotides used were included in Supplementary Data 1. The recombinant C-RGS4-FLAG plasmids were cloned as previously described50. These plasmids were transiently transfected in HEK293T, wild-type, and ATE1−/− HeLa cells using Lipofectamine 2000 (Invitrogen, 11668019). Cells were treated with MG132 (Enzo, BML-PI102).
Antibodies
The antibodies used in this study are as follows: rabbit polyclonal anti-RGS4 (Sigma Aldrich, AV30213, 1:3000) mouse monoclonal anti-ATE1 (Santa Cruz, sc-271219, 1:1000), mouse monoclonal anti-MYC(9E10) (Santa Cruz, sc-40, 1: 1000), mouse monoclonal anti-β-actin (Sigma Aldrich, A1978, 1:10000) and mouse monoclonal anti-FK2 (Enzo, BML-PW8810, 1:3000). The following list of secondary antibodies was used: Alexa fluor 488 goat anti-rabbit IgG (Invitrogen, A11034, 1:400), Alexa fluor 555 goat anti-mouse IgG (Invitrogen, A32727, 1:400), anti-rabbit IgG-HRP (Jackson Immuno Research, 111-035-003, 1:10000), and anti-mouse IgG-HRP (Jackson Immuno Research, 115-035-003, 1:10000).
Immunoblotting
Cells were harvested by centrifugation at 15,000 g for 2 min and resuspended with PBS. Then the cells were lysed in a 2x Laemmli sample buffer (277.8 mM Tris-HCI, pH 6.8, 4.4% LDS, 44.4% (v/v) glycerol; Bio-Rad, 1610747) with β-mercaptoethanol and boiled at 100 °C for 10 min. The samples were separated using SDS-PAGE and transferred onto polyvinylidene difluoride membranes (PVDF). Subsequently, the membranes were blocked with 4% skim milk for 30 min at room temperature followed by incubation with primary and secondary antibodies. To detect the signals, a mixture of SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher, 34080) or SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher, 34095) was used. Uncropped gels were included in the Source Data file.
Immunocytochemistry
Cells were cultured on poly-L-Lysine-coated coverslips in a 24-well plate and fixed using 4% paraformaldehyde for 15 min. After washing with PBS, the cells were permeabilized with 0.5% Triton X–100 for 15 min. For blocking, cells were washed and added with 2% BSA for 1 h followed by incubation with primary antibodies overnight at 4 °C. Subsequently, the samples were washed with PBS 3 times for 10 min and incubated with Alexa Fluor conjugated secondary antibodies for 30 min at room temperature. The coverslips were mounted on slide glasses using a hard-mounting solution with DAPI (VECTASHEILD, H-1500). Images of samples were obtained by confocal microscope (Zeiss, 510 Meta) and analyzed with Zeiss LSM Image Browser.
Co-immunoprecipitation
Cells were harvested and resuspended in a lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Triton X–100, 1 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail). The cell lysates were incubated on the rotator at 4 °C for 30 min, then cell debris was removed by centrifugation at 15,000 g for 10 min. Supernatants were collected to be pre-cleared with normal mouse IgG (Santa Cruz, sc-2025) and Protein A/G-Plus agarose beads (Santa Cruz, sc-2003). The samples were pre-cleared at 4 °C for 3 h on the rotator, then beads were eliminated by centrifugation at 15,000 g for 10 min. Supernatants were incubated with anti-Myc (9E10) antibody (1 μg/sample) overnight on the rotator at 4 °C, then added with beads. After binding for 2 h, the beads were washed with lysis buffer four times and precipitated by centrifugation at 2350 g for 2 min. The bound proteins were eluted at 2x Laemmli sample buffer by heating at 95 °C for 10 min, then separated by SDS-PAGE and immunoblotting.
Proximity ligation assay (PLA)
Cells were prepared, then fixed and permeabilized by immunocytochemistry methods, followed by steps proceeded with Duolink In Situ Red Starter Kit Mouse/Rabbit (Sigma Aldrich, DUO92101)51. The cells were blocked with a blocking solution for 1 h at room temperature. Subsequently, the samples were incubated with primary antibodies in an antibody dilution buffer overnight at 4 °C followed by washing with a wash buffer. Then the antibody solution with MINUS antibody and PLUS antibody diluted in dilution buffer was incubated with samples at 37 °C for 1 h. The antibodies in the samples were ligated by incubation with ligation solution at 37 °C for 30 min and amplified with an amplification mixture at 37 °C for 100 min. The cells were washed with wash buffers and mounted with a mounting medium with DAPI. Confocal images were obtained with a Zeiss microscope and analyzed with a Zeiss LSM Image Browser.
Statistical and Reproducibility
For quantification data of immunoblotting and immunocytochemistry analysis, each set of experiments was triplicated and performed three times. The data were shown as mean ± S.E.M. of three independent experiments. P-values were determined using a two-tailed student’s t-test (degree of freedom = n − 1). Statistical significance was determined as values of p < 0.05 (**p < 0.01; *p < 0.05).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-41770 and EMD-42071. The atomic coordinates for the deposited map have been deposited in the Protein Data Bank under accession codes 8TZV and 8UAU. Source data are provided with this paper. All other relevant data supporting the key findings of this study are available within the article, its Supplementary Information or from the corresponding authors upon request. The structural data used in this study are available in the PDB database under accession codes 7UXA, 8E3S and 8FZR. Source data are provided with this paper.
References
Kwon, Y. T. et al. An essential role of N-terminal arginylation in cardiovascular development. Science (1979) 297, 96–99 (2002).
Balzi, E., Choder, M., Chen, W. N., Varshavsky, A. & Goffeau, A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J. Biol. Chem. 265, 7464–7471 (1990).
Kashina, A. S. Protein arginylation: over 50 years of discovery. Methods Mol. Biol. 1337, 1–11 (2015).
Varshavsky, A. The N-end rule. Cell 69, 725–735 (1992).
Sriram, S. M., Kim, B. Y. & Kwon, Y. T. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 2011 12:11 12, 735–747 (2011).
Avcilar-Kucukgoze, I. & Kashina, A. Hijacking tRNAs from translation: regulatory functions of trnas in mammalian cell physiology. Front Mol. Biosci. 7, 388 (2020).
Leu, N. A., Kurosaka, S. & Kashina, A. Conditional Tek promoter-driven deletion of arginyltransferase in the germ line causes defects in gametogenesis and early embryonic lethality in mice. PLoS One 4, e7734 (2009).
Kurosaka, S. et al. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet 6, e1000878 (2010).
Lian, L. et al. Loss of ATE1-mediated arginylation leads to impaired platelet myosin phosphorylation, clot retraction, and in vivo thrombosis formation. Haematologica 99, 554–560 (2014).
Wang, J. et al. Arginyltransferase ATE1 is targeted to the neuronal growth cones and regulates neurite outgrowth during brain development. Dev. Biol. 430, 41–51 (2017).
Lee, M. J. et al. RGS4 and RGS5 are in vivo of the N-end rule pathway. Proc. Natl Acad. Sci. USA 102, 15030–5 (2005).
Cha-Molstad, H. et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 17, 917–929 (2015).
Zhang, Y. et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 9, 4373 (2018).
Dong, D. et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res 68, 498–505 (2008).
Rai, R. et al. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development 135, 3881–9 (2008).
Zhao, J. et al. alpha-Synuclein arginylation in the human brain. Transl. Neurodegener. 11, 20 (2022).
Pan, B. et al. Effects of glutamate arginylation on alpha-synuclein: studying an unusual post-translational modification through semisynthesis. J. Am. Chem. Soc. 142, 21786–21798 (2020).
Wang, J. et al. Protein arginylation targets alpha synuclein, facilitates normal brain health, and prevents neurodegeneration. Sci. Rep. 7, 11323 (2017).
Karakozova, M. et al. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science (1979) 313, 192–196 (2006).
Zhang, F., Saha, S., Shabalina, S. A. & Kashina, A. Differential arginylation of actin isoforms is regulated by coding sequence–dependent degradation. Science (1979) 329, 1534–1537 (2010).
Decca, M. B. et al. Post-translational arginylation of calreticulin: a new isospecies of calreticulin component of stress granules. J. Biol. Chem. 282, 8237–8245 (2007).
Goitea, V. E. & Hallak, M. E. Calreticulin and arginylated calreticulin have different susceptibilities to proteasomal degradation. J. Biol. Chem. 290, 16403–14 (2015).
Jiang, C. et al. Regulation of mitochondrial respiratory chain complex levels, organization, and function by arginyltransferase 1. Front Cell Dev. Biol. 8, 603688 (2020).
Abeywansha, T. et al. The structural basis of tRNA recognition by arginyl-tRNA-protein transferase. Nat. Commun. 2023 14:1 14, 1–12 (2023).
Kim, B. H. et al. Crystal structure of the Ate1 arginyl-tRNA-protein transferase and arginylation of N-degron substrates. Proc. Natl Acad. Sci. 119, e2209597119 (2022).
Wang, J. et al. Arginyltransferase ATE1 Catalyzes midchain arginylation of proteins at side chain carboxylates in vivo. Chem. Biol. 21, 331–337 (2014).
Wang, J. et al. Target site specificity and in vivo complexity of the mammalian arginylome. Sci. Rep. 2018 8:1 8, 1–12 (2018).
Wang, J. et al. Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chem. Biol. 18, 121–130 (2011).
Erbse, A. et al. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439, 753–6 (2006).
Van, V., Ejimogu, N.-E., Bui, T. S. & Smith, A. T. The structure of Saccharomyces cerevisiae arginyltransferase 1 (ATE1). J Mol Biol 167816 https://doi.org/10.1016/J.JMB.2022.167816 (2022).
Avcilar-Kucukgoze, I. et al. tRNAArg-derived fragments can serve as arginine donors for protein arginylation. Cell Chem. Biol. 27, 839–849.e4 (2020).
Maciejewski, M. W. et al. NMRbox: a resource for biomolecular NMR computation. Biophys. J. 112, 1529–1534 (2017).
Zhang, Y. et al. Mechanism for autoinhibition and activation of the MORC3 ATPase. Proc. Natl Acad. Sci. USA 116, 6111–6119 (2019).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Afonine, P. V. et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr D. Struct. Biol. 74, 814–840 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Hayne, C. K. et al. Structural basis for pre-tRNA recognition and processing by the human tRNA splicing endonuclease complex. Nat. Struct. Mol. Biol. 30, 824–833 (2023).
Trabuco, L. G., Villa, E., Schreiner, E., Harrison, C. B. & Schulten, K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 49, 174–180 (2009).
Tian, C. et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput 16, 528–552 (2020).
Aytenfisu, A. H., Spasic, A., Grossfield, A., Stern, H. A. & Mathews, D. H. Revised RNA dihedral parameters for the amber force field improve rna molecular dynamics. J. Chem. Theory Comput 13, 900–915 (2017).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput Chem. 26, 1668–1688 (2005).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph 14, 27–28, 33–38 (1996).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 44130 (2020).
Wriggers, W. Conventions and workflows for using Situs. Acta Crystallogr D. Biol. Crystallogr 68, 344–351 (2012).
Singharoy, A. et al. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. Elife 5, e16105 (2016).
Heo, A. J. et al. The N-terminal cysteine is a dual sensor of oxygen and oxidative stress. Proc. Natl Acad. Sci. USA 118, e2107993118 (2021).
Alam, M. S. Proximity Ligation Assay (PLA). Curr. Protoc. Immunol. 123, e58 (2018).
Acknowledgements
We thank Drs. Mehaffey and Ebmeier at the University of Colorado Boulder Mass Spectrometry Facility for help with collecting and analyzing samples. This work was supported by grants from NIH R00 CA241301 and R35 GM150678 to Y.Z., R01 GM133841, RM1 GM142002, and R01 CA240993 to D.J.T and Case Comprehensive Cancer Center (P30CA043703 to Y.Z. and D.J.T.) and the Basic Science Research Programs of the NRF funded by the Ministry of Science, ICT, and Future Planning (MSIP) (NRF-2020R1A5A1019023 and NRF-2021R1A2B5B03002614 to Y.T.K., and RS-2023-00249464 to C.H.J.). We are grateful to the Cryo-Electron Microscopy Core at the CWRU School of Medicine and K. Li and K. Whiddon for access to the sample preparation and cryo-EM instrumentation. Computational support was provided by the Case Western Reserve University High-Performance Computing Cluster. This publication was made possible by the Clinical and Translational Science Collaborative of Cleveland, UL1TR0002548 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research. This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant P41GM111135 (NIGMS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
X.L., W.H., S.B.K., D.F., T.A., J.L., and U.B. performed experiments and together with C.H.J., Y.T.K., D.J.T., and Y.Z. analyzed the data. C.H.J., D.J.T., and Y.Z. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
W.H. and D.J.T. provide consultation for, and D.J.T. has ownership interests in, Rappta Therapeutics, which focuses on areas of research unrelated to the present study. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Andrei Korostelev, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lan, X., Huang, W., Kim, S.B. et al. Oligomerization and a distinct tRNA-binding loop are important regulators of human arginyl-transferase function. Nat Commun 15, 6350 (2024). https://doi.org/10.1038/s41467-024-50719-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50719-w
- Springer Nature Limited