

Abstract
Synergistic combinations of immunotherapeutic agents can improve the performance of anti-cancer therapies but may lead to immune-mediated adverse effects. These side-effects can be overcome by using a tumor-specific delivery system. Here, we report a method of targeted immunotherapy using an attenuated Salmonella typhimurium (SAM-FC) engineered to release dual payloads: cytolysin A (ClyA), a cytolytic anti-cancer agent, and Vibrio vulnificus flagellin B (FlaB), a potent inducer of anti-tumor innate immunity. Localized secretion of ClyA from SAM-FC induces immunogenic cancer cell death and promotes release of tumor-specific antigens and damage-associated molecular patterns, which establish long-term antitumor memory. Localized secretion of FlaB promotes phenotypic and functional remodeling of intratumoral macrophages that markedly inhibits tumor metastasis in mice bearing tumors of mouse and human origin. Both primary and metastatic tumors from bacteria-treated female mice are characterized by massive infiltration of anti-tumorigenic innate immune cells and activated tumor-specific effector/memory T cells; however, the percentage of immunosuppressive cells is low. Here, we show that SAM-FC induces functional reprogramming of the tumor immune microenvironment by activating both the innate and adaptive arms of the immune system and can be used for targeted delivery of multiple immunotherapeutic payloads for the establishment of potent and long-lasting antitumor immunity.
Similar content being viewed by others

Introduction
Cancer immunotherapy, which aims to boost host immunity to eliminate cancer cells, has revolutionized the treatment of cancer patients1. However, only 20–30% of patients with cancer respond to immune checkpoint inhibitors. This can be due to a lack of pre-existing immunity or to involvement of alternative immunosuppressive pathways2,3. In light of this limitation, targeted anti-tumor approaches with advantages over conventional approaches might represent the next generation of cancer immunotherapies. These advantages include the capacity to activate both the innate and adaptive branches of immunity and increase the release of tumor antigens.
Tumor antigens can be divided into two major classes, tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs)4. TAAs are non-mutated self-antigens that are expressed on healthy cells and at elevated levels on tumor cells. TSAs, also referred to as neoantigens, are peptide epitopes generated by the non-synonymous mutation of malignant cells. Unlike TAAs, TSAs elicit strong tumor-specific immune responses in the absence of T cell-mediated central tolerance4,5. These antigens are the main targets of autologous tumor-specific T cells and can elicit a strong anti-tumor immune response4,6,7,8,9. However, even robust neoantigen-specific T cell responses typically fail to induce tumor regression. This is due, in part, to reduced presentation of neoepitopes by dendritic cells (DCs) to CD8+ T cells during priming10,11. One approach to promoting cross-presentation of tumor antigens by DCs is to enhance the release of damage-associated molecular patterns (DAMPs) such as the high mobility group box-1 (HMGB1) protein, calreticulin (CALR), or adenosine triphosphate (ATP), from stressed or damaged cancer cells undergoing immunogenic cell death (ICD)12,13. These factors act as immune adjuvants to further promote T-cell-mediated killing of cancer cells14.
Radiotherapy and some chemotherapeutic drugs such as oxaliplatin induce ICD of cancer cells, prompting them to release DAMPs, such as HMGB1, which in turn promotes cross-presentation by DCs via Toll-like receptor 4 (TLR4). This enhances activation of tumor antigen-specific T-cell responses and leads to tumor eradication15,16,17. However, the downregulation of MHC class I molecules, or the effects of immunosuppressive cells such as tumor-associated macrophages (TAMs), can induce loss of systemic tumor-specific T cells and subsequently reduce immunotherapeutic efficacy11,18,19,20. Therefore, in this study, our aim was to develop a strategy to increase the release of DAMPs and TAAs/TSAs from tumor cells, as well as induce TAM reprogramming, to improve both innate and adaptive immune responses against primary and metastatic tumors.
Recently, bacteria have emerged as a promising strategy for cancer therapy21,22. Live tumor-specific bacteria possess unique features, that offer advantages over conventional anti-cancer drugs; these include (1) tumor targeting and intratumoral penetration; (2) innate bacterial cytotoxicity; (3) ease of genetic manipulation to introduce therapeutic or imaging agents; and (4) biocompatibility via selection of probiotic or highly attenuated strains21,23,24,25,26,27,28. Attenuated Salmonella are the most studied bacterial species in the context of anti-cancer therapy21,29. However, such bacteria have shown inconsistent anti-tumor efficacy in different preclinical and clinical tumor models21,22,23,24,29,30,31,32. For this reason, genetic engineering techniques have been applied to load bacteria with anti-tumor agents as a way of enhancing their anti-tumor activity21,26,28,32,33,34,35.
Previously, we demonstrated that attenuated S. typhimurium defective in guanosine 5′-diphosphate-3′-diphosphate (ppGpp) synthesis (ΔppGpp S. typhimurium) induced the infiltration of abundant immune cells in the tumor microenvironment (TME)32,34. The ΔppGpp S. typhimurium (SAM) was engineered to express and secrete therapeutic payloads such as cytolysin A (ClyA) or heterologous bacterial flagellin (Vibrio vulnificus flagellin B, FlaB) in a controlled manner via the inducible bidirectional Tet system36. ClyA is a pore-forming cytotoxin expressed by Escheichia coli that contributes to cytotoxic activity toward mammalian cells37. As ClyA is known to form pores in cell membranes, which leads to ion imbalance and ultimately cell death38, we speculated that when ClyA is transported to cancer tissue, it might trigger ICD in cancer cells, which is characterized by the release of DAMPs and TAAs/TSAs. In addition, we reported previously that FlaB is a promising adjuvant that increases the polarization of M2 to M1 macrophages via the TLR5 signaling pathway32. The resulting reduction in the frequency of immunosuppressive TAMs may have the potential to inhibit tumor metastasis18,19,20,32.
Here, we ask whether SAM engineered to express dual payloads of ClyA and FlaB might potentiate the anti-tumor activity of SAM in primary and metastatic tumor models by increasing ICD and macrophage-activating capacity (Fig. 1a). Taking advantage of the inherent immunostimulatory nature of bacteria24,31,32, we show that secretion of ClyA within the TME increased tumor-specific T-cell-mediated cytotoxicity. Meanwhile, FlaB markedly inhibits the progression of both primary and metastatic tumors in mice without inducing systemic toxicity. Moreover, in this work, we demonstrate that treatment of tumors with SAM engineered to express ClyA establishes long-term immunological anti-tumor memory.

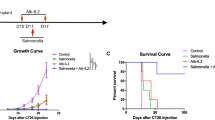
a Visual representation of the therapeutic mechanism underlying the mode of action of Salmonella (SAM) engineered to express ClyA and FlaB (SAM-FC). b Experimental scheme. Briefly, BALB/c mice were implanted subcutaneously (s.c) with CT26 cells. When tumors reached approximately 120 mm3 (day 0), the mice were randomly divided into five treatment groups: PBS; SAM-E; SAM-FR; SAM-CR; and SAM-FC. Mice received bacteria by intravenous injection (arrow). Bacteria-treated mice were then given Doxy (orally, 1.7 mg/kg) daily from day 3. ( + ) means Doxy was administered. The tumor-eradicated mice (survivors) were s.c rechallenged with CT26 cells on day 90. Naïve age-matched mice were implanted with CT26 cells used as controls. c Average growth curves of primary CT26 tumors [PBS (n = 24), SAM-E (n = 24), SAM-FR(+) (n = 21), SAM-CR (+) (n = 21), and SAM-FC(+) (n = 32)]. All mice were examined from three independent experimental replicates; *P = 0.0149, ****P < 0.0001; ns not significant; two-way ANOVA with Tukey’s multiple comparisons test. d Kaplan-Meier survival curves from (c) [*P = 0.0284, ***P = 0.0001 and ****P < 0.0001; Log-rank (Mantel-Cox) test]. e Average growth curves after CT26 tumor rechallenge [SAM-FR(+) (n = 6), SAM-CR(+) (n = 6), SAM-FC(+) (n = 16), and naïve mice (n = 9)]. All mice were examined from three independent experimental replicates; ****P < 0.0001; ns, not significant; two-way ANOVA with Tukey’s multiple comparisons test. f Experiment scheme. g Average growth curves of primary MC38 tumors [PBS (n = 9), SAM-E (n = 9), SAM-FR(+) (n = 10), SAM-CR(+) (n = 10), and SAM-FC(+) (n = 15)]. All mice were examined from two independent experimental replicates; **P = 0.0015, ***P = 0.0004, ****P < 0.0001; ns not significant; two-way ANOVA with Tukey’s multiple comparisons test. h Kaplan-Meier survival curves from (g) [*P = 0.0240, **P = 0.0017 in PBS vs SAM-CR(+), **P = 0.0014 in SAM-E vs SAM-FR(+), ****P < 0.0001; ns not significant; Log-rank in Mantel-Cox test]. i Average growth curves after MC38 tumor rechallenge [SAM-FR(+) (n = 7), SAM-CR(+) (n = 5 mice), SAM-FC(+) (n = 12), and naïve mice (n = 9 mice)]. All mice were examined from two independent experimental replicates; ***P = 0.0007 in SAM-FC(+) vs SAM-FR(+); ****P < 0.0001; ns, not significant; two-way ANOVA with Tukey’s multiple comparisons test.
Results
Design and characterization of engineered SAM in vitro and in vivo
The doxycycline (Doxy)-induced divergent expression plasmid pJH18, in which PTetA and PTetR show similar promoter strengths, was described previously36. Plasmids expressing payloads such as ClyA (C), FlaB (F), and Renilla luciferase variant 8 (Rluc8; R) were then constructed based on pJH18 and then transformed into SAM. The transformed SAM was named SAM-E (harboring empty pJH18), SAM-FR (harboring pJH18-FR), SAM-CR (harboring pJH18-CR), and SAM-FC (harboring pJH18-FC) (Fig. 1a and Supplementary Fig. 1). The TetR repressor (23 kDa) was expressed only in SAM transformed with pJH18 (Supplementary Fig. 2a). ClyA (34 kDa) and FlaB (43 kDa) were secreted from SAM-FC pellets only after Doxy induction (Supplementary Fig. 2b), as previously shown32,33,34,35,36,37,39,40. Expression of ClyA and FlaB (induced by Doxy ≥ 50 ng/ml) slowed the growth of bacteria in the stationary phase but did not change bacterial morphology (Supplementary Fig. 2c, d). ClyA and FlaB secreted from the transformed SAM exerted hemolytic effects and induced TLR5 simulation, respectively, only in the presence of Doxy (denoted by a “+”) (Supplementary Fig. 2e, f). The amounts and activities of ClyA and FlaB secreted from SAM-FC(+) were similar to those secreted from SAM-CR(+) or SAM-FR(+), indicating that the expression of one payload did not affect the expression of the other (Supplementary Fig. 2e–i). These results indicate that the ClyA and FlaB secreted by SAM-FC were fully functional.
Next, we measured the amounts of DAMPs in various tumor cells treated with bacterial supernatants to evaluate the extent of ICD. HMGB1 release, CALR expression on the cell surface, and ATP secretion were observed only in CT26, MC38, and 4T1 cells treated with supernatants from SAM expressing ClyA [SAM-CR(+) or SAM-FC(+)] (Supplementary Fig. 2j–l). Because HMGB1 is a ligand of TLR4, which plays a critical role in regulating both innate and adaptive immune responses, we evaluated DC activation after co-culture with CT26 cells treated with SAM-FC(+) supernatants (Supplementary Fig. 2 m). We found that the levels of DC activation makers (e.g., CD86, CCR7, IL-12p70, and IL-1β) increased significantly following treatment with SAM-FC(+) supernatant, but decreased markedly upon inhibition of HMGB1 and TLR4 by specific antibodies. These results indicate that ClyA-expressing SAM induced ICD in different types of cancer cells and increased DC activation by releasing HMGB1.
HMGB1 enhances the cross-presentation of tumor antigens by DCs by stimulating the TLR4 signaling pathway13,16. Therefore, we assessed whether SAM-CR(+) or SAM-FC(+) triggered expression of TAAs in tumor cells. The levels of TAAs, such as those derived from the CT26 proteins Aldh18a1, Mtch1, E2f8, and Glud1, increased significantly in CT26 cells treated with SAM-CR(+) or SAM-FC(+) supernatants (Supplementary Fig. 2n). This indicates that ICD induction by ClyA-expressing bacteria triggers release of tumor antigens from tumor cells.
In vivo bioluminescence imaging and biodistribution assays in SAM-FR(+)-treated CT26 tumor-bearing mice revealed that intravenously-injected SAM colonized and proliferated in tumor tissues. By contrast, the number of SAM in normal organs fell gradually, which is consistent with our previous observations (Supplementary Fig. 3a, b, and h)32,34. In situ production of anti-tumor payloads was also assessed in tumor tissues. Tumors colonized by SAM-FC(+) were characterized by increased flaB mRNA levels, which were 200-fold higher than those of the control groups (Supplementary Fig. 3c). ClyA expression was also identified in tumor tissues from the SAM-FC(+) group (Supplementary Fig. 3d). Expression of the SAM-FC(+)-associated plasmid was stably maintained in tumor tissues 48 h after Doxy induction (Supplementary Fig. 3i). This result is consistent with that of our previous study, which demonstrated that the pJH18 plasmid is well maintained in SAM during bacterial growth, probably because of bom gene activity, which prevents the division of plasmid-free bacteria36.
SAM-FC-mediated tumor regression in mouse tumor models
Next, we investigated the anti-tumor efficacy of SAM-FC in various tumor-bearing mouse models. In CT26 tumor-bearing mice, the strongest therapeutic effect was observed following treatment with SAM-FC(+), although treatment with plasmid free bacteria (SAM-E) also suppressed the tumors. Under this treatment condition with SAM-FC(+), the tumor was no longer detectable in 24 of 32 (75%) mice on day 12 after bacterial injection. We also observed complete tumor eradication in 12 of 21 mice treated with SAM-CR(+) (57.14%), and in 9 of 21 mice treated with SAM-FR(+) (42.86%) (Fig. 1b, c and Supplementary Figs. 4a, c, 5a), with no evidence of systemic toxicity (Supplementary Fig. 3e–h, and k-p) or body weight loss (Supplementary Fig. 3j). Only mice showing complete tumor eradication survived for over 90 days (Fig. 1d) after tumor challenge. Finally, treated animals that had remained CT26 tumor-free for 90 days were rechallenged by injection with twice the original number of CT26 cells into the contralateral flank (Fig. 1e, and Supplementary Figs. 4b, d, 5b). Notably, unlike the age-matched naïve controls and mice cured with SAM-FR(+), all of which developed tumors, mice cured with SAM-FC(+) or SAM-CR(+) remained tumor-eradication after rechallenge with CT26.
The therapeutic effect of SAM-FC( + ), SAM-CR(+), and SAM-FR(+) was also observed in MC38 tumor-bearing mice. Tumors were no longer detectable in 12 of 15 (80%) SAM-FC(+)-treated mice, in 5 of 10 (50%) SAM-CR(+)-treated mice, and in 7 of 10 (70%) SAM-FR(+)-treated mice on day 12. Moreover, the cured mice survived for over 90 days (Fig. 1f–h and Supplementary Fig. 4e, g). SAM-E significantly suppressed tumor growth but did not eradicate the tumors. In accordance with the CT26 model results, mice cured by SAM-FC(+) and SAM-CR(+) were resistant to rechallenge with MC38 cells (Fig. 1i and Supplementary Fig. 4f, h). These results strongly suggest that SAM engineered to express both of ClyA and FlaB could synergistically enhance a therapeutic anti-cancer vaccination effect (Supplementary Table 3). ClyA in particular may be essential for the activation of adaptive immunity and formation of protective immune memory following SAM therapy.
Characterization of immune responses induced by SAM-FC treatment
To assess the immune response elicited by SAM-FC treatment, CT26 tumors were excised from mice treated with the engineered SAM and subjected to immune cell analysis by flow cytometry (Fig. 2a). We found that treatment with SAM expressing FlaB [SAM-FR(+) and SAM-FC(+)] increased the number of M1-like macrophages and the concentrations of IL-1β and tumor necrosis factor (TNF)-α in the TME to a greater extent than the other treatments (Fig. 2b, Supplementary Fig. 6a, c, o, and p, and Supplementary Fig. 7a–c). As expected, SAM-FC(+), SAM-FR(+), and SAM-CR(+) reduced the M2-like macrophage population significantly compared with SAM-E and PBS controls (Fig. 2c, and Supplementary Fig. 7a–c). These results indicate that M2-to-M1 polarization was induced by treatment with FlaB-expressing SAM. The number of neutrophils was significantly increased in all mice treated with any of the bacteria (Supplementary Fig. 6b), suggesting that this increase was induced by SAM itself.

a Experimental schedule for SAM treatment of CT26 subcutaneous tumor (s.c)-bearing mice. Mice received the indicated amounts of SAM by intravenous (i.v) injection when their tumors had reached approximately 120 mm3 (day 0). Bacteria-treated mice were then given Doxy (orally, 1.7 mg/kg) daily from day 3. ( + ) means Doxy was administered. Tumors, spleens, tumor-draining lymph nodes (TdLNs), and blood were isolated on day 9 for flow cytometry (FACS) analysis. Ex vivo isolated tumor-infiltrating lymphocytes were restimulated with PMA/ionomycin in the presence of brefeldin A. b–g Frequency of isolated intratumoral M1-like macrophages (F4/80+CD86+) (b), M2-like macrophages (F4/80+CD206+) (c), activated DCs (CD11c+CD86+MHCIIHi) (d), Tregs (CD3+CD4+CD25+FOXP3+) (e), activated CD4+ T cells (CD4+FOXP3−IFN-γ+) (f), activated CD8+ T cells (GzmB+CD8+) (g) (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; ns not significant; unpaired two-tailed t-test). h, i Frequencies of L-2Ld MuLV gp70 peptide tetramer-bound (Tet+) CD8+ T cells in peripheral blood (left), TdLNs (middle), and tumors (right) (h); and exhausted/activated CD8+ T cells (PD-1+CD8+) (i) n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; ns, not significant; unpaired two-tailed t-test). j, k Frequencies of central memory CD4+ T cells (CD4+CD44HiCD62Hi) (j) and central memory CD8+ T cells (CD8+CD44HiCD62Hi) (k) in TdLNs (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; ns not significant; unpaired two-tailed t-test). l Experimental scheme for SAM-FC treatment of CT26 tumor-bearing BALB/c mice with T cell depletion. The mice were injected intravenously with 2 × 107 cfu SAM-FC(+). T cells were depleted by intraperitoneal administration of anti-CD4 (αCD4) or anti-CD8 (αCD8) antibodies (200 µg/mouse, i.p injection, twice per week for 5 times). IgG isotype was used as a control. m Average tumor growth curves (left) and representative images (right) of CT26 tumor-bearing BALB/c mice with or without T cell depletion after SAM-FC(+) treatment as described in (l) (n = 5 mice/group, from one experiment; ****P < 0.0001; ns, not significant; two-way ANOVA with Tukey’s multiple comparisons test).
Intratumoral growth of SAM expressing ClyA [SAM-CR(+) and SAM-FC(+)] increased the activation of DC populations (MHCIIHi IL-1β+ CD11c+ CD86+) in CT26 tumors (Fig. 2d and Supplementary Figs. 6d, 7a–c). This suggests that ClyA increased the ability of DCs to present tumor antigens. Additionally, tumors treated with SAM-FC(+) or SAM-CR(+) had significantly increased populations of activated CD4+ T (IFN-γ+ CD4+ FOXP3- and Ki-67+ CD4+ FOXP3−) and CD8+ T [granzyme (Gzm) B+ CD8+, Ki-67+ CD8+, and IFN-γ+ CD8+] cells, and a decreased population of regulatory T cells (Tregs) (CD3+ CD4+ FOXP3+ CD25+) (Fig. 2e–g and Supplementary Figs. 6e–g, 7a–c). These findings were further supported by immunofluorescence staining, which showed an increase in the number of anti-tumor M1-like macrophages, CD4+ T cells, and CD8+ T cells, but a decrease in the number of pro-tumor M2-like macrophages and Tregs, in the tumor tissues of mice treated with SAM-FC(+) (Supplementary Fig. 6k–n). These T cell responses were specific for TAAs, as indicated by the increase in the number of H-2LdMuLV gp70 peptide (SPSYVYHQF) tetramer-bound CD8+ T cells (Tet+ CD8+) in the peripheral blood, tumor-draining lymph nodes (TdLNs), and tumors of CT26-bearing mice after treatment with SAM-FC(+) or SAM-CR(+)41,42,43 (Fig. 2h and Supplementary Figs. 7d–f, 12m). This result was consistent with the cytokine profiles showing significantly higher amounts of IFN-γ, but lower amounts of IL-4, in the TME of SAM-FC treated mice than in the controls (PBS, SAM-E) (Supplementary Fig. 6q, r). Interestingly, the frequencies of programmed cell death protein 1 (PD-1)+ and cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4)+ T cells were significantly higher within the tumors of SAM-FC(+)-treated mice than within those of mice subjected to the other treatments, indicating that these T cells had either been activated or exhausted44,45 (Fig. 2i and Supplementary Figs. 6h–j, 7a–c). This result also indicates that SAM-FC(+) could be combined with PD-1 blockade to reinvigorate the tumor-specific CD8+ T cell responses.
Since the anti-tumor vaccination effect in CT26 or MC38-bearing mice was induced by SAM-FC(+) or SAM-CR(+), we next characterized memory T-cell populations in the TdLNs of mice treated with engineered SAMs (Fig. 2j, k and Supplementary Fig. 7a–c). We found that the frequency of central memory CD4+ T (CD4+ CD44Hi CD62Hi) and CD8+ T (CD8+ CD44Hi CD62Hi) cells increased significantly in SAM-FC(+)- or SAM-CR(+)-treated mice. A T-cell-dependent anti-tumor response was also verified in mice with T cell depletion using specific neutralizing antibodies. The anti-tumor response in SAM-FC(+)-treated mice was abrogated substantially by the depletion of CD4+ T and CD8+ T cells (Fig. 2l, m). This result is consistent with the results of the tumor rechallenge studies (Fig. 1e, i). Together, these findings demonstrate that, compared with all other treatment conditions, SAM-FC induced the optimal level of anti-tumor immunity, which implicated both the innate and adaptive immune responses and resulted in a long-lasting therapeutic effect.
SAM-FC suppresses metastases and primary tumors
Cancer metastasis induces immune tolerance mechanisms that reduce the efficacy of immunotherapy, resulting in high mortality rates18. To explore whether bacterial treatment reshaped the immune microenvironment of the metastatic organ, we established diverse mouse models.
Firstly, immunocompetent BALB/c mice with 4T1 tumors and spontaneous lung metastasis were generated by inoculating them with 4T1 cells into either the mammary fat pad or subcutaneously (Fig. 3a). The mice treated with SAM-FC(+) exhibited the strongest reduction in 4T1 primary tumor size, followed by SAM-CR(+), SAM-FR(+) and SAM-E on day 15 (Fig. 3b and Supplementary Fig. 9e, f). This finding indicates that ClyA-secreting bacteria inhibit the growth of 4T1 primary tumors more effectively than FlaB-secreting bacteria. However, complete tumor eradication was not observed with any treatment, including SAM-FC(+), indicating that although SAM-FC bacteria suppresses 4T1 tumors, its effectiveness is less marked than on CT26 tumors (Fig. 1b–e). By contrast, FlaB-secreting bacteria were superior to ClyA-secreting bacteria in inhibiting 4T1 lung metastasis. The mice treated with SAM-FC(+) and SAM-FR(+) exhibited the strongest inhibition in 4T1 lung metastasis. Treatment with SAM-E or SAM-CR(+) was more effective in reducing lung metastasis than treatment with PBS (Fig. 3c and Supplementary Fig. 9a). These findings suggest that FlaB-secreting bacteria are more potent in inhibiting metastasis than ClyA-secreting bacteria.

a Experimental scheme. Briefly, 4T1 cells were implanted into the mammary fat pad of BALB/c mice. When tumors reached approximately 140 mm3, mice received bacteria through i.v injection (day 0). Doxy (+) and CSF-1R antibody (αCSF-1R) were administered as shown. Pimary tumors and metastatic lungs were collected on day 15 and 35, respectively (b, c) and flow cytometry was performed on day 9 (d, e). b Average growth curves of 4T1 primary tumors (n = 6 mice examined from two independent experimental replicates; *P = 0.0144, ****P < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test). c Individual images (left) and metastatic nodule count (right) of lungs (n = 6 examined from two independent experimental replicates; ****P < 0.0001; ns not significant; unpaired two-tailed t-test). d Flow cytometry gating of M1 (F4/80+ CD86+) and M2 (F4/80+ CD206+) macrophages. e Ratio of M1/M2 macrophages in lungs (left) and tumors (right) (n = 6 mice examined from two independent experimental replicates; ****P < 0.0001; ns, not significant; unpaired two-tailed t-test). f In vivo and ex vivo bioluminescent imaging of MDA-MB-231-FLuc-GFP lung metastasis. Left, images of the mice on day 0 (pre-treatment) and on day 14 (post-treatment) and the excised lungs on day 14; right, quantitation of bioluminescence levels in the lungs. g Frequency of macrophages phagocytosing tumor cells (F4/80+GFP+) in the metastatic lungs of (f) (n = 3 mice/group, from one experiment, unpaired two-tailed t-test). h Experimental scheme. i Average growth curves of subcutaneous tumors (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test). j Kaplan-Meier survival curves (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; Log-rank (Mantel-Cox) test). k Metastatic scores of livers. Metastatic nodules on the liver surface were counted at the experimental end point. The scores were classified as follows: 0, 0%; 1, <20%; 2, 20%–50%; 3, >50% (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; unpaired two-tailed t-test).
We assessed whether the anti-metastasis effect of FlaB-secreting bacteria was mediated by functional macrophage polarization46. In BALB/c mice with orthotopic 4T1 primary tumors and lung metastasis (Fig. 3a), SAM-FC(+) and SAM-FR(+) showed similar inhibition of lung metastasis, while SAM-E and SAM-CR(+) showed a modest effect (Fig. 3c). This inhibition was consistent with the increased ratio of M1 (F4/80+ CD86+)/M2(F4/80+ CD206+) macrophages in the metastatic lung of mice treated with FlaB-secreting bacteria (Fig. 3d, e). A similar pattern of macrophage polarization was also observed in mice with 4T1 subcutaneous tumors (Supplementary Fig. 9b, c). These results were consistent with the shift of macrophage toward M1 phenotype in MC38 liver metastasis inhibited by treatment with SAM-FC(+) (Fig. 4c). In addition, the result of M1 polarized macrophages was consistent with the observed increase in the population of NK cells (CD3−CD49+) in the lungs, which might synergistically promote anti-metastatic outcomes (Supplementary Fig. 9h, j). Note that the increase in NK cells was not observed in the primary tumors treated with SAM-FR(+) or SAM-FC(+) (Supplementary Fig. 9j)47,48,49.

a Experimental scheme. C57BL/6 mice were implanted with MC38 cells simultaneously via the subcutaneous (s.c) and intrasplenic routes (SC + liver tumors) (day 0), and sequentially intravenously (i.v) injected with SAM-FC (2 × 107 cfu) on day 6 after tumor implantation. Bacteria-treated mice were then given Doxy (1.7 mg/kg, orally once daily) starting on day 3. The liver metastatic tumors and subcutaneous tumors were harvested on day 9 for flow cytometry analysis and immunofluorescence staining; (+) means Doxy was administered, (−) means no Doxy. b Frequency of CD45+ total leukocytes in the liver. Representative liver images (left upper panels). Flow cytometry plots of CD45+/− cells after live cell gating; the CD45+ cells are shown within the gates (left bottom). Quantitation of CD45+ cells (right) (n = 3 mice/group, from one experiment; unpaired two-tailed t-test). c–e Different macrophage subsets in the liver. c Classification of M1-like (CD80+ CD206−) and M2-like (CD80− CD206+) macrophages after gating on CD45+F4/80+ cells (left panels). Percentages of M1-like and M2-like subtypes within the total macrophage population (right panel) (n = 3 mice/group, from one experiment; ns not significant; two-way ANOVA with Tukey’s multiple comparisons test). FasL expression by M1- (d) and M2-(e) like macrophages (n = 3 mice/group, from one experiment; unpaired two-tailed t-test). f, g Frequencies of CD8+ T cells in different treatment groups (f) and the expression of Fas on CD8+ T cells (g) (n = 3 mice/group, from one experiment; unpaired two-tailed t-test). Please also see flow cytometry gating strategies in Supplementary Fig. 10m. h Immunofluorescence staining of CD8+ T cells isolated from the MC38 subcutaneous tumors of mice with subcutaneous tumor and liver metastasis treated with PBS or SAM-FC(−/+). The subcutaneous tumors were harvested and analysed on day 9, as described in (a). Scale bar, 50 µm.
To further explore the role of macrophages in the inhibition of 4T1 metastasis, we neutralized macrophages using the specific antibody αCSF-1R50. Neutralizing macrophages with αCSF-1R markedly reduced the M1/M2 ratio in both primary and metastatic lung tissues (Fig. 3d, e). αCSF-1R treatment also attenuated the inflammatory cytokine response (IL-1α and TNF-α) in the tumor (Supplementary Fig. 9g) and suppressed primary tumor growth and metastasis in the SAM-FC(+)-treated group (Fig. 3a–c and Supplementary Fig. 9e, f). Notably, SAM-FC(+) was able to significantly reduce metastasis in immunodeficient BALB/c athymic nu–/nu– mice bearing human MDA-MB-231-FLuc-GFP or 4T1-FLuc2 tumors, indicating that innate immune cells are more crucial for the inhibition of metastasis than T cells (Fig. 3f and Supplementary Fig. 9d). This finding was further supported by results showing increased phagocytosis of F4/80+ macrophages in mice treated with SAM-FC(+) (Fig. 3g). Together, these results indicate that FlaB-secreting bacteria induce functional M2-to-M1 macrophage polarization, which is essential for the inhibition of metastasis by phagocytosis.
We next analyzed the immunologic phenotypes in orthotopic 4T1 primary tumors and lung metastases treated with SAMs. The results showed that the mice treated with SAM-FC(+) and SAM-CR(+) resulted in more pronounced decreases in the number of Treg cells in primary tumors and metastatic lungs than treatment with PBS, SAM-E or SAM-FR(+) (Supplementary Fig. 9h, l). SAM-FC(+) and SAM-CR(+)-treated mice also exhibited increases in the CD8+ T cell population in primary tumors but not in lungs (Supplementary Fig. 9h, m). Consistent with this, ClyA-secreting bacteria significantly suppressed 4T1 primary tumors in immunocompetent BALB/c mice but not in immunodeficient BALB/c athymic nu–/nu– mice, which lack T-cell mediated immunity (Fig. 3b and Supplementary Fig. 9d). Therefore, we speculated that T-cell immunity induced by ClyA-secreting bacteria might be critical for inhibiting the growth of 4T1 primary tumors. Additionally, no significant changes in the numbers of neutrophils or activated DCs were observed after any of the SAM treatments (Supplementary Fig. 9h, i, and k). This indicates that secreted ClyA is more effective than FlaB in modulating Treg and CD8+ T cell responses in 4T1 primary tumors. Collectively, ClyA and FlaB, secreted from SAM, acted synergistically or in a complementary manner to reprogram tumor immune microenvironment, effectively suppressing both 4T1 primary tumor and its metastasis.
In separate experiments, we generated liver metastases by subcutaneous implantation and intrasplenic injection of MC38 cells (into C57BL/6 mice) (Fig. 3h)18 or by intraperitoneal injection of HepG2-FLuc (into BALB/c athymic nu–/nu– mice). SAM-FC(+) treatment significantly suppressed liver metastasis of MC38 and HepG2-FLuc tumors (Fig. 3i–k and Supplementary Fig. 10n, o). Also, we investigated the mechanism underlying the ant-tumor effects of SAM-FC in C57BL/6 mice with subcutaneous tumors and/or liver metastasis (SC or SC + liver) (Fig. 4a). After intravenous injection of SAM-FC, bacterial number increased in the livers of SC + liver group but decreased in the SC group, while liver weights were increased in both SC and SC + liver group (Supplementary Fig. 10a–d). This indicates that SAM-FC migrates to and proliferates in metastatic organs, thereby triggering immune cell responses. This finding was consistent with the increased leukocyte (CD45+) number in the metastatic liver of SAM-FC-treated SC + liver mice. The percentages of leukocytes in the livers of PBS, SAM-FC(−), and SAM-FC(+) groups were 6.6%, 11.5%, and 15.5%, respectively (Fig. 4b and Supplementary Fig. 10m), suggesting that ClyA and FlaB secreted by SAM-FC(+) were more effective in promoting leukocyte infiltration in metastatic lesions than bare bacteria [SAM-FC(−)]. The leukocyte population comprised mainly M1-like macrophages (F4/80+ CD86+) with low-level Fas ligand (FasL) expression, CD8+ T cells (CD45+ CD3+ CD8+) with low-level Fas expression, and proliferating CD4+ T cells (Ki-67+ CD4+ FOXP3−) (Fig. 4c, d, f–h and Supplementary Fig. 10l, m). This suggested that ClyA and FlaB expression strongly enhanced the anti-tumor activity of SAM by facilitating the recruitment and activation of immune cell cells in both primary tumors and metastatic livers. Notably, the frequencies of FasL-expressing M2-like macrophages (F4/80+ CD206+), PD-1 ligand 1 (PD-L1)-expressing myeloid-derived suppressor cells (MDSCs; PD-L1+ CD11b+ Gr-1+), and Tregs (CD4+ FOXP3+ CD25+) were significantly lower in the livers of the mice treated with SAM-FC(+) than in those of mice treated with SAM-FC(−) (Fig. 4c, e and Supplementary Fig. 10j, k, m). Moreover, SAM-FC(+) treatment also increased T cell infiltration into subcutaneous tumors significantly (Fig. 4h), which may have further increased tumor eradication with no evidence of toxicity (Fig. 3h–k and Supplementary Fig. 10e–i). Collectively, these data reinforce the notion that SAM-FC(+) treatment remodulated systemic immunity, thereby suppressing both metastatic and primary tumor growth and significantly prolonging survival without systemic toxicity.
SAM-FC enhances the HMGB1-mediated cross-presentation of TSAs
HMGB1 is secreted during the late stages of ICD, during which it binds to its receptor (TLR4) to accelerate processing of the phagocytic cargo in DCs, which ultimately promotes antigen presentation to T cells13,16,17,51. Using splenocytes and bone marrow (BM) derived DCs, we examined the potential role of HMGB1 in DC maturation and T cell activation with or without DC involvement (Supplementary Fig. 11a, b). We observed previously that TLR4 is expressed not only in DCs54 but also in splenic T cells (Supplementary Fig. 2m, 11c). This suggests that HMGB1 not only induces maturation of CD11c+ DCs (increased levels of CD86+, MHCIIHi, and CCR7+) but also directly stimulates CD3+ T cells (increased expression of TCRβ+, Ki-67+, and CD25+) by binding to the TLR4 receptor, even in the absence of DCs (Supplementary Fig. 11d–i). Notably, T cell activation markers were more elevated in recombinant HMGB1 (rHMGB1) + CD3+ T cells than in rHMGB1 + CD3+ T cells + CD11c+ DCs (Supplementary Fig. 11i). However, without the antigen-presenting process of DCs, these activated T cells lack anti-tumor capabilities. By contrast, HMGB1-stimulation of DCs, which increases antigen-presentation to T cells, results in the formation of T cells with effective anti-tumor responses. These findings highlight the dual role of HMGB1 in immune cell activation and responses52,53,54.
Because HMGB1 was released from tumor cells treated with SAM-FC in vitro, we next evaluated the role of HMGB1 in vivo in T-cell activation by dendritic cells in CT26 tumor-bearing mice. Neutralization of HMGB1 in mice was achieved by intraperitoneal administration of an anti-HMGB1 antibody (αHMGB1) (Fig. 5a). The therapeutic efficacy of SAM-FC(+) was more significantly reduced in mice treated with αHMGB1 than in those treated with the isotype control (Fig. 5b and Supplementary Fig. 12a). Consistent with this, significant decreases in various immune cell populations were observed in SAM-FC(+)-treated mice following HMGB1 neutralization. These included total DCs (CD11c+CD86+), proliferating CD4+ T cells (Ki-67+ CD4+FOXP3−), and proliferating CD8+ T cells (Ki-67+CD8+) within the spleens, activated DCs (CD11b+CD86+MHCIIHi), and activated CD8+ T cells (GzmB+CD8+) in tumors, and effector memory CD4+ T (CD3+CD4+CD44+CD62L−) and CD8+ T (CD3+CD4+CD44+CD62L+) cells in TdLNs, as well as tumor-specific CD8+ T cells (Tet+ CD8+) in TdLNs and peripheral blood. By contrast, the Treg cell (CD3+CD4+FOXP3+CD25+) population in tumor tisues was notably increased. (Fig. 5c–h and Supplementary Figs. 7a–f, 12b–e).

a Experimental scheme of HMGB1 neutralization in mice treated with SAM-FC. BALB/c mice were implanted subcutaneously (s.c) with 5 × 105 CT26 cells. Tumor-bearing mice were injected intravenously with PBS alone or with SAM-FC (day 0) when tumor volume reached approximately 120 mm3. An anti-HMGB1 antibody (αHMGB1) was administrated intraperitoneally (i.p) twice a week (starting on day 2). An isotype antibody (IgG) was injected as a negative control. Tumors, spleens, tumor-draining lymph nodes (TdLNs), and peripheral blood were obtained on day 12. The splenocytes were co-cultured with a pool of four TSAs derived from the CT26 proteins Mtch1, Aldh18a1, E2f8, and Glud1. The tumor-specific immune response was evaluated by flow cytometry and in ELISA assays. b Average tumor growth curves (n = 10 mice examined from two independent experimental replicates; ****P < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test). Frequency of intratumoral activated DCs (CD11c+ CD86+ MHCIIHi) (c), intratumoral Treg (CD3+CD4+FOXP3+CD25+) cells (d), activated intratumoral CD8+ T (GzmB+CD8+) cells (e), effector memory CD4+ T cells (CD3+CD4+CD44+CD62L−) in TdLNs (f), CD8+ T cells (CD3+CD4+CD44+CD62L−) in TdLNs (g), tumor specific CD8+ T (Tet+CD8+) cells in TdLNs (h) (n = 7 mice examined from two independent experimental replicates; ****P < 0.0001; unpaired two-tailed t-test). i Experimental scheme for evaluating the tumor-specific antigen (TSA)-specific T cell responses. CT26-bearing mice were treated with PBS, SAM-FC(−) or SAM-FC(+). Splenocytes were isolated on day 9 and co-cultured with TSA peptides overnight. The TSA-specific T cell response was then evaluated. Frequency of activated DCs (CD11c+CD86+MHCIIHi) (j), activated CD4+ T (CD45+CD3+IFN-γ+CD4+) cells (k), activated CD8+ T (CD45+CD3+IFN-γ+CD8+) cells (l) (n = 5 mice/group, from one experiment; unpaired two-tailed t-test). m The IFN-γ to IL-4 ratio produced by splenocytes; the cytokine concentrations were measured by ELISA (n = 5 mice/group, from one experiment; unpaired two-tailed t-test). Dimethyl sulfoxide (DMSO) and PMA/ionomycin were used as negative and positive controls, respectively.
To further verify whether SAM-FC induced TSA-specific T-cell responses, splenocytes from SAM-FC(+)-treated mice bearing CT26 tumors were co-cultured with a pool of four TSA peptides derived from the CT26 proteins Aldh18A1, E2f8, Mtch1, and Glud1 (Fig. 5i and Supplementary Table 4)4. Activated DCs (MHCIIHi CD11c+ CD86+), CD4+ T cells (IFN-γ+TNF-α+ CD4+), and CD8+ T cells (IFN-γ+ Ki-67+ CD8+) increased significantly in the SAM-FC(+)-treated group, and returned to baseline after αHMGB1 treatment (Fig. 5j–m and Supplementary Figs. 12f, g, l). These findings were supported by the results of the cytotoxicity and IFN-γ ELISpot assays (Supplementary Fig. 12h, i). Notably, cross-reactive T cell responses were activated more significantly by TSA peptides than by TAA peptides (Supplementary Fig. 12j, k). Moreover, the IFN-γ (Th1) to IL-4 (Th2) ratio increased significantly in the SAM-FC(+) group, and was reduced following HMGB1 neutralization (Fig. 5m), indicating that SAM-FC promotes the Th1 response, which requires HMGB1. Collectively, these data demonstrate that SAM-FC induced a robust T-cell-mediated anti-tumor immune response by promoting TSA cross-presentation, a process in which HMGB1 plays a primary immunoadjuvant role in the tumor-specific immune response.
An anti-tumor immune response is induced in bilateral tumors by unilateral intratumoral injection of SAM-FC
Next, we explored whether intratumoral injection of SAM-FC into unilateral tumors suppresses tumor formation at distal sites. Mice injected with CT26 in both flanks were treated with SAM in a unilateral fashion. After intratumoral injection of bioluminescent SAM (SAM-lux) and SAM-FR at one tumor site (proximal tumor), we observed bacterial trafficking to the other tumor (distal tumor) (Fig. 6a, b). Eventually, the bioluminescent signals of SAM-lux or SAM-FR(+) were observed in both proximal and distal tumors but were undetectable in normal organs. This suggests that SAM expressing anti-tumor payloads can migrate from one site to the other and localize to multiple tumors by intratumoral injection into a single tumor. Furthermore, the biodistribution of SAM-FC(+) was similar in both tumors, but negligible in normal organs (1 × 103 colony forming unit [cfu]/g) (Fig. 6c). Both proximal and distal tumors significantly regressed in the SAM-FC(+) group compared with the PBS and SAM-FC(−) groups (Fig. 6d and Supplementary Fig. 13a, b). Analysis of untreated distal tumors in the SAM-FC(+) group revealed an increased frequency of proliferating effector CD4+ (Ki-67+ CD4+ FOXP3−) and CD8+ (Ki67+ CD8+) T cells, but a decreased frequency of Tregs (CD4+ FOXP3+ CD25+), compared with the PBS and SAM-FC(−) groups (Fig. 6e and Supplementary Fig. 7a–c). These data indicate that intratumoral injection of SAM-FC(+) in the unilateral side successfully targeted distal tumors and induced robust innate and adaptive immune responses at these sites, thereby promoting systemic tumor eradication and prolonging survival (Supplementary Fig. 13c).

CT26 cells (1 × 106) were implanted subcutaneously (s.c) into both hind flanks of BALB/c mice. When the tumors reached approximately 120–150 mm3, mice received an intratumoral (i.t) injection of SAM-lux (4 × 108 cfu), SAM-FR (2 × 108 cfu), or SAM-FC (2 × 108 cfu) into a tumor in the left flank. a Representative bioluminescence images of a mouse treated with SAM-lux (n = 3 mice/group, from one experiment); images were taken before and after bacterial treatment. b Representative bioluminescence images of a mouse treated with SAM-FR (n = 3 mice/group, from one experiment); images were taken after intravenous injection of coelenterazine, 12 h after the oral adminstration of Doxy on the indicated day. The images of mice in the Doxy (−) group were taken 12 h before Doxy adminstration. c Biodistribution of SAM-FC. After i.t injection of bacteria, the indicated organs or tissues were harvested on day 1, 3, and 5 for viable bacterial count. (n = 3 mice/group, from one experiment; ns not significant; two-way ANOVA with Tukey’s multiple comparisons test). d Average growth curves of injected and uninjected distal tumors. Bacterial i.t injection (2 × 108 cfu) was performed as shown (upper panel). The volumes of treated (left bottom) and untreated (right bottom) tumors were measured every 3 days, starting on day 0. PBS, untreated control group (n = 5 mice/group, from one experiment; **P = 0.0027, ****P < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test). Please also see Supplementary Fig. 13. e Frequency of different immune cells within uninjected distal tumors. Uninjected distal tumors were obtained on day 9 and analyzed. Left, proliferating conventional CD4+ T cells (Ki-67+CD4+FOXP3−); middle, proliferating CD8+ T cells (Ki−67+CD8+); right, Tregs (CD3+CD4+FOXP3+CD25+) (n = 5 mice/group, from one experiment; ***P = 0.0001, ****P < 0.0001; unpaired two-tailed t-test). Please also see Supplementary Fig. 7a–c.
Discussion
In this study, we exploited the immunostimulatory properties of attenuated bacteria expressing the anti-tumor payloads ClyA and FlaB to effectively eradicate tumors via reprogramming of the tumor immune microenvironment. First, SAM expressing ClyA and FlaB demonstrated strong anti-tumor effects in diverse tumor-bearing mouse models. Second, the localized secretion of ClyA from bacteria promoted the release of TSAs/TAAs and DAMPs by inducing the ICD of cancer cells, which eventually established long-term anti-tumor memory. Third, SAM expressing FlaB promoted M2-to-M1 polarization in the TME via TLR4 and TLR5 signaling pathways, which markedly inhibited tumor metastasis12,18,27,32,55,56. Moreover, we demonstrated that the intratumoral injection of engineered bacteria exerted a tumor-suppressive effect in both SAM-treated and untreated tumors, which was similar to that elicited by intravenous injection of bacteria28.
We demonstrated that in situ production of ClyA via tumor-targeting SAM induced a potent T cell-mediated anti-tumor effect. Because ClyA induces cell death via the perforation of the cell membrane, many studies investigated the potential of using ClyA as an anti-tumor payload in bacteria-mediated cancer therapy25,34,36,38,55. Cholesterol, which is an important component of the plasma membrane, maybe a potent activator of ClyA38. This is especially true for cancer cells, which are enriched in cholesterol as it is critical for maintaining their softened cortical structure57. Previous studies show that cholesterol-regulated lytic ClyA activity is critical for membrane pore formation and subsequent induction of cancer cell death via apoptosis and necrosis25,55,57,58,59. Here, we showed that engineered SAM promoted ICD via the interaction between ClyA and tumor cell cholesterol. This led to the release of DAMPs (including HMGB1, CALR, and ATP), which promoted the processing and presentation of TAAs/TSAs, and eventually induced a potent anti-tumor immune response. This immune response not only suppressed tumor growth but also prolonged survival and prevented cancer recurrence. In addition, we clarified the molecular mechanisms underlying the induction of potent anti-tumor immunity by engineered SAM. First, we showed that ClyA induced the release of HMGB1 from dying cancer cells. Second, the binding of HMGB1 to its receptor TLR4 promoted the maturation of DCs and improved DC-mediated cross-presentation of tumor antigens to T cells. Third, improved DC-mediated T cell priming elicited more potent tumor eradication. Since activation of DCs by SAM-FC was abrogated significantly by neutralization of HMGB1 or TLR4, HMGB1, a major factor in DAMPs-mediated tumor-specific antigen presentation12,13,17,60, might act as an immunoadjuvant to boost TLR4-dependent DC activation and ultimately the efficacy of anti-tumor T cell responses.
Although HMGB1 induces TLR4-dependent DC activation, it may not be sufficient to trigger adaptive immunity in the absence of TSAs11,12,13. Therefore, the presence of TSAs in the TME plays a key role in the final maturation of professional antigen-presenting cells (mainly DCs) to improve the efficacy of anti-tumor immunity. This study demonstrates that treatment with ClyA-secreting bacteria not only leads to HMGB1 release, but also increases the availability of TSA peptides (including those derived from Aldh18a, Mtch1, E2f8, and Glud1), which induce potent T cell immunity. We surmised that a pool of non-self-peptide TSAs has the potential to alter the Th1/Th2 balance in favor of Th1, and subsequently promote stronger anti-tumor T cell responses than self-peptide TAAs. Subsequently, we demonstrated that, in various murine tumor models, treatment with ClyA-secreting SAM induced potent and long-term anti-tumor immunity, which led to the formation of tumor-specific immunological memory and protected mice from recurrent tumors. Indeed, almost all CT26 or MC38 tumor-bearing mice treated and cured with ClyA-secreting SAM were resistant to secondary rechallenge with the primary tumors; this was not achieved with FlaB-secreting SAM treatment. Moreover, the established long-term memory was specific for the primary CT26 tumor antigens as opposed to antigens derived from the 4T1 murine breast cancer cell line implanted into the same mouse strain (Supplementary Fig. 8a, b). Similar results were obtained using MC38 tumor-eradicated mice, in which only B16F10 cell-derived tumors but not MC38 cell-derived tumors grew (Supplementary Fig. 8c, d). These results are consistent with increased production of proinflammatory cytokines such as TNF-α, IL-1β, and IFN-γ, and a reduction in the levels of anti-inflammatory cytokines such as IL-4 (Supplementary Fig. 6o–r)24,27.
Recent research has demonstrated the essential role of TAMs, including anti-tumorigenic M1- and pro-tumorigenic M2-like macrophages, in controlling tumor metastasis19,56,61. M2-like macrophages possess unique features required for metastasis initiation and are regulated by integrated signals from immune-activating and immune-suppressive ligands, as well as cytokines such as IL-4, IL-10, IL-13, and IL-3519,20,56,61. Here, we found that among all treatment conditions used, bacteria secreting FlaB (i.e., SAM-FC or SAM-FR) were most effective at reducing both metastasis and tumoral growth in the mouse models. This therapeutic effect was consistent with the decrease in the M2-like, and the increase in the M1-like macrophage population within the TME. We also demonstrated that SAM-FC bacteria reduced the expression of Fas on CD8+ T cells, and FasL on hepatic M2- and M1-like macrophages, similar to the effect of radiotherapy, which is known to enhance the efficacy of immunotherapy against hepatic metastasis18. Moreover, a significant reduction in the frequencies of activated MDSCs expressing PD-L1, as well as Tregs, was also observed in mice treated with SAM-FC. Collectively, our data reveal a mechanism, whereby SAM-FC can alter hepatic peripheral tolerance, resulting in enhanced suppression of liver and subcutaneous tumor growth, and prolonged survival.
Because of the high level of PD-L1 expression on MDSCs within liver metastases, future studies should explore the combined use of anti-PD-L1 blockade and SAM-FC to induce TME remodeling. Although we explored the anti-tumor therapeutic potential of bacteria secreting ClyA by focusing on induction of ICD to release DAMPs (e.g., HMGB1, CALR, and ATP) and TAAs/TSAs (e.g., Aldh18a, Mtch1, E2f8, and Glud1) from tumor cells, we did not evaluate the role of ATP or CALR in anti-tumor immunity. We hypothesize that exposure of CALR on the cell surface, as well as ATP secretion, also plays important roles in DC maturation12,16, however, this will need to be explored in future studies.
In summary, engineering Salmonella to express and secrete dual anti-cancer payloads represents an approach to harnessing anti-tumor immunity for the conversion of “cold” tumors into “hot” ones, without inducing systemic toxicity. This study reports a rational design for using bacteria secreting ClyA and FlaB as immunoadjuvants (by increasing DAMP and TSA levels) to boost anti-tumor T-cell responses. Moreover, although SAM secreting either ClyA or FlaB alone was effective at eradicating tumors in C57BL/6 or BALB/c mice, respectively, the use of both anti-tumor payloads together induced more robust and durable anti-tumor responses against both primary and metastatic cancers and established long-term immunological memory. The effect of combined therapy with SAM-FC bacteria and immune checkpoint blockers on aggressive and poorly immunogenic tumor models is currently under study. The mechanisms underlying the immunoadjuvant effects of engineered SAMs will be elucidated in future studies with the aim of increasing the potential of cancer immunotherapy.
Methods
Ethic statement
All in vitro and animal care and experimental procedures were implemented in accordance with the guidelines of Chonnam National University Institutional Animal Care and Use Committee (NO. CNU IACUC-H-2016-15) and the National Centre of the Replacement, Refinement, and Reduction on Animals in Research36,62.
Study design
The main objective of this study was to develop a strategy for targeted cancer immunotherapy based on treatment with attenuated S. typhimurium engineered to express ClyA and FlaB. The attenuated Salmonella (SAM) were transformed with the plasmid pJH18-FC encoding clyA and flaB to generate SAM-FC. Tumor targeting of SAM-FC was evaluated by measuring viable bacterial counts and bioluminescence imaging. Expression and functional activity of ClyA and FlaB were assessed by performing western blotting, quantitative RT-PCR, hemolysis test, and the NF-κB binding assay. The therapeutic efficacy of the engineered bacteria was evaluated in mice bearing primary or metastatic tumors of mouse and human origin. The formation of protective memory after treatments was evaluated by rechallenging cured mice with tumor cells. The functional involvement of immunological factors such as DAMPs, TAAs/TSAs, and tumor-specific T-cells was examined by co-treating with neutralizing antibodies of HMGB1, CD4, or CD8 T cells in tumor-bearing mice, or by co-culturing splenocytes from mice treated with SAM-FC and/or Doxy with a pool of four TAA/TSA peptides. The immune responses elicited by SAM-FC were examined by FACS analysis, immunofluorescence staining, and enzyme-linked immunosorbent assay. Systemic toxicity after bacterial treatment was examined by evaluating clinical chemistry parameters.
Plasmids, bacterial transformation and DNA sequencing
The payload gene fragments were amplified with primers containing restriction enzyme sites by polymerase chain reaction (PCR). The pelB-FlaB gene fragment (1194 base pairs) was amplified with primers FlaB-PtetA-F(StuI) and FlaB-PtetA-R(SacI) against pBAD-pelB-FlaB plasmid32 as a template. The ClyA gene fragment (912 base pairs) was amplified with primers ClyA-PtetA-F(KpnI) and ClyA-PtetA-R(SalI) or primers ClyA-PtetR-F(StuI) and ClyA-PtetR-R(SacI) against pBAD-ClyA plasmid33 as a template. The Rluc8 gene (933 base pairs) was amplified with Rluc8-PtetR-F(KpnI) and Rluc8-PtetR-R(HindIII) primer against pBAD-Rluc8 plasmid33,63. The fragments were cut with the specific restriction enzymes and ligated into the same enzyme sites of pJH18 backbone plasmid35,36. The resulting plasmids are named pJH18-FR, pJH18-CR, and pJH18-FC, in which letters F, C, and R refer to genes encoding FlaB, ClyA, and Rluc8, respectively. All plasmids were confirmed by Sanger sequencing (Macrogen, South Korea) and has been deposited in the NCBI database. The simple maps of plasmids and primer sequences are shown in Supplementary Fig. 1 and Supplementary Table 1, respectively. These plasmids were transformed into S. typhimurium strains SAM by electroporation (Gene Pulser Xcell, Bio-Rad, USA) (Supplementary Table 2). In addition, auto-luminescent SAM carrying the lux operon (SAM-lux) were generated by P22HT int transduction33. The transformed bacteria were cultured at 37 °C in Luria-Bertani (LB) broth containing ampicillin (100 µg/ml) and stored at −80 °C as 25% glycerol stocks.
Measurement of TetR expression in absence of Doxy
The bacteria were freshly cultured overnight in ampicillin-containing LB broth. Bacterial pellets (8 × 108 cfu) were dissolved in the sample buffer and their proteins were separated by SDS-PAGE. TetR protein ( ~ 23 kDa) was detected with its specific antibody (10 µg/ml) in western blot analysis. The untransformed bacteria (SAM) were used as a control. M, size marker.
Cancer cell lines
CT26 (mouse colon carcinoma, CRL-2638), 4T1 (mouse breast cancer, CRL-2539), and HEK293T (human epithelial cell, CRL-3216) cell lines were purchased from the American Type Culture Collection (ATCC, USA). MC38 (murine colon adenocarcinoma, Kerafast ENH204-FP) and Luciferase/tdTomato dual-reporter MC38 (murine colon adenocarcinoma, Alstem LRL04) (MC38-Fluc-RFP) cell lines were purchased from Kerafast or Alstem Cell Advancements, respectively. The MDA-MB-231-FLuc-GFP (human triple-negative breast cancer, SC044, Gen Target, USA) and HepG2-FLuc (human hepatocellular carcinoma cells stably expressing firefly luciferase, HB-8065) cell lines were kindly provided by Dr. Mee Sun Yoon (Chonnam National University, Republic of Korea). CT26, MC38, and 4T1 were cultured in high-glucose Dulbecco’s Modified Eagles Medium (DMEM) (Welgene, Korea) or in Roswell Park Memorial Institute (RPMI-1640) medium (Welgene). MDA-MB-231-FLuc-GFP cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI) (Welgene, Korea). HepG2-FLuc cells were cultured in Minimum Essential Medium (MEM) (Welgene). All media contained 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and all cells were cultured at 37 °C in an incubator with 5% CO2. The cell lines were mycoplasma tested by the Waterborne Virus bank (Seoul, Korea) prior to experiments.
Expression of flaB gene in tumors colonized by SAM-FC
CT26 tumor-bearing BALB/c mice were injected intravenously with SAM-FR or SAM-FC (2 × 107 cfu), followed by oral administration of Doxy daily starting on day 3. After Doxy induction on day 3, total RNAs were obtained from tumor tissues treated with bacteria. The amount of flaB mRNA was measured by RT-PCR, normalized against that of the bacterial housekeeping gene aroC, and expressed as fold change. Total RNAs from tumor tissues of mice treated with SAM-E or SAM-FC cultured in vitro with Doxy [SAM-FC(+) in vitro] were used as negative and positive controls, respectively.
Western blotting to detect expression of ClyA and FlaB in in vitro cultured bacteria
Western blotting was used to detect FlaB and ClyA proteins within SAM. SAM was transformed with the plasmids and cultured overnight in LB medium containing ampicillin. The cultured bacteria were inoculated into fresh medium (1:100 dilution) and cultured further. On reaching an optical density of ~0.6 at 600 nm (OD600), 0–500 ng/ml of Doxy was added, and the bacteria were further cultured. After 5 h, the bacteria were pelleted by centrifugation at 140 g for 5 min. After washing with phosphate-buffered saline (PBS), the bacterial pellets were dissolved in a 2X sample buffer containing 0.2% β-mercaptoethanol (Sigma–Aldrich, USA) and incubated for 5 min at 95 °C. Bacterial supernatants were collected after centrifugation. The 1 OD600 bacterial pellets and supernatants (1 OD600 equivalents) were loaded into each lane of a 15% SDS-PAGE gel. After separation by electrophoresis, the proteins were transferred to a nitrocellulose membrane (GE Healthcare, MA, USA). The membranes were incubated in blocking buffer (5% [w/v] skim milk in Tris-buffered saline buffer containing 0.1% tween 20 [Sigma-Aldrich, Germany]; TBS-T) for 1 h at room temperature. After decanting the blocking buffer, an anti-FlaB antibody (diluted 1:20,000 in TBS-T) or an anti-ClyA antibody (diluted 1:250 in TBS-T) were added to the membranes, which were incubated for 2 h. After removing the primary antibodies by washing three times with TBS-T, secondary antibodies conjugated to horseradish peroxidase (HRP) were added to the membranes, which were incubated for 1 h. The membranes were washed three times with TBS-T and the chemiluminescent HRP substrate (Merck Millipore, USA) was added. The specific protein bands were detected using the ChemiDocTM XRS+ system imager (Bio-Rad, USA).
All of the antibodies used in this study are listed in Supplementary Table 5.
Assessment of ClyA and FlaB activity in in vitro cultured bacteria
The activity of ClyA secreted from the transformed SAM was measured on a red blood agar plate. Briefly, 1 μl of bacterial supernatant was spotted onto a blood agar plate and incubated overnight at 37 °C. Areas of blood cell lysis appeared as spots on the agar.
The activity of FlaB secreted from the transformed SAM was measured in human TLR5 (hTLR5)-transfected cells after the cells were treated with bacterial supernatants, as previously described32,64. Briefly, 2 × 105 HEK293T cells were transfected with p3xFlag-hTLR5 (100 ng), pNF-κB-Luc (100 ng), and pCMV-β-Gal (50 ng), and then mixed with 5 μl of Effectene (Qiagen, USA). At 24 h post-transfection, the cells were treated with bacterial supernatants (5 μg). Lipopolysaccharide-free recombinant FlaB proteins (400 ng in PBS, a gift of Dr. Joon Haeng Rhee, Chonnam National University Medical School) and PBS were used as positive and negative controls, respectively. After 18 h of incubation, the luciferase activity of each cell was measured using the MicroLumat-Plus LB 96 V luminometer (Berthold, USA). β-galactosidase activity in each cell was also measured after addition of 2-nitrophenyl-β-D-glucopyranoside (1.33 mg/ml). Plasmid transfection efficiency was normalized against that of untransfected cells.
Western blot images of HMGB1 from tumor cells
Tumor cells were treated with the supernatant (400 µg/ml total protein) from the bacteria cultured with or without Doxy (300 ng/ml) for 5 h. CT26, MC38, or 4T1 cells (5 × 105) were treated for 12 h with the bacterial supernatants. After SDS-PAGE separation, the cell culture supernatants were western blotted with an anti-HMGB1 antibody (1 µg/ml).
Establishment of tumor mouse models and bacterial injection
Female (6-week-old) BALB/c, C57BL/6, and BALB/c athymic nu–/nu– mice were purchased from Orient (Seongnam, Korea) and maintain in specific pathogen-free animal facility for one week prior to performing any experiments. The mice were grouped and housed 5 mice per plexiglass cage (22.3 × 26.8 × 12.0 cm), maintained 12 h/12 h at ligh/dark cycle in a temperature-controlled room (22 °C) and 40–60% humidity, with free access to water and food (PMI Nutrition International, LLC 4001 Lexington Avenue North, Arden Hills, MN 55126). Cages were arranged with full autoclave wood fiber (Northeastern Product Corp., Warrensburg, NY, 12885) and changed by clean cages 2–3 times/week. No enrichment material was used inside the cage. At the moment of the experiments, mice weighed approximately 19–20 grams and aged 7 weeks. The animals were randomly selected from different cages to house them in one clean cage for tumor implantation, and each mouse was used for experiment only once. All experimental processes were implemented from 9:30 to 6:00 pm. Euthanasia processes were implemented under deep anesthesia using isoflurane.
For the subcutaneous flank tumor models, CT26 (106), MC38 (5 × 105) or 4T1 (106) cells were injected subcutaneously (in 50 µl PBS) into BALB/c or C57BL/6 mice, which were anesthetized with 2% isoflurane prior to the injections. The tumor volume reached 100–120 mm3 at 8–10 days post-injection and received indicated types of engineered bacteria (2 × 107 cfu) via intravenous injection. Bacteria-treated mice were then given Doxy (orally, 1.7 mg/kg) every day from day 3; (+) means Doxy was administered. The CT26- or MC38-tumor-eradicated mice (survivors) were subcutaneously rechallenged with 2 × 106 CT26 cells or 1 × 106 MC38 cells, respectively, into the left flank on day 90. Naïve age-matched BALB/c or C57BL/6 mice were implanted with CT26 cells (1 × 106) or MC38 cells (5 × 105), respectively, were used as controls. To generate the 4T1 othortopic tumor model, 4T1 cells (1 × 106) were implanted into the mammary fat pad of BALB/c mice. When the tumor volume reached 120–140 mm3 at 18–20 days post-injection, the tumor-bearing mice were subjected to bacterial treatment.
An experimental mouse model harboring both subcutaneous and liver metastatic tumors was subsequently generated18. Briefly, MC38 cells were administered to mice simultaneously via both the subcutaneous (5 × 106 cells) and intrasplenic (5 × 105 cells) routes (SC + liver). 8 days after tumor implantation, the tumor-bearing mice were treated with PBS or SAM-FC (1.5 × 107 cfu) (day 0).
To generate the xenograft tumor models, MDA-MB-231-FLuc-GFP (1 × 107) cells were suspended in 100 µL of phosphate-buffered saline and Matrigel (BD Biosciences, San Jose, CA) at a 1:1 ratio and implanted into the mammary fat pad of BALB/c thymic nu-/nu- mice. 14 days after tumor implantation (Day 0), SAM-FC(−) or SAM-FC(+) (1 × 107 cfu) were i.v injected into mice.
For the modeling of human liver metastasis, 3 × 105 HepG2-FLuc cells were injected intraperitoneally into BALB/c athymic nu–/nu– mice (anesthetized with 2% isoflurane)65. After 2 days, SAM-FC (1 × 107 cfu) was injected intravenously (day 0), and then Doxy was given (day 3). In vivo bioluminescence imaging was performed on day 0 and 23.
For bacterial injection, SAM transformants were cultured overnight and then inoculated into fresh LB medium containing ampicillin (1:100 dilution), and further incubated until an OD600 of 2–2.5 was reached (early stationary phase). After centrifugation at 140 g for 5 min, bacterial pellets were washed twice with PBS. Bacteria (2 × 107 cfu in 100 μl PBS) were injected intravenously into the mice via the tail vein (day 0). To induce expression of cargo genes in the SAM transformants, 1.7 mg/kg/day Doxy (Sigma–Aldrich, USA) was administered via oral gavage to mice, once daily, 1 h after fasting, starting on day 3 after bacterial treatment. Tumor volume (mm3) was calculated every three days using the formula: (length × height × width) / 2. Mice with tumors that reached 1500 mm3 in volume were sacrificed following the guidelines of the Chonnam National University Institutional Animal Care and Use Committee. In mice that were found to exceed 1500 mm3, they were immediately euthanized under close monitoring by a veterinarian, and there was no distress. Mice that exhibited symptoms of pain, discomfort, or distress were immediately euthanized. Most of the data analysis and experiments were blinded to prevent bias.
DC maturation and the T-cell activation assay
Immature DCs (imDCs) were generated from the bone marrow (BM) of BALB/c mice, as described previously66. Briefly, BM cells were obtained from the femurs and tibiae of mice and cultured in RPMI-1640 medium (Gibco-BRL, USA) supplemented with 10% FBS and 1% penicillin-streptomycin in the presence of 20 ng/ml recombinant murine granulocyte-macrophage colony stimulating factor (GM-CSF) (R&D Systems, USA) and 10 ng/ml recombinant murine interleukin (IL)-4 (R&D Systems). On days 2 and 4, half of the medium was replaced with fresh medium containing cytokines. On day 6, the bound cells were detached from the culture dish using a cell scraper. The cells were then incubated with magnetic beads coated with anti-CD11c+ antibodies (Miltenyi Biotec, USA) for 30 min. After washing, CD11c+ imDCs were sorted using MACS Separators (Miltenyi Biotec) in accordance with the manufacturer’s instructions. To obtain mature DCs (mDCs), CD11c+ imDCs were further cultured for 48 h.
The activity of DCs was measured after co-culture with CT26 cells treated with SAM supernatant. Briefly, SAM was cultured after Doxy induction (300 ng/ml) for 5 h, and their supernatants were obtained by centrifugation and filtration. CT26 cells (5 × 105) were cultured overnight in 12-well plates and then treated for 12 h with bacterial supernatants (400 µg/ml total protein). To neutralize the released HMGB1, 10 µg/ml anti-HMGB1 antibody (ab18256, Abcam, US) was administrated to the cells 3 h before co-culture with 2 × 105 imDCs. A rabbit IgG (ab171870, Abcam) was used as a negative control. For TLR4 blocking, imDCs were treated with 10 µg/ml TLR4/MD-2 complex monoclonal antibody (MTS510, Invitrogen, USA) or the isotype control rabbit IgG (ab171870, Abcam) for 2 h before co-culture with the tumor cells. After a 24 h incubation, expression of DC markers (CD86, CCR7) was evaluated by flow cytometry, and cytokine productions (IL-12-p70, IL-1β) in the DC culture supernatants were measured in an enzyme-linked immunosorbent assay (ELISA). CO, control (untreated cancer cells + imDC cells).
T-cell activation was measured in the absence or presence of imDCs and rHMGB1. Briefly, T cells or DCs were isolated from the spleens or BMs of BALB/c mice using CD3+ and CD11c+ magnetic beads, respectively. Then, the same numbers of DCs and T cells (5 × 105) were co-cultured with or without rHMGB1 (20 μg/ml) for 24 h. The activation markers of T cells (TCRβ, Ki-67, and CD25) were measured by flow cytometry with specific antibodies. Also, the expression of TLR4 was measured in T cells by flow cytometry.
Expression of tumor associated antigens (TAAs) in CT26 cells after treatment with SAM-FC
Bacteria were cultured with or without Doxy (300 ng/ml) for 5 h. CT26 cells were treated with the bacterial supernatants for 12 h. Antibodies specific for CT26 TAAs including Aldh18a1 (0.4 µg/ml), Mtch1 (1 µg/ml), E2f8 (0.1 µg/ml), and Glud1 (0.14 µg/ml) were used for western blotting.
In vivo bioluminescence imaging
In vivo bioluminescence imaging was also performed using IVIS lumina (Perkin Elmer) to determine metastatic involvement; in vivo bioluminescence signals of MC38-FLuc-RFP, MDA-MB-231-FLuc-GFP, and HepG2-FLuc cancer cells were induced by intraperitoneal injection of 150 mg/kg D-luciferin.
At 12 h post-treatment with bacteria (2 × 107 cfu) carrying the Rluc8 reporter gene (SAM-FR), tumor-bearing mice were orally administered Doxy. To produce bioluminescence signals in vivo, coelenterazine (0.7 mg/kg body weight in 200 µl of PBS; Biotium, Hayward, CA) was injected intravenously via the tail vein. SAM-lux bacteria (4 × 107 cfu) were used to monitor in vivo bacterial migration after intratumoral injection into one of the two tumors in dual tumor models. The bioluminescent signals within the area of interest were measured in maximum photon per second per centimeter square per steradian (p/s/cm2/sr) units35.
Real-time polymerase chain reaction (RT-PCR) to detect bacterial FlaB gene expression in tumors
Tumor-bearing mice were treated with Doxy on day 3 after bacterial treatment. After 12 h, tumor tissues were obtained and soaked in 1 ml of RNAprotect Bacteria Reagent (Qiagen, Germany) at 4 °C. Then, total RNA was isolated using the RNEasy Mini Kit (Qiagen, Germany), according to the manufacturer’s protocol. After quantifying total RNA, cDNA was generated from 1 μg of total RNA using the TOPscript cDNA synthesis Kit (Enzynomics, Korea). RT-PCR was performed using the TOPreal qPCR 2X premix multiplex probe (Enzynomics, Korea) in the presence of CYBR green. RT-PCR products were quantified on a Rotor-Gene Q RT-PCR cycler (Qiagen, Germany) by following the manufacturer’s protocol. The following PCR conditions were used: 10 min at 95 °C, followed by 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 15 s for 40 cycles. The following PCR primer sets were used: (1) for the Salmonella housekeeping aroC gene: Forward, 5′-TCGCCGATCTCCACGCCTT-3′; Reverse, 5′-GCGCGAAAGTGACGGTGATG-3′); and (2) for flab: Forward, 5′-GAGCGTCTGTCTTCAGGTT-3′; Reverse, 5′-GTTGTAGGATGTTGGTGGTC-3′. The mRNA levels of flaB were normalized against those of aroC, as previously described32.
Detection of ClyA proteins in tumors by western blotting
Tumor tissues were homogenized in protein extraction solution (20 mM Tris-Cl pH 6.8, 150 mM NaCl, 5 mM EDTA, 1% NP-40, and 1× Xpert protease inhibitor cocktail solution [GenDEPOT, USA]) and passed through a 0.25-µm filter. Proteins (120 µg) were separated on a 15% SDS-PAGE gel and then transferred to a nitrocellulose membrane (GE Healthcare, Germany). Western blotting was performed with an anti-ClyA antibody (diluted 1:500 in TBS-T).
Bacterial biodistribution in tumor-bearing mice
After bacterial injection, organs or tissues from tumor-bearing mice were harvested, weighed, and homogenized in PBS using the IKAT 10 Basic ULTRA-TURRAX homogenizer (IKA Dispersers, Germany). The samples were serially diluted in PBS and spread on an LB agar plate containing ampicillin. After culture overnight at 37 °C, the colonies were counted to determine the number of bacteria (expressed as cfu per g of organ) in each sample.
Evaluation of plasmid loss in tumors colonized by SAM-FC
CT26 tumor-bearing BALB/c mice were injected intravenously with SAM-FR or SAM-FC (2 × 107 cfu), followed by oral administration of Doxy daily starting on day 3. The bacteria in tumor tissues were isolated at the indicated times and cultured on agar plates with (+) or without (−) ampicillin. Bacterial colonies were counted after overnight culture.
Clinical chemistry parameter
Blood samples were consistently collected during the same time interval in the afternoon (15:00 to 16:00) via heart puncture using heparinized syringes. The blood was transferred into serum separator gel tubes (Microtainer, BectonDickinson, Franklin Park, NJ) and centrifuged at 9300 g for 30 min at 4 °C for serum separation. Following centrifugation, the serum was immediately used for biochemical analyses. The serum activity of aspartate aminotransferase and alanine aminotransferase, along with concentrations of blood urea nitrogen, creatinine, C-reactive protein, and procalcitonin, were measured using an automated analyzer (Hitachi Instruments, Tokyo, Japan) in accordance with the manufacturer’s instructions. Standard controls were run prior to each determination, and the values obtained for the various biochemical parameters consistently fell within the expected ranges.
Mouse antibody depletion experiments
To deplete specific T-cell subsets, mouse anti-CD4 (GK1.5, Bio X Cell, USA) or mouse anti-CD8α (2.43, Bio X Cell, USA) antibodies in PBS (10 mg/body weight kg) were injected intraperitoneally into mice 1 day before bacterial therapy; the anti-CD4 and anti-CD8α antibody injections were repeated twice weekly throughout the study period. A rat IgG2b antibody (10 mg/body weight kg, LTF-2, Bio X Cell, USA) was used as an isotype control. Depletion of CD4+ and CD8+ T cells was confirmed by flow cytometry analysis of mouse spleens.
To neutralize macrophages, anti-mouse CSF1R (CD115) (20 mg/body weight kg, BE0213, Bio X Cell, USA) monoclonal antibody in 200 µl PBS were i.p injected into tumor-bearing mice. Antibody injections were repeated 5 times twice weekly throughout the study period. A rat IgG2a isotype antibody (20 mg/body weight kg, Bio X Cell, USA) was used as an isotype control.
To neutralize HMGB1, a rabbit anti-HMGB1 antibody (25 µg/injection, ab18256, Abcam, UK) in PBS was injected intraperitoneally into mice 12 h after Doxy induction, and further injected twice weekly throughout the study. A rabbit IgG isotype antibody was used as a control (25 µg/injection, ab171870, Abcam, UK). At 2 weeks post-antibody treatment, tumor growth was measured and splenocytes were subjected to flow cytometry analysis.
Cytokine detection by ELISA
The concentrations of cytokines IL-12p70, IL-1β, IL-4, and interferon (IFN)-γ were measured using ELISA kits (Invitrogen).
Flow cytometry
Immunophenotyping of tumors was described previously67. Briefly, solid tumors, TdLNs, spleens, and well perfused metastatic livers were extracted from mice on day 9 or 12 after bacterial treatment and soaked in 2 ml of isolation buffer (RPMI 1640 medium containing 5% FBS, 1% L-glutamine, 1% penicillin-streptomycin, and 10 mM HEPES). After mechanical homogenization, the cell preparations were treated with 1 mg/ml collagenase type IV (Roche, Switzerland) and 50 μg/ml DNase I (Roche, Switzerland) and then incubated for 45 min at 37 °C. Next, 2 ml of each sample were mixed with an equal volume of 1× lysis buffer (BioLegend, USA) and incubated for 4 min at 37 °C to remove red blood cells. The cells were then filtered through 40-μm cell strainers (BD Falcon, USA). Finally, the cells were washed sequentially with isolation buffer and FACS buffer (2% FBS in DPBS). Cells were then incubated for 10 min at room temperature with Fc blocking buffer (BioLegend) to prevent non-specific binding. The samples were then stained for 30 min on ice with specific fluorescent-dye-conjugated antibodies according to manufacturer’s instruction (listed in Supplementary Table 5). The samples were acquired on a FACS Canto flow cytometer (BD Biosciences, USA) and data were analyzed using FlowJo software (Becton, Dickinson & Company, USA). A minimum of 106 cellular events were recorded per sample.
To detect tumor-antigen-specific cytokine production by T cells, tumor-infiltrated lymphocytes (TILs) were stimulated for 6 h at 37 °C in a CO2 incubator with a 2 µl/ml 12-myristate 13-acetate (PMA)/ionomycin cell activation cocktail in the presence of Golgi Plug (containing brefeldin A) (BD Biosciences). The cells were then stained with specific antibodies according to manufacturer’s instruction (listed in Supplementary Table 5).
Evaluation of anti-tumor splenocyte responses
Activation of T cells in response to specific tumor antigens was measured as described previously4. Briefly, splenocytes were collected from mice 9 days after bacterial treatment. Next, 106 splenocytes were transferred to each well of a 24-well plate containing RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin, and 20 ng/ml recombinant mouse (rm)IL-2 (R&D systems, USA). The cells were then incubated overnight with a mixture tumor peptide antigen derived from the proteins Aldh18a1, Mtch1, E2f8, and Glud1 (2 µg of each/ml). The splenocytes were then subjected to intracellular IFN-γ staining and the lactate dehydrogenase (LDH) release assay. The cell supernatants were used for cytokine concentration quantification by ELISA. During intracellular IFN-γ staining, the stimulated splenocytes were treated for 5 h with a protein transport inhibitor (containing Brefeldin A; BD Biosciences). Next, 15% dimethyl sulfoxide (Sigma-Aldrich, USA) and a 2 µl/ml PMA/ionomycin cell activation cocktail (with Brefeldin A) (BioLegend) was used as negative and positive controls, respectively. The splenocytes were harvested and stained with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (0.5 µl per106 cells, Invitrogen), and antibodies specific for mouse CD45.2 (0.25 µg per 106 cells, Clone 104, BioLegend), CD3 (0.25 µg per 106 cells, clone 17 A, eBioscience, USA), CD4 (0.25 µg per 106 cells, clone GK 1.5, BioLegend), and CD8a (1 µg per 106 cells, clone 5.3–6.7, BioLegend). The cells were then permeabilized with FOXP3 fixation buffer (eBioscience) and fixed with 1% paraformaldehyde (in DPBS), according to the manufacturer’s instructions. After washing, the cells were stained with anti-mouse antibodies against IFN-γ (0.125 µg per 106 cells, clone XMG1.2, eBioscience) and FOXP3 (0.5 µg per 106 cells, clone FJK-16s, eBioscience). The samples were acquired on a FACS Canto flow cytometer (BD Biosciences) and the data were analyzed using FlowJo software (Becton, Dickinson & Company, USA); 20,000 cellular events (within the target gate) were recorded for each sample.
Anti-tumor T-cell cytotoxicity assays
After stimulation with the tumor antigen peptide mix, splenocytes were mixed with CT26 tumor cells (in a 1:1 ratio) in a Costar 96-well plate (Corning, USA) and further cultured for 6 h at 37 °C. The concentration and activity of LDH released by dead cells into the culture medium were measured using the CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Promega, USA).
IFN-γ ELISpot assay
Briefly, 5 × 105 splenocytes were seeded into each well of a 96-well filtration ELISpot plate (Merck, HAMAS5410) and stimulated overnight with a 2 µg/ml peptide mixture containing Aldh18a1, Mtch1, E2f8, and Glud1 epitopes. The peptide diluted in dimethyl sulfoxide (Sigma–Aldrich) and 10 ng/ml concanavalin A (Sigma–Aldrich) were used as negative and positive controls, respectively. Next, 2 days after culture, IFN-γ production was measured using a mouse IFN-γ ELISpot Kit (BD Bioscience, 551083), in accordance with the manufacturer’s instructions. The 3-amino-9-ethylcarbazole (AEC) substrate set (BD Biosciences, 551951) was used to develop the ELISpot plates. IFN-γ production was quantified using the CTL ImmunoSpot Analyzer and ImmunoSpot Professional Software version 5.0 (Cellular Technology, Shaker Heights, OH, USA). The ELISpot data were presented as IFN-γ spot-forming cells (SFCs) per 500,000 splenocytes.
CD8+ T-cell tetramer staining
To confirm the presence of CT26-tumor-antigen-specific CD8+ T cells, peripheral blood mononuclear cells (PBMCs) were extracted from the peripheral blood of mice treated with PBS, SAM-FC(−), SAM-FC(+), or SAM-FC(+) + αHMGB1 on day 12 after treatment. The PBMCs were stained with an APC-conjugated H2Ld MuLV gp70 tetramer-SPSYVYHQF (50 µl/test, TB-M521-2, MBL) according to the manufacturer’s instructions. Tetramer staining was performed together with PE-conjugated anti-mouse CD8α (0.125 µg per 106 cells, clone 5.3–6.7, Cat#12-0081-82, eBioscience) antibody staining and analyzed by flow cytometry.
Synergism evaluation of SAM-FC
We measured the Coefficient of Drug Interaction (CDI) to analyze whether tumor suppression by the dual-drug effect was additive or synergistic, as previously described68. The CDI was calculated using the equation CDI = {(AB/control) / [(A/control) × (B/control)]}, where A and B are the effects of single drugs, and AB is the combined effect of the dual-drug. CDI values less than, equal to, or greater than 1 indicate that dual-drugs are synergistic, additive, or antagonistic, respectively. Specifically, a CDI value below 0.7 indicates a significant synergistic interaction between the drugs. The data were obtained from the average tumor volumes for each group on day 15 (CT26 tumor model) or day 12 (MC38 tumor model), as shown in Fig. 1c, g. The ratios of average tumor volumes in the SAM-FC(+), SAM-FR(+), and SAM-CR(+) groups relative to the control group (SAM-E) were designated as ‘AB’, ‘A’, and ‘B’, respectively.
Statistical analysis
Statistical analysis was performed using Prism 9.0 software (GraphPad, USA). P-values < 0.05 were considered significant. Details relating to the specific statistical tests used are outlined in the Figure legends. Student’s t-test or one-way ANOVA were used for direct comparison of single variables. Two-way ANOVA with Tukey’s correction was used for multiple comparisons. Survival analysis was performed using a Kaplan–Meier curve and a long-rank (Mantel-Cox) test. All data are shown as the mean ± standard error of the mean (s.e.m).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DNA sequencing data have been deposited at the NCBI database under the accession code PRJNA1122980. Source data are provided as the source data file with this paper. All remaining data can be found in the Article, Supplementary and Source Data files. Source data are provided with this paper.
References
Limeta, A., Ji, B., Levin, M., Gatto, F. & Nielsen, J. Meta-analysis of the gut microbiota in predicting response to cancer immunotherapy in metastatic melanoma. JCI Insight 5, e140940 (2020).
Sun, J.-Y. et al. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms, predictive factors, and future perspectives. Biomark. Res. 8, 1–10 (2020).
Haslam, A. & Prasad, V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2, e192535–e192535 (2019).
D’Alise, A. M. et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun. 10, 1–12 (2019).
Xie, N. et al. Neoantigens: promising targets for cancer therapy. Signal Transduct. Target. Ther. 8, 9 (2023).
Cai, Z. et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol. Cancer 20, 1–13 (2021).
Ding, Z. et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct. Target. Ther. 6, 1–12 (2021).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Holm, J. S. et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 13, 1–17 (2022).
Lhuillier, C. et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Investig. 131, e138740 (2021).
Kroemer, G., Galassi, C., Zitvogel, L. & Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022).
Bianchi, M. E. et al. High‐mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 280, 74–82 (2017).
Sabado, R. L., Balan, S. & Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 27, 74–95 (2017).
Suzuki, Y. et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell CarcinomaAntigen-specific T-cell response following chemoradiotherapy. Cancer Res. 72, 3967–3976 (2012).
Apetoh, L. et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Yamazaki, T. et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 21, 69–78 (2014).
Yu, J. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 27, 152–164 (2021).
Guo, Z. et al. M2 macrophages promote NSCLC metastasis by upregulating CRYAB. Cell Death Dis. 10, 1–11 (2019).
Lee, C.-C. et al. Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 9, 1–18 (2018).
Kang, S.-R., Nguyen, D.-H., Yoo, S. W. & Min, J.-J. Bacteria and bacterial derivatives as delivery carriers for immunotherapy. Adv. Drug Delivery Rev. 181, 114085 (2021).
Nguyen, D.-H., Chong, A., Hong, Y. & Min, J.-J. Bioengineering of bacteria for cancer immunotherapy. Nat. Commun. 14, 3553 (2023).
Harimoto, T. et al. A programmable encapsulation system improves delivery of therapeutic bacteria in mice. Nat. Biotechnol. 40, 1–11 (2022).
Gurbatri, C. R., Arpaia, N. & Danino, T. Engineering bacteria as interactive cancer therapies. Science 378, 858–864 (2022).
Cheng, K. et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug-and-Display technology. Nat. Commun. 12, 1–16 (2021).
Gurbatri, C. R. et al. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci. Transl. Med. 12, eaax0876 (2020).
Duong, M. T.-Q., Qin, Y., You, S.-H. & Min, J.-J. Bacteria-cancer interactions: bacteria-based cancer therapy. Exp. Mol. Med. 51, 1–15 (2019).
Chowdhury, S. et al. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat. Med. 25, 1057–1063 (2019).
Forbes, N. S. Engineering the perfect (bacterial) cancer therapy. Nat. Rev. Cancer 10, 785–794 (2010).
Toso, J. F. et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 20, 142–152 (2002).
Kim, J.-E. et al. Salmonella typhimurium suppresses tumor growth via the pro-inflammatory cytokine interleukin-1β. Theranostics 5, 1328 (2015).
Zheng, J. H. et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 9, eaak9537 (2017).
Nguyen, V. H. et al. Genetically engineered Salmonella typhimurium as an imageable therapeutic probe for cancer. Cancer Res. 70, 18–23 (2010).
Tan, W. et al. Targeting of pancreatic cancer cells and stromal cells using engineered oncolytic Salmonella typhimurium. Mol. Ther. 30, 662–671 (2022).
Jiang, S.-N. et al. Engineering of bacteria for the visualization of targeted delivery of a cytolytic anticancer agent. Mol. Ther. 21, 1985–1995 (2013).
Nguyen, D.-H. et al. Optimized doxycycline-inducible gene expression system for genetic programming of tumor-targeting bacteria. Mol. Imaging Biol. 24, 82–92 (2022).
Wai, S. N. et al. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 115, 25–35 (2003).
Sathyanarayana, P. et al. Cholesterol promotes cytolysin A activity by stabilizing the intermediates during pore formation. Proc. Natl Acad. Sci. 115, E7323–E7330 (2018).
Lei, S.-P., Lin, H., Wang, S.-S., Callaway, J. & Wilcox, G. Characterization of the Erwinia carotovora pelB gene and its product pectate lyase. J. Bacteriol. 169, 4379–4383 (1987).
Kusuma, S. A. F. et al. Improving of pelB-secreted MPT64 protein released by Escherichia coli BL21 (DE3) using Triton X-100 and Tween-80. J. Adv. Pharm. Technol. Res. 13, 171 (2022).
Buhrman, J. D. et al. Augmenting antitumor T-cell responses to mimotope vaccination by boosting with native tumor antigens. Cancer Res. 73, 74–85 (2013).
McMahan, R. H. et al. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J. Clin. Investig. 116, 2543–2551 (2006).
Jordan, K. R., McMahan, R. H., Kemmler, C. B., Kappler, J. W. & Slansky, J. E. Peptide vaccines prevent tumor growth by activating T cells that respond to native tumor antigens. Proc. Natl Acad. Sci. 107, 4652–4657 (2010).
Philip, M. & Schietinger, A. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022).
Yost, K. E., Chang, H. Y. & Satpathy, A. T. Recruiting T cells in cancer immunotherapy. Science 372, 130–131 (2021).
Yuan, P. et al. Highly sensitive imaging of tumor metastasis based on the targeting and polarization of M2-like macrophages. J. Am. Chem. Soc. 145, 7941–7951 (2023).
Zhou, J., Zhang, S. & Guo, C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int. Immunopharmacol. 101, 108374 (2021).
Bellora, F. et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc. Natl Acad. Sci. 107, 21659–21664 (2010).
Krneta, T. et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leucoc. Biol. 101, 285–295 (2017).
Swierczak, A. et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol. Res. 2, 765–776 (2014).
Kang, R., Zhang, Q., Zeh, H. J., Lotze, M. T. & Tang, D. HMGB1 in cancer: good, bad, or both? Clin. Cancer Res. 19, 4046–4057 (2013).
Del Prete, A. et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 20, 432–447 (2023).
Martinez‐Usatorre, A. & De Palma, M. Dendritic cell cross‐dressing and tumor immunity. EMBO Mol. Med. 14, e16523 (2022).
Fu, C. et al. Plasmacytoid dendritic cells cross-prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc. Natl Acad. Sci. 117, 23730–23741 (2020).
Qin, W. et al. Bacteria‐elicited specific thrombosis utilizing acid‐induced Cytolysin A expression to enable potent tumor therapy. Adv. Sci. 9, 2105086 (2022).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 1–22 (2022).
Lei, K. et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat. Biomed. Eng. 5, 1411–1425 (2021).
Ryan, R. et al. Bacterial delivery of a novel cytolysin to hypoxic areas of solid tumors. Gene Ther. 16, 329–339 (2009).
Lai, X.-H. et al. Cytocidal and apoptotic effects of the ClyA protein from Escherichia coli on primary and cultured monocytes and macrophages. Infect. Immun. 68, 4363–4367 (2000).
Peng, P., Hu, H., Liu, P. & Xu, L. X. Neoantigen-specific CD4+ T-cell response is critical for the therapeutic efficacy of cryo-thermal therapy. J. Immunother. Cancer 8, e000421 (2020).
Duan, Z. & Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 6, 1–21 (2021).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. J. Cereb. Blood Flow. Metab. 40, 1769–1777 (2020).
Ngo, H. T.-T. et al. Reprogramming a doxycycline-inducible gene switch system for bacteria-mediated cancer therapy. Mol. Imaging Biol. 26, 148–161 (2024).
Lee, S. E. et al. A bacterial flagellin, Vibrio vulnificus FlaB, has a strong mucosal adjuvant activity to induce protective immunity. Infect. Immun. 74, 694–702 (2006).
Kamiya, T., Chang, Y.-H. & Campana, D. Expanded and activated natural killer cells for immunotherapy of hepatocellular carcinoma. Cancer Immunol. Res. 4, 574–581 (2016).
Chu, T.-H. et al. Potent anti-myeloma efficacy of dendritic cell therapy in combination with pomalidomide and programmed death-ligand 1 blockade in a preclinical model of multiple myeloma. Cancer Immunol. Immunother. 70, 31–45 (2021).
Daemen, S., Chan, M. M. & Schilling, J. D. Comprehensive analysis of liver macrophage composition by flow cytometry and immunofluorescence in murine NASH. STAR Protoc. 2, 100511 (2021).
Lu, L. et al. Synergistic cytotoxicity of ampelopsin sodium and carboplatin in human non-small cell lung cancer cell line SPC-A1 by G 1 cell cycle arrested. Chin. J. Integr. Med. 23, 125–131 (2017).
Acknowledgements
We thank Dr. Joon Haeng Rhee of Chonnam National University Medical School for valuable discussions. We thank Mr. Myeongjun Kim and Mr. Jeong Hwan Kwon for technical support at the imaging facility. We also thank Dr. Linh Mai Tran for supporting the animal experiments. This work was supported by the Bio & Medical Technology Development Program of National Research Foundation of Korea (NRF) (No. NRF-2020M3A9G3080282), NRF grants (No. 2020R1A5A2031185, RS-2024-00343402), and Korea Drug Development Fund funded by Ministry of Science and IT (MSIT), and Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare (HN22C0637, RS-2022-00167101(1711194341), Republic of Korea). Y.H. was supported by the Bio & Medical Technology Development Program of NRF (No. NRF-2020M3A9G3080330). Figures 1a, 1b, 1f, 2a, 2l, 3a, 3h, 4a, 5a, 5m, 6a, 6b, 6d, supplementary Fig. 10a, Supplementary Fig. 11a were created by BioRender with confirmation of publication and licensing rights.
Author information
Authors and Affiliations
Contributions
J.M, Y.H., and D.N. conceived the project and designed the experiments. D.N. performed gene cloning experiments to generate engineered SAM. D.N. performed in vitro and ex vivo experiments. D.N., S.Y., H.N., T.C., K.N., K.T., and S.K. performed animal experiments. D.N., J.M., Y.H., and S.H. performed and analyzed flow cytometry analysis. J.M., Y.H., S.H., and D.N. wrote the original draft, review, and editing. J.M. and Y.H. supervised the research. All authors contributed to the final manuscript version.
Corresponding authors
Ethics declarations
Competing interests
A patent for a novel DNA construct aimed at the diagnosis and treatment of cancer (KR102252939) has been registered by J.M., Y.H., and S.Y. as the inventors. In addition, a patent application for an innovative anticancer therapy using engineered bacteria expressing flagellin and pore-forming proteins (KR1020210153355) has been submitted by J.M., Y.H., S.Y., and D.N. as its inventors. CNCure Biotech, Inc. possesses exclusive licenses for these intellectual properties. This IP stands to gain financial benefits upon further validation. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nguyen, DH., You, SH., Ngo, H.TT. et al. Reprogramming the tumor immune microenvironment using engineered dual-drug loaded Salmonella. Nat Commun 15, 6680 (2024). https://doi.org/10.1038/s41467-024-50950-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50950-5
- Springer Nature Limited