

Abstract
The legume albumin-1 gene family, arising after nodulation, encodes linear a- and b-chain peptides for nutrient storage and defense. Intriguingly, in one prominent legume, Clitoria ternatea, the b-chains are replaced by domains producing ultra-stable cyclic peptides called cyclotides. The mechanism of this gene hijacking is until now unknown. Cyclotides require recruitment of ligase-type asparaginyl endopeptidases (AEPs) for maturation (cyclization), necessitating co-evolution of two gene families. Here we compare a chromosome-level C. ternatea genome with grain legumes to reveal an 8 to 40-fold expansion of the albumin-1 gene family, enabling the additional loci to undergo diversification. Iterative rounds of albumin-1 duplication and diversification create four albumin-1 enriched genomic islands encoding cyclotides, where they are physically grouped by similar pI and net charge values. We identify an ancestral hydrolytic AEP that exhibits neofunctionalization and multiple duplication events to yield two ligase-type AEPs. We propose cyclotides arise by convergence in C. ternatea where their presence enhances defense from biotic attack, thus increasing fitness compared to lineages with linear b-chains and ultimately driving the replacement of b-chains with cyclotides.
Similar content being viewed by others


Introduction
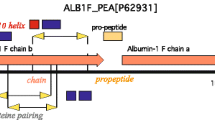
The legume albumin-1 gene family is significant in the evolutionary history of nitrogen-fixing legumes, having appeared after the development of rhizobial symbioses, serving nutrient storage and insecticidal functions in seeds, and being implicated in nodule biology. Legume albumin-1 genes are restricted to the subfamily Faboideae within the Fabaceae, and exhibit expression typically confined to seeds, roots, and nodules1,2. Albumin-1 precursors comprise a signal peptide, followed by a ‘b-chain’ peptide of the knottin class, a short linker sequence, and an ‘a-chain’ peptide implicated as a nutrient storage protein (Fig. 1a)3. Multiple functions have been described for the b-chain, including insecticidal activity (membrane disruption/depolarization, vacuolar ATPase inhibition), peptide signaling (soybean leginsulin), and roles in root nodule biology (A1-nodulins)2,4,5,6,7. Thus, albumin-1 genes may provide fitness advantages in the Faboideae as a nutrient reservoir, nitrogen-rich seed protector, and nodule biology modulator2,6.

a Schematic representation of the Pisum sativum albumin-1 precursor with lengths in amino acids noted on the right of the bar diagram representation of the precursor. The PA1 precursor encodes a signal peptide (‘SP’ - yellow), the a-chain (blue), and the b-chain peptide called PA1b (pink). Within the PA1b domain cystines (orange circles) and disulfide connectivity (orange lines connecting cystines) are given. N- and C-termini of PA1b are noted on both the bar diagram and the 3D representation illustrated below the bar diagram (purple 3D structure - adapted from PDB: 1P8B). Domain lengths are proportional to that observed in their respective sequences. b Schematic representation of the Clitoria ternatea Cter_M albumin-1 precursor with lengths in amino acids noted on the right of the bar diagram. The Cter_M precursor encodes a signal peptide (‘SP’ – yellow), an a-chain (blue), and the cyclotide Cter_M (pink). Within the Cter_M domain cystines (orange circles) and disulfide connectivity (orange lines connecting the cystines) are given. As depicted on both the bar diagram and 3D representation, the N- and C-termini of Cter_M are composed of Gly and Asn residues respectively and go on to form the ligation point leading to cyclic Cter_M (blue 3D structure adapted from PDB: 2LAM). c Example C. ternatea ‘UQ01’ phenotype. d Simplified phylogenetic tree of tribe Phaseoleae members adapted from the Fabaceae phylogenetic study of da Silva et al. 2023 showing relationships and divergence time estimates of last common ancestor at nodes in Mya37. The common name and number of non-redundant putative albumin-1 loci are given in parentheses. Red circle at basal-most branch represents the remainder of the phylogeny that includes tribes Loteae (ex. Lotus), Trifolieae (ex. Medicago), and Fabeae (Pisum).
Clitoria ternatea (butterfly pea) is a basal Phaseoleae taxon in subtribe Clitoriinae that produces >80 ultra-stable cyclic peptides called cyclotides, and variously called Cter, cliotide, or ctr peptides, which are encoded by albumin-1 genes by apparently replacing the b-chain domain with a cyclotide domain (Fig. 1a, b)8,9,10,11,12. Examination of other Clitoria species shows no evidence of cyclotides, suggesting they are restricted to C. ternatea, thus making albumin-1 evolution in this plant exceptional10.
It is unclear how the usurping of albumin-1 genes by cyclotide domains occurred, but structural similarities between b-chain peptides and cyclotides, namely the six Cys residues that form the disulfide knot and cystine spacing of the C1XXXC2 and C4XC5 motifs, suggest C. ternatea cyclotides arose from b-chain peptides to resemble cyclotides encoded in ‘dedicated’ (i.e. cyclotide specific) genes from Rubiaceae, Solanaceae, and Violaceae plants13. A comparison of precursor structures from cyclotide-bearing plant families highlights the unique structure of cyclotide-encoding albumin-1 genes from C. ternatea versus non-legume ‘dedicated’ cyclotide precursors (Supplementary Fig. 1). Key differences between cyclotides and b-chains are that cyclotide domains before cyclization terminate in Asn or Asp residues as these are required for cyclization, cyclotides exhibit a highly conserved Glu (C1XEXC2; Supplementary Fig. 2) whereas b-chains do not2,14. Cyclotides are posited to have emerged through convergence and biosynthetic parallelism due to their varied precursor structures and disjointed phylogenetic distribution15,16,17. Cyclization between the N- and C-termini of the cyclotide domain and certain residues in the inter-cysteine loops segregates cyclotides into topological classes. A cis-Pro residue in loop 5 confers a backbone twist and defines the Möbius topological group, whereas lack of the loop 5 Pro residue and at least one Gly residue in loop 2 defines the bracelet topological group; a third group called hybrid lacks the loop 5 Pro but contains at least one Gly residue in loop 2, thus hybrid cyclotides share traits of both classes (Supplementary Fig. 2)13,18. Cyclotides have attracted great interest because of their applications as stable peptide scaffolds in medicine and as eco-friendly pesticides in agriculture8,9,10,17.
Biosynthesis of mature cyclotides requires the activity of an asparaginyl endopeptidase (AEP). Members of this protease family typically function as peptide hydrolases19 but certain structural characteristics for select AEPs in cyclotide-bearing plants confer peptide ligase activity20,21. These ligase-type AEP features include two ‘ligase activity determinant’ (LAD1 and LAD2) motifs and the ‘marker of ligase activity’ (MLA), allowing for functional prediction based on sequence22,23.
Unlike typical albumin-1/b-chain expression, C. ternatea cyclotides are expressed throughout the plant, displaying expression patterns linked to biophysical properties, namely their isoelectric point and net charge values10. This plant-wide distribution, combined with the known insecticidal activity of cyclotides, implies that specific biophysical attributes may be fine-tuned to safeguard plant organs from biotic threats or indicate expanded roles for cyclotides8,10,17. C. ternatea cyclotides are approved and marketed as active ingredients in the insecticidal preparation Sero-XTM17. In contrast, typical Medicago truncatula albumin-1 genes are expressed in root tissue and N-fixing nodules (Alb-1 nodulins)2. M. truncatula AG41 and Pisum sativum PA1b are insecticidal b-chains located in nodules and seeds respectively, suggesting diverse roles for b-chain derivatives in different lineages2,3. Before the current study it remained unclear if b-chain peptides are present in C. ternatea, and what the impact of their conversion into cyclotide domains has had on the albumin-1 gene family.
Using a chromosome-level assembly of C. ternatea, we investigated the albumin-1 and AEP gene families and compared them to grain legumes. We identified 163 loci encoding albumin-1 precursors widely distributed across the genome, concentrated in four genomic islands, indicating a multi-fold expansion of the albumin-1 gene family in contrast to other Faboideae. Unlike the conventional b-chains which are replaced, cyclotides display a broader range of biophysical attributes, widespread expression in plant tissues, and co-evolution with ligase-competent AEPs. We present an evolutionary model elucidating the acquisition of cyclotide biosynthesis in C. ternatea.
Results
Clitoria ternatea genome assembly and annotation
We selected a self-fertile Clitoria ternatea ‘Milgarra’ accession (UQ01) previously examined by RNA-seq (Fig. 1c)10 and produced long-read PacBio Hifi data with 15.5-times coverage of the haploid (1n = 8) C. ternatea genome estimated to be 1.89 gigabase pairs (Gbp) in size by flow cytometry (Supplementary Table 1; Supplementary Data 1). Chromatin confirmation capture next-generation (Hi-C) sequencing resulted in a 1.72 Gbp assembly in 828 molecules and boosted coverage to ~67× (Supplementary Table 1). Pseudochromosomes from contigs were built with the 3D-DNA/Juicer pipeline for contact-map-guided refinement to improve the quality of the resulting genome assembly (Supplementary Fig. 3)24. An assembly consisting of eight pseudochromosomes containing 97% of assembled bases was produced and named Ct_HiC3, which comprises 1,721,557,839 bp in 859 molecules. Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis with the Fabales_obd10 non-redundant gene set revealed 97.5% of the gene set is present in Ct_HiC3 as complete orthologs (Supplementary Table 2), indicating a high level of completeness25.
Annotation of Ct_HiC3 guided by six organ transcriptomes resulted in 159,932 gene models (Supplementary Fig. 4)26. Manual curation of loci sharing homology to albumin-1 and AEP gene models was performed to refine the error prone automated annotation pipeline (see Data Availability for combined automated and refined annotation set). (Supplementary Fig. 4). Misannotated and overlapping albumin-1 and AEP gene models were observed in the original annotation set and were manually curated using known genes as guides (Supplementary Information - Method 1). For genes encoding albumin-1 and AEP loci, we depreciated dubious overlapping models while retaining sensical models and adding models discovered by TBLASTN, resulting a total of 160,040 gene models (Supplementary Information - Method 1)27,28. The curated set of gene models was captured as a predicted transcriptome and predicted proteome for BUSCO assessment to show >90% representation (Supplementary Table 2).
C. ternatea albumin-1 loci are expanded across the genome and exhibit increased diversity
We discovered 163 albumin-1 loci, including three putative pseudogenes with non-canonical Cys patterns that confounded characterization. Notably, no albumin-1 locus could be identified as containing a b-chain peptide in the genome, thus supporting the lack of b-chains in C. ternatea as suggested from transcriptomic analysis10. We performed RNA-seq quantification of organ transcriptomes mapped to a curated albumin-1/AEP gene set (Supplementary Data 2) and observed negligible expression (transcripts per million ratio (TPM) < 1) for these pseudogenes. Three additional sequences encoding an otherwise canonical cyclotide domain but missing a single Cys residue (Cter_nC series) were observed, one previously reported (Supplementary Data 2)10. Five loci were identified as encoding acyclotides (Cter_ac series), which lack an AEP-ligase recognition site but otherwise resemble cyclotides (Supplementary Data 2). The remaining 152 loci encode domains predicted to form cyclic knotted gene products representing ~93.3% of the albumin-1 loci identified in Ct_HiC3; 22 exhibited negligible expression (Supplementary Data 2).
Albumin-1 gene family comparisons, excluding pseudogene models and using manually curated gene models in select cases, between C. ternatea (n = 160), Cajanus cajan (n = 7), Glycine max (n = 3), Phaseolus acutifolius (n = 19), and Vigna unguiculata (n = 4) revealed that the albumin-1 gene family in C. ternatea is larger by factors of ~23, ~53, ~8, and 40× compared to C. cajan, G. max, P. acutifolius, and V. unguiculata respectively (Fig. 1d; Supplementary Data 3)29,30,31. Sequences in the 152 cyclotide-encoding loci revealed 85 unique cyclotide domains compared to the previously recorded 67 nucleotide-encoded peptides in CyBase (cybase.org.au)14. Overall, 35 cyclotide sequences were discovered in Ct_HiC3 that are absent from previous studies and/or CyBase (Supplementary Table 3)8,10,11. The 85 cyclotide species increased diversity in the loops between canonical Cys residues yet no encoded C-terminal Asp residues were observed, demonstrating that C-terminal Asp residues observed in proteomics data must arise exclusively from Asn deamination (Fig. 2a, b)12. Among the unique cyclotide species, 28 are found at multiple loci, with 11 occurring twice, seven thrice or four times, and one six times. Cliotide_T11 and Cter_R were each observed in nine loci. The cyclotides fall into three topological categories: bracelet (52), Möbius (10), and hybrid (23). The isoelectric point (pI) and charge at pH 5.5, relevant to the vacuole and secretory system, were calculated and plotted for cyclotides and b-chains (Fig. 2c). C. ternatea cyclotides exhibit a broader range (pI [3.67 to 10.21]; pH5.5 net charge [−1.9 to 7.6]) of these biophysical properties compared to b-chain peptides (pI [3.02 to 7.04]; pH 5.5 net charge [−4.7 to 1.6]; Supplementary Data 2 and 3). Notably, the pI and charge range for cyclotides is 6.54 and 9.5 respectively, versus 4.03 and 6.3 for b-chains. Thus, cyclotides have greater familial diversity for pI and net charge than other Phaseoleae b-chains.

Alignments of cyclotide domains were used as input, positions in alignment that occur as gaps in >90% of the input sequences were omitted, and positions in the alignment are numbered in the center of the wheel. Small circles with single letter amino acid codes are stacked around the wheel. Circle fill color represents residue category: orange = Cys, blue = cationic, red = anionic, green = hydrophobic, yellow = polar uncharged. Distance from center scales with decreasing rates of occurrence at each position around the circle. Filled black boxes with numbers appear between cystine residues and denote the loop number. a Diversity wheel of the 85 unique canonical cyclotide species encoded in the Ct_HiC3 assembly. b Diversity wheel of the 67 canonical cyclotide species present in CyBase.org.au evidenced by nucleotide sequencing prior to this work. c Scatter plot of calculated values for pI versus charge at pH 5.5 of b-chain peptides from C. cajan, G. max, P. acutifolius, and V. unguiculata, overlaid on the distribution of the same parameters for Ct_HiC3 cyclotides. PA1b, a functionally characterized b-chain from Lathyrus oleraceus [Lam.] (syn. Pisum sativum, garden pea) is shown as the exemplar Trifolieae insecticidal peptide. Each source species or peptide is given in the legend with the representative symbol for each.
We searched for the origins of the albumin-1 gene family C. ternatea through co-linearity analysis between G. max, P. acutifolius, and V. unguiculata, excluding C. cajan where gene models were not available. Albumin-1 genes were found on all Ct_HiC3 pseudochromosomes except Ct5 (Fig. 3), whereas albumin-1 genes were observed only on Glyma.08 and Glyma.13 of G. max, Phacu.CVR.002 and Phacu.CVR.011 of P. acutifolius, and Vigun11 of V. unguiculata (Supplementary Fig. 5; Supplementary Data 2 and 3)29,30,31. Putative syntenic loci between C. ternatea and the examined species were only observed on Ct1 and Phacu.CVR.011, comprising 925.6 kbp and 21 albumin-1 loci on Ct1, versus 230.7 kbp and 18 albumin-1 loci on Phacu.CVR.011. Support for synteny was poor because the Phacu.CVR.011 interval identified exclusively contained albumin-1 loci, thus lacking syntenic context (Supplementary Fig. 5). The inability to identify syntenic albumin-1 loci and wide versus narrow gene family distribution between C. ternatea and the Phaseolinae suggests albumin-1 genes rapidly evolve between lineages.

Pseudochromosomes, labeled Ct1 through Ct8, are depicted as purple shaded round-ended bars proportional to their size in megabase pairs and labeled with their size within the bar. Horizontal black bars across pseudochromosomes are locations of albumin-1 loci with loci information in black bound shapes. Loci names are given in non-bold font, encoded peptides are in bold and identified by peptide name, appended with ‘-n’ that represents which copy of the peptide is encoded by the relevant albumin-1 locus. Information after the peptide name is coded as depicted in the boxed legend: yellow circles note domains encoding mature cyclotides and give the topological category to which the cyclotide belongs (B = bracelet, H = hybrid, and M = Möbius), boxes containing “A” and “U” label acyclotide and unusual domains respectively. Cter_ac and Cter_nC are the prefixes for acyclic and minus-one Cys residue C. ternatea b-chain peptides respectively. Loci with the prefix Alb1l before a numerical designation are albumin-1-like loci that do not encode a b-chain or cyclotide-like sequence and are putative pseudogenes. Predicted pI is color-coded as shown in adjacent rectangles with predicted charge values at pH 5.5 printed in each rectangle. Horizontal orange bars across pseudochromosomes are AEP loci, with loci information in adjacent, orange-bound boxes. Green trapezoids on pseudochromosomes denote locations of the four albumin-1 enriched islands Ct1_4.53Mbp, Ct1_225.4kbp, Ct6_31.7Mbp, and Ct7_12.1Mbp.
Four regions enriched with cyclotide encoding genes were discernible in Ct_HiC3 and termed albumin-1 islands. Two such islands are found on Ct1, one on Ct6, and one on Ct7, with the sizes of the islands being ~4.53, ~225.4, ~31.7, and ~12.1 Mbp respectively, containing 29, 12, 48, and 28 albumin-1 loci respectively. We term these islands Ct1_4.53Mbp, Ct1_225.4kbp, Ct6_31.7Mbp, and Ct7_12.1Mbp, which when combined contain ~77% of the 152 loci predicted to encode mature cyclotides (Fig. 3). The most compact albumin-1 island, Ct1_225.4kbp, contains loci encoding two predicted retrotransposon sequences (gag-pol Ct1g_CT149963; Ty3-gypsy protein Ct1g_CT149967) and exhibits a repeating pattern of Cter_36-Cter_49-encoding gene pairs with Cter_36 loci displaying TPM < 0.7 (Fig. 3; Supplementary Data 2). The remaining cyclotide-encoding albumin-1 genes occur in smaller groups on Ct2, Ct3, and Ct4 encoding four, six, and 16 loci respectively, whilst Ct8 hosts a single albumin-1 encoding acyclotide cliotide_T22 (Ct8g_CT160055; Fig. 3). We observed a trend linking physical albumin-1 location in Ct_HiC3 to biophysical properties (pI, net charge at pH 5.5) and topology (bracelet, Möbius, or hybrid) of the encoded cyclotides (Fig. 3; Supplementary Data 2), suggesting localized tandem duplications within the genome and encompassing several albumin-1 loci are common.
Evolutionary signatures associate with increased cyclotide diversity
We examined albumin-1 loci encoding mature cyclotides in the four albumin-1 islands for non-neutral, positive, and purifying selection signals using pairwise and overall Z-tests (dN – dS). Previous examination indicated lower conservation in cyclotide domains compared to a-chains10. Two sequence sets were analyzed: (i) sequences encoding the cyclotide domain plus three C-terminal residues (cyclotide-CTR) and (ii) the remaining sequence downstream of the CTR containing the a-chain (a-chain; Supplementary Information - Method 2). Overall Z-tests for each island revealed significant non-neutral support for cyclotide-CTR gene sets, except for Ct6_31.7Mbp, which showed no positive selection support and significant purifying selection (Supplementary Data 4). The a-chain set also exhibited significant non-neutral and purifying selection support (Supplementary Data 4). Pairwise Z-tests within islands indicated non-neutrality between cyclotide-CTRs (Supplementary Data 5) and significant positive selection (Supplementary Data 6) between loci in Ct6_31.7Mbp and Ct7_12.1 Mbp when the calculated pI was <4 or >7 for one or both sequences. This finding suggests selective pressure influencing the expansion of the cyclotide pI range, as depicted in Fig. 2c; however, low or no expression of some loci weakens support of this trend (Supplementary Data 2). Additionally, sequences encoding the same cyclotide exhibited significant values for purifying selection compared to nearby loci (Supplementary Data 7). At the three loci encoding Cter_A, the cyclotide domain of Ct1g_CT159945(Cter_A-3) is being maintained as Cter_A (p = 0.0253) compared to the nearby loci Ct1g_CT159935(Cter_A-1) and Ct1g_CT159939(Cter_A-2). Similarly, among the Cter_R encoding domains of Ct1_4.53Mbp, Ct1g_CT126925(Cter_R-3) is being maintained as Cter_R compared to Ct1g_CT126919(Cter_R-1) and Ct1g_CT126924(Cter_R-2) with p = 0.0135 and p = 0.0067 respectively. The two Cter_B loci, Ct6g_CT005312(Cter_B-1) and Ct6g_CT160018(Cter_B-2) in Ct6g_31.7 Mbp display purifying selection (p = 0.0337). In the a-chain sets, similar trends were observed or weakly evident in comparison to cyclotide-CTR regions (Supplementary Data 8–10). Together, these trends of positive selection supporting biophysical property expansion, purifying selection to retain multiple loci of identical cyclotides, duplication, and diversification suggest this cyclotide-encoding albumin-1 gene family can perform varied and perhaps emerging biological roles.
Cyclotide distribution suggests expanded albumin-1 gene functions
Cyclotides and acyclotides occur throughout C. ternatea organs10,11. To support the biological relevance of previously known sequences, we validate the presence of select mature peptides in tissues throughout the plant by MALDI-MSI. We attempted to capture the expression patterns of cyclotides using MALDI-MSI but found that the large number of peptides of similar mass confounded analysis for some but not all masses (Supplementary Figs. 6, 7). Cyclotide distribution shows tissue-specific patterns, including subtle differential distribution of Met-oxidized Cter_M in shoots, and distinct tissues/cell layers of roots and nodules (Fig. 4, Supplementary Fig. 8). Example genome-identified cyclotides were observed in extracts of roots using MS-MS and validate Cter_60 and Cter_47 (Supplementary Fig. 9, Supplementary Fig. 10, Supplementary Data 11, Supplementary Table 4), demonstrating that some genome-identified sequences are detectable.

Each panel is divided into labeled rectangles bounding 10 µm cross sections of (1) mature leaf, (2) shoot, (3) secondary root, (4) primary root, (5) young nodule, and (6) senescent nodule cross section. In all panels, scale bar in (2) is 9 mm, MS imaging is based on [M + H]+ mass at 50 µm pixel size. In panels with MS data (b–h), a color scale bar indicating signal intensity for each molecular species is given, ranging from 0–100% signal, with values > 100% corresponding to dark red and value above bar relates to the upper bound of signal values for the indicated mass, all detected mass values were rounded to the third decimal place. a Optical image of organ sections before application of matrix used to acquire MS imaging data. b Distribution of 3227.452 ± 0.039 Da mass consistent with 3227.4528 Da theoretical mass of cyclic Cter_R. c Distribution of 3058.205 ± 0.04 Da mass consistent with 3058.2048 Da theoretical mass of cyclic Cter_M. d Distribution of 3074.211 ± 0.04 Da mass consistent with 3074.1961 Da theoretical mass of cyclic Cter_M with oxidized methionine (Cter_M_ox). e Distribution of 3098.392 ± 0.04 Da mass consistent with 3098.3848 Da theoretical mass of cyclic Cter_P. f Distribution of 3068.252 ± 0.04 Da mass consistent with 3068.2468 Da theoretical mass of linear Cter_70. g Distribution of 3084.328 ± 0.04 Da mass consistent with 3084.3698 Da theoretical mass of cyclic c_T1. h Distribution of 3249.425 ± 0.042 Da mass consistent with 3249.4598 Da theoretical mass of cyclic Cter_28.
Identification of an ancestral proteolytic AEP and ligase AEP cluster
Eleven AEP loci were identified, and gene models were refined through alignments with reported C. ternatea AEPs to reveal a total of ten full-length AEP loci (Supplementary Data 12). Gene symbols applied to these Ct_HiC3 AEPs followed historical numbering when the full-length model matched previously described fragments but used the CtAEP prefix, thus CtAEP6 and CtAEP10 here are full-length AEPs consistent with the fragments reported for butelase-6 and CtAEP1010,11,20.
The origin of ligase-type AEPs in the genome is relevant to the evolution of cyclotides in C. ternatea, and we used co-linearity analysis to identify ancestral AEPs in G. max, P. acutifolius, and V. unguiculata syntenic to the characterized ligase butelase-1/CtAEP110,20. A ~ 92.5 kbp region on Ct3 was identified containing four CtAEP loci syntenic to AEP-encoding loci on Glyma04, Glyma06, Phacu.CVR.009, and Vigun09 where the syntenic homologs occur as singletons (Fig. 5a)29,30,31. Pairwise identity percentages between syntenic and C. ternatea AEPs reveal that CtAEP15 exhibits greater similarity to syntenic Phaseoleae AEPs than to other CtAEPs in the ~92.5 kbp region (Fig. 5b). Alignments of LAD and MLA regions indicate that AEPs encoded near CtAEP15 resemble canonical ligase AEPs, specifically the hydrophobic MLA and high identity at LAD residues, whereas CtAEP15 shares features with the hydrolase CtAEP2 (Fig. 5c)32. The features present for each LAD and MLA of each AEP in the genome are given in Supplementary Data 12. In planta tests of AEP function were performed in Nicotiana benthamiana employing transient co-expression with the [T20K]kB1 precursor, derived from Oldenlandia affinis, as the substrate (Fig. 5d, e)33,34,35. CtAEP15 displayed increased linear products compared to mock and was similar to CtAEP2, confirming CtAEP15 as a hydrolase (Fig. 5d, e). CtAEP1 exhibited peptide ligase activity as previously known, with CtAEP10 activity similar to CtAEP1 demonstrating CtAEP10 is a canonical ligase AEP. CtAEP13 co-expression was not distinguishable from mock indicating CtAEP13 was non-functional, in accordance with CtAEP13 having negligible expression and a 10aa deletion (TPM = 1.95, Fig. 5d, e, Supplementary Fig. 11, Supplementary Fig. 12, Supplementary Data 12). CtAEP10, CtAEP15, and CtAEP16 amplified by RT-PCR matched the gene models of Ct_HiC3, supporting the conclusion that CtAEP10 is a canonical ligase AEP. Expression levels of CtAEP1 were higher than CtAEP10 except in roots, suggesting subfunctionalization of these AEPs post neofunctionalization of a progenitor ligase.

a Ct3 region containing an AEP duplication hotspot (Ct3 92.5 kilobase pair region between CtAEP13 and CtAEP15) syntenic to hydrolase-type AEPs in C. ternatea (blue), G. max (green), P. acutifolius (magenta), and V. unguiculata (black). Size of syntenic intervals for each species are given in bound boxes below the horizontal bar. Syntenic loci within the region are given as open arrowed boxes with orientation of transcription following arrow direction and labeled with the gene identifier. Loci encoding AEPs are given as filled boxes and labeled with gene symbols for C. ternatea. Unannotated open triangles represent non-syntenic genes annotated by FGENESH + + in C. ternatea. b Matrix of pairwise identity percent values for MUSCLE-aligned AEP zymogen without signal peptides. c Protein sequence alignments for ligase activity determinants 2 and 1 (LAD2 and LAD1 respectively), and the marker of ligase activity (MLA) between known hydrolase-type AEP CtAEP2 and ligase-type AEP CtAEP1. Manhattan plots below MLA residues are shaded in red (hydrophilic) and blue (hydrophobic) denote hydrophobicity patterns for the respective MLA sequence. d Relative mean amounts of each peptide product class normalized against internal control, shaded to indicate yellow = unprocessed, purple = linear, and green = cyclic. Bars for each CtAEP series are composed of stacked colored bars proportionate to the relative amounts of product type, and error bars representing SD are given for each biological replicate (n = 4, except for CtAEP2 where n = 3). e MALDI-TOF analysis of peptides produced in N. benthamiana transient expression assays using Oak1[T20K]kB1 precursor as substrates co-expressed with green fluorescent protein (mock), CtAEP2, CtAEP15, CtAEP1, CtAEP10, and CtAEP13. Representative traces are shown with masses plotted along the x-axis of m/z. Peaks are labeled above with codes describing the molecular species at each peak. As in d purple shaded peaks are linear products (linear minus N-terminal Gly, and linear), green shaded peaks are cyclic products, orange shaded peaks are internal control used to determine relative amounts, yellow shaded peaks note linear products not processed (unprocessed) by AEP.
Discussion
Examples of albumin-1 sequences are known from certain taxa in tribes Cicereae, Fabeae, Trifolieae, Phaseoleae, and in the unplaced Cladrastis clade2,36,37. The albumin-1 gene families of cyclotide-producing and non-producing taxa exhibit distinct evolutionary trajectories, evident in the C. ternatea genome by multi-fold family expansion, replacement of b-chains with cyclotides, and the broader biophysical properties and expression patterns compared to typical b-chains. The observation that multiple albumin-1 loci encode the same cyclotide suggests these sequences may be recent duplications or are conserved. Albumin-1 genes of Medicago (Trifolieae) are mostly limited to root and seed expression yet display family expansion (n = 52) across seven of eight chromosomes, similar to C. ternatea, suggesting genome-wide distribution is not linked to organism-wide expression or broader biophysical properties. Unlike other Faboideae, C. ternatea albumin-1 genes encode predominantly cyclotides and acyclotides, and not PA1b-like peptides. No distinct syntenic albumin-1 relationships were identified, thus we are unable to determine the syntenic ancestral albumin-1 gene in relatives. Compared to the other albumin-1 islands we describe, Ct6_31.7 Mbp contains the most diverse range of albumin-1 loci in terms of encoded cyclotides, acyclotides, and putative pseudogenes (Fig. 3). This observation suggests Ct6_31.7Mbp either contains the oldest albumin-1 loci, having had the opportunity to diversify and undergo gene birth and death, or Ct6_31.7 Mbp exists in a state of rapid change and requires further study to understand why Ct6_31.7Mbp is the most diverse.
Despite albumin-1 differences between C. ternatea, Phaseolinae, and Trifolieae, the insecticidal function of the albumin-1 gene family is retained, exemplified in entomological assays and the formulation of C. ternatea cyclotides into the commercial insecticide Sero-XTM,8,17. The archetypical insecticidal b-chain peptide, PA1b, and a similar b-chain from M. truncatula (AG41 peptide) are restricted to seeds and share the C4RC5 motif required for activity against insect cells2,38. In contrast, C. ternatea cyclotides are expressed throughout the plant, lack the C4RC5 motif, and are insect- and bacterial membrane disrupting peptides8,39. These observations suggest that b-chain and their cyclotide derivatives function in plant defense but employ different mechanisms. The detection of albumin-1 products in root and nodule tissue is another similarity with M. truncatula, and it is possible cyclotides or b-chain peptides may have a role in plant-microbe interactions similar to nodule-specific cysteine rich peptides, warranting further study in this context (Fig. 4b, e–h, Supplementary Fig. 8)1,2,40.
Our analysis supports the enzymes CtAEP1, CtAEP10, and CtAEP13 as derivatives of the ancestral CtAEP15. The data support an evolutionary model whereby duplication events followed neofunctionalization of an ancestral duplicate into a peptide ligase. Typical hydrolase AEPs exhibit some limited transpeptidation in vitro and in planta resulting in peptide ligation, suggesting recruitment of an AEP ligase occurred after the appearance of an Asn or Asp residue in the C-terminus of an ancestral albumin-1 b-chain19,41. This ancestral canonical ligase AEP underwent two additional duplication events resulting in three AEPs: CtAEP1, CtAEP10, and CtAEP13. Alignments show that CtAEP13 contains a 10aa deletion, a possible basis for its non-functionality, and exhibits a low TPM value suggesting the locus is undergoing pseudogenization. The most parsimonious model is that duplication of the CtAEP15 ancestor facilitated the biosynthesis of cyclic molecules derived from b-chain peptides through AEP neofunctionalization.
Of known cyclotide-bearing plants, C. ternatea is unique in the replacement of an existing defense peptide domain with cyclotide domains. The data indicates albumin-1 loci that encode cyclotides are evolutionarily favored, being members of a comparatively expanded and diversified gene family versus other Faboideae. We propose an evolutionary model of genome-wide b-chain succession to cyclotide domains via: (i) acquisition of a C-terminal Asx residue; (ii) native weak ligase activity of hydrolytic AEPs; (iii) the enablement of cyclotide biosynthesis by AEP duplication and neofunctionalization; (iv) cyclotide-encoding albumin-1 gene duplication and diversification to perform various roles (Supplementary Fig. 4)41. This work thus provides an evolutionary explanation for the origin of the ultra-stable family of plant cyclotides and supports their functional differentiation from conventional b-chains.
Methods
Plant material, DNA extraction, and genomic sequencing
A line isolated by single seed descent from Clitoria ternatea ‘Milgarra’ (Queensland Department of Primary Industries) was maintained in a controlled environment with a mean temperature of 26 °C at 16 h day-length under LED illumination (Valoya Oy, Finland; AP67 spectrum) in potting soil. Shoots were harvested and used as input for Hi-C sequencing using the Arima 4 library preparation and sequencing pipeline at The John Curtin School of Medical Research—Biological Research Facility (Australian National University, Canberra, Australia). Additional shoots were harvested and ground extensively with a mortar and pestle under liquid nitrogen then processed as per manufacturer’s protocol of the Circulomics Nanobind Plant Nuclei Big DNA Kit (PacBio, USA). High molecular weight DNA (10–15 kbp) was sequenced on a PacBio Sequel II with an 8 M SMRT Cell by the Australian Genome Research Facility (Australia).
Genome assembly and annotation to create Ct_HiC3
All reads were quality checked using FastQC software (v0.12.1 Babraham Institute, United Kingdom) and output reports used to define parameters for trimming and filtering steps. Cutadapt (v3.5) was used with sequential commands on PacBio Hifi raw data with the following parameters: -g AAGCAGTGGTATCAACGCAGAGTACT; -a AGTACTCTGCGTTGATACCACTGCTT; -u 10; -u -10 42. For Hi-C raw data, Trimmomatic (v0.39) was used with parameter set: HEADCROP:20 MINLEN:3043. Trimmed and filtered data were assembled using Hifiasm (v0.16.1) with the following parameters: -k57 -z20 -l0 --hg-size 1800m44,45. The assembled contigs and Hi-C data were used with 3D-DNA (Phasing branch 201008) and the Juicer pipeline as outlined by Dudchenko et al. before visualization and manual adjustment with Juicebox (v2.20.00)24. Following manual adjustment of the refined pseudochromosomes, the assembly was reconstituted as described in the pipeline documentation. RNA-seq data from six plant organs was used with StringTie (v2.2.1) to create a set of transcripts, followed by TransDecoder (v5.5.0) to obtain longest open reading frames and predicted proteins as per standard software protocol46,47. A UniProt proteome annotation set was obtained using BLASTP (BLAST+ v2.13.0) with parameters: uniprot_sprot.fasta -evalue 1e-5 -max_target_seqs 1 -outfmt 627. The resulting files were prepared for input into the FGENESH + + (Softberry Inc.) pipeline for gene prediction using the Phaseolus vulgaris gene set28. Gene annotations were applied to the genomic assembly and visualized in Geneious Prime (v2023.2.1, Biomatters Inc., New Zealand). See Supplementary Information - Method 1 for manual refinement of albumin-1 and AEP gene family loci.
Albumin-1 and asparaginyl endopeptidase family quantification, comparisons, and co-linearity analysis
Genomes and proteomes of G. max (Wm82.a4.v1), P. acutifolius (v1.0), and V. unguiculata (v1.1) were obtained through Phytozome (phytozome-next.jgi.doe.gov, v13) for use in calculating peptide masses and predicted biophysical properties, and for use in the MCScanX pipeline along with the Ct_HiC3 genome and annotation set (Supplementary Information - Method 3)48. Expression estimates were obtained against the curated set of gene models for both albumin-1 and AEP gene families combined (Supplementary Information - Method 4). Protein sequences from co-linear AEP loci were aligned with MUSCLE in Geneious Prime.
MALDI-MSI and MS/MS analysis
Plant material was cultured as described above. Leaf, shoot, secondary root, primary root, young nodule, senescent nodules were harvested fresh before embedding in SCEM under a hexane-dry ice mixture to freeze samples before using the film sectioning method of Kawamoto & Kawamoto (SECTION-LAB Co. Ltd., Japan) followed by cryo-sectioning into 10 µm sections49. Sections were mounted on glass slides and air-dried without rinsing to preserve peptides in the section. Sections were scanned with a Tissue Scanner II before spraying with 2,5-dihydroxybenzoic acid (30 mg/mL in ethanol) as the matrix. Matrix was applied with a SunCollect Sprayer (SunChrom, Germany) set for one layer at 10 µL/min, one layer at 20 µL/min, and eight layers at 40 µL/min. Matrix-coated samples were recrystallized with 5% isopropanol in a sealed petri dish at 55 °C for 3 min on a hotplate. Imaging was performed on a Bruker TIMS TOF Flex MALDI-2 system (Bruker, USA) calibrated using ESI-L Tune Time (Agilent, Australia, G1969-85000) in positive mode across m/z 1000-8000. Mass imaging data was handled with SCiLS Lab software (v2024a 12.00.15110, Bruker, USA, v2024a) with default settings for Bruker TIMS TOF Flex data and normalized using the root mean squared method. Images for specific m/z values were exported as TIFF files.
For MS/MS analysis, peptide extraction employed lyophilized plant organs (100 mg; n = 1 of each tissue type) consisting of leaves and shoots, roots, and nodules that were ground with a mortar and pestle separately and incubated overnight in 50% ethanol supplemented with 1% formic acid. Supernatants from each organ type were filtered through a 25 µm nylon mesh filter were lyophilized and reconstituted in 100 nM ammonium bicarbonate pH 8.0, only root extract was taken for cyclotide discovery. The MS/MS experiments were performed using a Shimadzu UPLC coupled with an AB Sciex 5600 TripleTOF High-resolution mass spectrometer using information dependent acquisition scanning on an Agilent Zorbax C18 300-Å column with dimensions of 100 × 2.1 mm, 1.8 μm. ProteinPilot (v5.0.2, AB Sciex) was used for the identification of proteins by searching the MS/MS data against the peptide sequences in Supplementary Data 2.
Asparaginyl endopeptidase validation and in planta assays
Coding sequences for CtAEP10, CtAEP15, and CtAEP16 were amplified using RT-PCR employing the SuperScript III kit (ThermoFisher Scientific, Australia) and Phusion polymerase kit (NEB, Australia). Amplicons contained attB flanks and were cloned into pDS221 before Sanger sequencing was performed to validate the gene models for the above AEPs50. See Supplementary Information - Method 5 for detailed methods including primers used for cloning CtAEP sequences and in planta assays. In brief, cloned AEPs, GFP, and cyclotide precursors were moved into pEAQ for transient co-expression of tested AEPs and cyclotide precursors33,35. Relative abundance estimates were calculated against spiked-in peptide control and data processed as previously described.
Statistics and reproducibility
Statistical tests involving nucleotide diversity were performed as detailed in Supplementary Method 2 (Supplementary Information). All tests performed on protein-coding nucleotide sequences were z-tests for selection, with tests of neutrality being two-tailed, and tests for positive or purifying selection being one-tailed. Statistical analysis of relative peptide amounts from in planta expression assays (Supplementary Method 5) were two-tailed Students t-tests. Reproducibility for in planta assays was assured through repeated experiments using different substrates to show consistent outcomes between tested enzymes and built in controls for variation by distribution of treatments across different plants and leaves and not performed in a blinded fashion. No data were excluded from analyzes except for a single replicate of CtAEP2 and noted in the source data and Fig. 5d legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw genomic sequence reads are deposited at the National Center for Biological Information (NCBI) Sequence Read Archive (SRA) under PRJNA1033355. Previously released RNA-seq data used in this study is available through NCBI-SRA under PRJNA286977. The Ct_HiC3 assembly is available via NCBI Genome under JAXHEB000000000. The entire set of gene models with refined albumin-1 and AEP models is available along with corresponding genome sequence via The University of Queensland eSpace Ct_HiC3 [https://doi.org/10.48610/757738c]. Albumin-1 and AEP gene model information available through NCBI-Genebank with accession numbers listed in Supplementary Data 2 and Supplementary Data 12 respectively. MS/MS data are available at The University of Queensland eSpace [https://doi.org/10.48610/0f8519a]. Source data and metadata (README files) for the source are available in the public repository [https://doi.org/10.48610/757738c]. All data deposited are available unrestricted.
References
Liu, Z. et al. Single-nucleus transcriptomes reveal spatiotemporal symbiotic perception and early response in Medicago. Nat. Plants 9, 1734–1748 (2023).
Karaki, L. et al. Genome-wide analysis identifies gain and loss/change of function within the small multigenic insecticidal Albumin 1 family of Medicago truncatula. BMC Plant Biol. 16, 63 (2016).
Higgins, T. J. et al. Gene structure, protein structure, and regulation of the synthesis of a sulfur-rich protein in pea seeds. J. Biol. Chem. 261, 11124–11130 (1986).
Hanada, K., Nishiuchi, Y. & Hirano, H. Amino acid residues on the surface of soybean 4-kDa peptide involved in the interaction with its binding protein. Eur. J. Biochem 270, 2583–2592 (2003).
Watanabe, Y. et al. A peptide that stimulates phosphorylation of the plant insulin-binding protein. Isolation, primary structure and cDNA cloning. Eur. J. Biochem 224, 167–172 (1994).
Gressent, F., Da Silva, P., Eyraud, V., Karaki, L. & Royer, C. Pea Albumin 1 subunit b (PA1b), a promising bioinsecticide of plant origin. Toxins (Basel) 3, 1502–1517 (2011).
Hashidume, T. et al. Identification of soybean peptide leginsulin variants in different cultivars and their insulin-like activities. Sci. Rep. 8, 16847 (2018).
Poth, A. G., Colgrave, M. L., Lyons, R. E., Daly, N. L. & Craik, D. J. Discovery of an unusual biosynthetic origin for circular proteins in legumes. Proc. Natl Acad. Sci. USA 108, 10127–10132 (2011).
Nguyen, G. K. et al. Discovery and characterization of novel cyclotides originated from chimeric precursors consisting of albumin-1 chain a and cyclotide domains in the Fabaceae family. J. Biol. Chem. 286, 24275–24287 (2011).
Gilding, E. K. et al. Gene coevolution and regulation lock cyclic plant defence peptides to their targets. N. Phytol. 210, 717–730 (2016).
Kalmankar, N. V., Venkatesan, R., Balaram, P. & Sowdhamini, R. Transcriptomic profiling of the medicinal plant Clitoria ternatea: identification of potential genes in cyclotide biosynthesis. Sci. Rep. 10, 12658 (2020).
Poth, A. G. et al. Discovery of cyclotides in the Fabaceae plant family provides new insights into the cyclization, evolution, and distribution of circular proteins. Acs Chem. Biol. 6, 345–355 (2011).
Park, S. et al. Cyclotide evolution: insights from the analyses of their precursor sequences, structures and distribution in violets (Viola). Front Plant Sci. 8, 2058 (2017).
Wang, C. K., Kaas, Q., Chiche, L. & Craik, D. J. CyBase: a database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res 36, D206–D210 (2008).
Mylne, J. S. et al. Cyclic peptides arising by evolutionary parallelism via asparaginyl-endopeptidase-mediated biosynthesis. Plant Cell 24, 2765–2778 (2012).
Gruber, C. W. et al. Distribution and evolution of circular miniproteins in flowering plants. Plant Cell 20, 2471–2483 (2008).
Oguis, G. K., Gilding, E. K., Jackson, M. A. & Craik, D. J. Butterfly Pea (Clitoria ternatea), a cyclotide-bearing plant with applications in agriculture and medicine. Front Plant Sci. 10, 645 (2019).
Craik, D. J., Daly, N. L., Bond, T. & Waine, C. Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 294, 1327–1336 (1999).
Saska, I. et al. An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. J. Biol. Chem. 282, 29721–29728 (2007).
Nguyen, G. K. T. et al. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 10, 732–738 (2014).
Harris, K. S. et al. Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase. Nat. Commun. 6, 10199 (2015).
Jackson, M. A. et al. Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases. Nat. Commun. 9, 2411 (2018).
Hemu, X. et al. Structural determinants for peptide-bond formation by asparaginyl ligases. Proc. Natl Acad. Sci. USA 116, 11737–11746 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res 10, 516–522 (2000).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Solovyev, V., Kosarev, P., Seledsov, I. & Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7, S10 11–S10 12 (2006).
Valliyodan, B. et al. Construction and comparison of three reference-quality genome assemblies for soybean. Plant J. 100, 1066–1082 (2019).
Moghaddam, S. M. et al. The tepary bean genome provides insight into evolution and domestication under heat stress. Nat. Commun. 12, 2638 (2021).
Liang, Q. et al. A view of the pan-genome of domesticated Cowpea (Vigna unguiculata [L.] Walp.). Plant Genome, e20319 https://doi.org/10.1002/tpg2.20319 (2023).
Serra, A. et al. A high-throughput peptidomic strategy to decipher the molecular diversity of cyclic cysteine-rich peptides. Sci. Rep. 6, 23005 (2016).
Sainsbury, F., Thuenemann, E. C. & Lomonossoff, G. P. pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J. 7, 682–693 (2009).
Poon, S. et al. Co-expression of a cyclizing asparaginyl endopeptidase enables efficient production of cyclic peptides in planta. J. Exp. Bot. 69, 633–641 (2018).
Jackson, M. A. et al. Plant-based production of an orally active cyclotide for the treatment of multiple sclerosis. Transgenic Res 32, 121–133 (2023).
Louis, S. et al. Broad screening of the legume family for variability in seed insecticidal activities and for the occurrence of the A1b-like knottin peptide entomotoxins. Phytochemistry 68, 521–535 (2007).
da Silva, F. G., de Moraes, R. M., Martins, L. S., Ramos, R. D. & Silva, G. C. Plastid marker-based phylogeny reveals insights into relationships among Papilionoideae species. Genet Resour Crop Ev https://doi.org/10.1007/s10722-023-01637-1 (2023).
Da Silva, P. et al. Molecular requirements for the insecticidal activity of the plant peptide pea albumin 1 subunit b (PA1b). J. Biol. Chem. 285, 32689–32694 (2010).
Nguyen, K. N. et al. Immunostimulating and Gram-negative-specific antibacterial cyclotides from the butterfly pea (Clitoria ternatea). Febs J. 283, 2067–2090 (2016).
Mathesius, U. Are legumes different? Origins and consequences of evolving nitrogen fixing symbioses. J. Plant Physiol. 276, 153765 (2022).
Gillon, A. D. et al. Biosynthesis of circular proteins in plants. Plant J. 55, 505–515 (2008).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Cheng, H. Y. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332 (2022).
BJ, H. TransDecoder. https://github.com/TransDecoder/TransDecoder (2022).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Kawamoto, T. & Kawamoto, K. Preparation of thin frozen sections from nonfixed and undecalcified hard tissues using Kawamoto’s film method (2020). Methods Mol. Biol. 2230, 259–281 (2021).
Du, J. et al. A bifunctional asparaginyl endopeptidase efficiently catalyzes both cleavage and cyclization of cyclic trypsin inhibitors. Nat. Commun. 11, 1575 (2020).
Acknowledgements
Support for this project was secured through the following: Clive and Vera Ramaciotti Facility for Producing Pharmaceuticals in Plants (D.J.C., Perpetual, Australia), Australian Research Council (ARC) Centre for Innovations in Peptide and Protein Science (K.B., D.J.C., CE200100012), ARC Linkage Project Eco-friendly Pesticides for Crop Protection (D.J.C., LP190100958) in partnership with Innovate Ag (Australia), ARC Discovery Project Understanding the Mechanisms of Peptide Cyclisation (E.K.G., DP200101299), and National Health and Medical Research Council Investigator Grant to D.J.C. (2009564). We thank Prof Jan Suda (Charles University, Prague, Czech Republic) for kindly performing genome size estimates using flow cytometry. Sequencing was supported by the National Collaborative Research Infrastructure Strategy funded Bioplatforms Australia with the use of the services and facilities of the Australian Genome Research Facility and the Biomolecular Resource Facility, Canberra. Computational resources were provided by the Australian FGENESH + + service supported by the Australian Biocommons and its partners and the Pawsey Supercomputing Research Centre with funding from the Australian Government and the Government of Western Australia. Additional computing resources were obtained through The University of Queensland’s Bunya high-performance computing cluster. The pEAQ system was graciously provided by Prof George Lomonossoff of the John Innes Centre and Plant Bioscience Ltd. (Norwich, United Kingdom).
Author information
Authors and Affiliations
Contributions
E.K.G., D.J.C., and K.B. conceived the project. E.K.G. and C.H. prepared samples and acquired genomic data. E.K.G. and K.A.F. assembled and annotated the genomic data. E.K.G. and W.L.H. performed gene family analyzes. L.T.T.N. and B.R.H. assisted in MALDI-MSI sample preparation and interpretation. K.Y. performed MS-MS analysis and M.A.J. performed AEP activity assays. E.K.G. wrote the manuscript and was revised by all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Marek Mutwil, Jonathan Chekan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gilding, E.K., Jackson, M.A., Nguyen, L.T.T. et al. Hijacking of N-fixing legume albumin-1 genes enables the cyclization and stabilization of defense peptides. Nat Commun 15, 6565 (2024). https://doi.org/10.1038/s41467-024-50742-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50742-x
- Springer Nature Limited