

Abstract
Imbalance of proinflammatory and anti-inflammatory responses plays a crucial role in the progression of abdominal aortic aneurysms. ILF3, a known modulator of the innate immune response, is involved in cardiovascular diseases. This study aims to investigate the role of ILF3 in abdominal aortic aneurysm formation. Here, we use multi-omics analyzes, transgenic male mice, and multiplex immunohistochemistry to unravel the underlying involvement of ILF3 in abdominal aortic aneurysms. The results show that macrophage ILF3 deficiency attenuates abdominal aortic aneurysm progression, while elevated macrophage ILF3 exacerbates abdominal aortic aneurysm lesions. Mechanistically, we reveal that macrophagic ILF3 increases NF-κB activity by hastening the decay of p105 mRNA, leading to amplified inflammation in macrophages. Meanwhile, ILF3 represses the anti-inflammatory action by inhibiting the Keap1-Nrf2 signaling pathway through facilitating the ILF3/eIF4A1 complex-mediated enhancement of Keap1 translational efficiency. Moreover, Bardoxolone Methyl treatment alleviates the severity of abdominal aortic aneurysm lesions in the context of elevated ILF3 expression. Together, our findings underscore the significance of macrophage ILF3 in abdominal aortic aneurysm development and suggest its potential as a promising therapeutic target for abdominal aortic aneurysms.
Similar content being viewed by others


Introduction
Abdominal aortic aneurysm (AAA) is a complex fatal vascular disease characterized by irreversible enlargement of the aortic diameter, accompanied by focal inflammation, turbulent proteolytic activities, and extracellular matrix (ECM) degradation1,2,3. Inflammatory cell infiltration and intense inflammatory response play crucial roles in AAA initiation and progression1,4. Recently, there has been an emphasis on the role of immune regulation in maintaining the inflammatory balance during AAA progression5. Clinical investigations have found that aneurysm size in patients with AAA is dependent on the imbalance of pro-inflammatory and anti-inflammatory responses6. Moreover, serum levels of the pro-inflammatory cytokine interleukin 6 (IL-6) were significantly increased in AAA patients, in contrast to a notable reduction in the anti-inflammatory cytokine Interleukin-10 (IL-10)2. Similar findings have been observed in human AAA explant cultures and murine models5,7. Recent microarray-based gene expression studies have confirmed that AAA is a chronic inflammatory disease, with the actions of activated macrophage subsets playing a significant role7. Macrophage infiltration into the adventitia serves as a seminal event in the onset of AAA pathogenesis, which mediates inflammatory homeostasis by producing chemokines and cytokines that is critical to aneurysmal progression8,9. However, the specific molecular mechanism of macrophage infiltration and inflammation in aneurysm dilatation are not yet fully delineated. Besides, no explicit therapy targeting the immunoinflammatory response is currently available to impede excessive inflammatory response and remodeling of the abdominal aortic wall.
Interleukin enhancer-binding factor 3 (ILF3) is an RNA-binding protein initially identified as a positive regulator of IL2 gene expression10. Subsequent reports have indicated that ILF3 is also involved in dendritic cell maturation, innate immune response, and carcinoma progression11,12. More broadly, there is evidence that ILF3 is associated with cardiovascular diseases, such as stroke and myocardial infarction13,14. It has been reported that ILF3 is involved in the enhanced vascular smooth muscle cell apoptosis due to the overexpression of circACTA2 via reducing CDK4 mRNA stability15. Nevertheless, no studies have been conducted to explore the role of ILF3 in AAA.
The current study identified ILF3 as the target gene because of its high expression and specificity with macrophages in AAA lesions, as determined through bioinformatic analyzes and immunoassays. By employing conditional macrophage ILF3-deficient and overexpression mouse strains, we revealed the detrimental role of macrophage ILF3 in AAA development, with macrophage ILF3 being positively correlated to the intensity of focal vascular inflammatory response. Mechanistic studies further demonstrated that macrophage ILF3 acts as a dual modulator in the inflammatory cascade, where it both accelerates p105 mRNA instability to potentiate NF-κB-driven pro-inflammatory cascades and enhances Keap1 translational efficiency via the ILF3/eIF4A1 complex to dampen the Keap1-Nrf2-mediated anti-inflammatory responses. Furthermore, interventions targeting ILF3 gene expression, as well as pharmacological interventions using BM, were found to effectively mitigate the exacerbated progression of AAA caused by high ILF3 expression. These findings thus help elucidate the mechanisms of aortic aneurysm formation and provide a framework to guide the future development of treatments for patients with AAA by targeting ILF3.
Results
ILF3 is elevated in human and murine AAA tissues
To pinpoint the candidate genes potentially associated with AAA, we downloaded publicly available data from the Gene Expression Omnibus (GEO) database (GSE57691 and GSE12591), and DEGs were acquired via GEO2R analysis. 715 genes displayed the same trend in human and mouse AAA samples compared with normal controls, among which 410 genes were downregulated and 305 were upregulated (Fig. 1A). Among the top differentially expressed genes shown in the heatmap, ILF3 expression was significantly elevated in patients with AAA (Fig. 1B, and Supplementary Fig. 1A) and Ang-II-induced AAA mice (Fig. 1C), indicating a vital role of ILF3 in AAA progression. Besides, western blot (Figs. 1D, E), quantitative PCR (Fig. 1F), and immunohistochemical staining results (Supplementary Fig. 1B) showed that the expression of ILF3 was augmented in human AAA (patients’ information in Supplementary Table 1). Similar results were also seen in Ang-II-induced mouse AAA compared with the control group (Figs. 1G–I; and Supplementary Fig. 1C).

A Venn diagram showing the overlap of differentially expressed genes (DEGs) between human and mouse samples. B Heatmap showing gene expression in human control and AAA aorta tissues (p < 0.05, Foldchange>1.2). C Heatmap showing gene expression in mouse control and angiotensin II–induced AAA aorta tissues (p < 0.05, Foldchange>1.2). D and E Western blot analysis of ILF3 levels in aortas of controls and patients with AAA (n = 10 per group). F Relative mRNA levels of ILF3 in aorta tissues from controls and patients with AAA (n = 10 per group). The control sample was set as 1. G–H Western blot analysis of ILF3 levels in aortas from mouse control and angiotensin II–induced AAA samples (n = 7 per group). I RT-PCR of ILF3 mRNA in aorta tissues from control and angiotensin II–induced AAA mouse samples (n = 7 per group). The control sample was set as 1. J ILF3+ macrophages from human AAA and non-aneurysmal samples were projected onto the t-SNE plot. The color intensity of each dot corresponds to the average gene expression across all cells. K Dot plot representing the ILF3 expression of macrophage clusters in human AAA and non-aneurysmal samples. The color of the dot represents the ILF3 expression level (average expression), and the size of the dot corresponds to the percentage of cells expressing ILF3 (percent expressed) across the human AAA and non-aneurysmal samples. L FACS analysis quantified the proportion of ILF3+ macrophages within lesions of ApoE−/− mice subjected to Ang II infusion at specified time (n = 4 per group). M Representative confocal images of ILF3 (green) colocalized with CD68 (a marker of macrophage, red) in aorta tissues from controls and AAA lesions of humans. Nuclei were stained with DAPI (blue), and yellow represents merged images (Scale bar=25 μm; n = 8 per group). N Representative confocal images of ILF3 (green) colocalized with CD68 (marker of macrophage, red) in aorta tissues from ApoE−/− mice treated with Ang II for different intervals (Scale bar=25 μm; n = 8 per group). Data are represented as mean ± SD. For comparisons between two groups, a Student’s t-test with two-tailed analysis (1B–1C, 1E–1F, 1H–1I). For comparisons between more than two groups, one-way ANOVA followed by Tukey’s post hoc multiple comparisons test was used (1L). Boxplots were created using the first and third quartiles to define the bounds of the box. The minima are defined as the first quartile minus 1.5 times the inter-quartile range (IQR), and the maxima are defined as the third quartile plus 1.5 times the IQR. Outliers are displayed as individual points (1E–1F, 1H–1I and 1L).
Macrophage ILF3 is augmented during AAA progression
To discern ILF3’s spatial role in AAA lesions, we utilized Seurat for analyzing human single-cell RNA sequencing data (GSE166676) from the GEO database. Through UMAP and canonical markers, we classified cells into seven types (Supplementary Figs. 1D, E); the representative genes in each cell type are shown in Supplementary Fig. 1F. GO analysis showed that the immune response, particularly mononuclear cell activation and focal adhesion, were elevated in ILF3+ cells compared with ILF3− clusters (Supplementary Fig. 1G). Furthermore, a substantial increase in ILF3-positive macrophages was observed in human AAA lesions (Figs. 1J, K; Supplementary Figs. 1H–K), underscoring the critical role of macrophage ILF3 in AAA progression. To further explore ILF3’s function in macrophages within AAA, we established AAA models at different pathological stages using Ang II induction over varied timeframes. Flow cytometry demonstrated a progressive rise in ILF3-positive macrophages with prolonged Ang II exposure (Fig. 1L; Supplementary Fig. 2A). Additionally, increased ILF3 expression in both human and mouse AAA samples was verified, with clear macrophage colocalization noted in immunofluorescence assays (Figs. 1M, N). Notably, ILF3 protein levels in bone marrow-derived and peritoneal macrophages from wild-type C57BL/6 J mice showed a time and dose-dependent increase upon Ang II treatment (Supplementary Figs. 2B–E). Collectively, these findings compellingly suggest that the marked elevation of ILF3 in macrophages plays a crucial role in AAA development.
Macrophage ILF3 overexpression exacerbates AAA formation
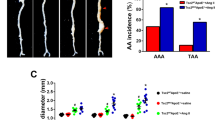
Observing elevated ILF3 levels in AAA, particularly in macrophages, we investigated macrophage ILF3 overexpression’s impact on AAA using ILF3 conditional overexpression mice (ILF3Flox/FloxLysMCre+; hereafter referred to as ILF3M-Tg) and its littermates (ILF3Flox/FloxLysMCre-; hereafter referred to as ILF3M-WT) in an ApoE−/− background (Supplementary Figs. 3A–3D). Following a 4-week saline infusion, no aneurysms were observed in either group (Fig. 2A). However, under Ang II induction, both groups experienced a significant rise in systolic blood pressure with Ang II, with no differences between the ILF3M-TgApoE−/− and ILF3M-WTApoE−/− mice (Supplementary Table 2). Notably, ILF3M-TgApoE−/− mice displayed more pronounced aortic dilation compared with ILF3M-WTApoE−/− mice (Fig. 2A). The incidence of AAA formation (Fig. 2B) and the rupture rate of AAA (Fig. 2C) in ILF3M-Tg ApoE−/− mice was markedly higher than that of ILF3M-WT ApoE−/− group through the 4-week Ang II exposure. Among those subjected to Ang II infusion, 68.75% (22/32) of ILF3M-Tg ApoE−/− mice died, apparently higher than the mortality rate of ILF3M-WT ApoE−/− (37.50%, 12/32; Fig. 2D). Vascular ultrasound imaging and maximal external aortic diameter detection further corroborated these findings, revealing more pronounced dilation in the ILF3M-Tg ApoE−/− mice post-Ang II infusion (Fig. 2E; Supplementary Fig. 3E). Histological analysis indicated no elastin disruption under baseline conditions; however, ILF3 overexpression in macrophages significantly exacerbated elastin degradation in Ang II-induced AAA models (Figs. 2F, G).

The 8-week-old male ILF3M-Tg ApoE−/− and ILF3M-WTApoE−/− mice were infused with Ang ll or saline (sham group). A Schematic representation of study design and representative images showing the morphology of the whole aorta from all groups showing the macroscopic characteristics of aneurysms. B–D The AAA incidence (B) rupture rate (C) and Kaplan-Meir survival curve (D) across different groups (sham group, n = 6; Ang II group, n = 32). Statistical analyses employed a chi-square test with a two-tailed analysis for (B, C) and the Log-rank test for (D). E Representative ultrasound images of aorta across different groups (sham group, n = 6; Ang II group, n = 12). F and G Representative hematoxylin and eosin (HE) and Verhoeff-Van Gieson (VVG) staining (F) and elastin degradation grading (G). (sham group, n = 5; Ang II group, n = 12; Scale bar=100 μm). 8-week-old male ILF3M-Tg and ILF3M-WT mice were infused with CaCl2 or saline (sham group). H and I Schematic representation of study design and representative photographs (H) of saline-treated and CaCl2-induced aneurysm and maximal abdominal aortic diameter (I; sham group, n = 6; Cacl2 group, n = 12). Data are represented as mean ± SD. For comparisons between two groups, an unpaired Student’s t-test with two-tailed analysis (2G and 2I). Boxplots were created using the first and third quartiles to define the bounds of the box, with the median shown in a circle. The minima are defined as the first quartile minus 1.5 times the inter-quartile range (IQR), and the maxima are defined as the third quartile plus 1.5 times the IQR. Violin plots display the density distribution of the data points through smoothed histograms.
To further validate the role of macrophage ILF3 in AAA, we evaluated the effects of macrophage ILF3 overexpression in CaCl2-induced AAA model mice. Consistent with the results observed in Ang-II-induced AAA model mice, ILF3M-Tg mice were more susceptible to abdominal aortic dilation with increased maximal abdominal aortic diameter in CaCl2-induced AAA model (Figs. 2H, I). Moreover, HE and VVG staining showed that elastic lamellae of ILF3M-Tg mice were disrupted and degraded to a greater extent than that of ILF3M-WT mice (Supplementary Figs. 3F, G). Collectively, these results underscore the pivotal role of macrophage-derived ILF3 in promoting AAA formation and progression in both CaCl2- and Ang II-induced models.
Macrophage ILF3 deficiency attenuates AAA formation
To identify whether macrophage ILF3 deficiency is protective against AAA formation, we constructed macrophage-ILF3 knockout mice (ILF3flox/floxLysMCre+; hereafter referred to as ILF3M-KO) in ApoE−/− mice (Supplementary Figs. 4A, B). Upon four weeks of Ang II or saline infusion, ILF3M-KOApoE−/− mice exhibited reduced aortic dilation in response to Ang II, while saline infusion had no discernible effect on either group (Fig. 3A). Importantly, blood pressure measurements remained consistent between the two genotypes, both pre- and post-Ang II infusion (Supplementary Table 3). Upon Ang II treatment, ILF3M-KOApoE−/− mice showed a decreased incidence of AAA (Fig. 3B) and a reduced rupture rate (Fig. 3C) compared to ILF3M-WTApoE−/− mice. And the mortality rate of ILF3M-WTApoE−/− mice post-Ang II infusion was 40.63% (13/32), evidently higher than the 15.79% (6/38) observed in ILF3M-KOApoE−/− (Fig. 3D). Moreover, after Ang II infusion, ILF3M-KOApoE−/− mice exhibited a discernible reduction in aortic dilation relative to ILF3M-WTApoE−/− mice (Supplementary Fig. 4C; Fig. 3E). Histological assessments using HE and VVG staining revealed that ILF3-deficient mice presented with moderate elastic degradation (Fig. 3F, G). Using CaCl2-induced AAA model, we also found that ILF3M-KO mice were less susceptible to abdominal aortic dilation with mitigated elastic degradation (Figs. 3H, I; Supplementary Figs. 4D, E). Overall, these results showed that macrophage ILF3 deficiency attenuated AAA formation, thereby highlighting its promise as a therapeutic target for AAA management.

The 8-week-old male ILF3M-KO ApoE−/− and ILF3M-WTApoE−/− mice were infused with Ang ll or saline (sham group). A Schematic representation of study design and representative images showing the morphology of the whole aorta from all groups showing the macroscopic characteristics of aneurysms. B–D The AAA incidence (B) rupture rate (C) and Kaplan-Meir survival curve (D) across different groups (sham group, n = 6; ILF3M-WTApoE−/− + Ang II group, n = 32; ILF3M-KO ApoE−/− + Ang Ii, n = 38). Statistical analyses employed a chi-square test with a two-tailed analysis for (B, C) and the Log-rank test for (D). E Representative ultrasound images of aorta across different groups (sham group, n = 6; Ang II group, n = 12). F and G Representative hematoxylin and eosin (HE) and Verhoeff-Van Gieson (VVG) staining (F) and elastin degradation grading (G). (sham group, n = 5; Ang II group, n = 12; Scale bar=100 μm). 8-week-old male ILF3M-KO and ILF3M-WT mice were infused with CaCl2 or saline (sham group). H and I Schematic representation of study design and representative photographs (H) of saline-treated and CaCl2-induced aneurysm and maximal abdominal aortic diameter (I) sham group, n = 6; CaCl2 group, n = 12). Data are represented as mean ± SD. For comparisons between two groups, an unpaired Student’s t-test with two-tailed analysis (3G and 3I). Boxplots were created using the first and third quartiles to define the bounds of the box, with the median shown in a circle. The minima are defined as the first quartile minus 1.5 times the inter-quartile range (IQR), and the maxima are defined as the third quartile plus 1.5 times the IQR. Violin plots display the density distribution of the data points through smoothed histograms.
Macrophage ILF3 contributes to Ang-II-induced inflammatory cell infiltration and the inflammatory response
To uncover the mechanism underlying the potential role of macrophage-derived ILF3 on AAA, we analyzed the enrichment results based on the scRNA-seq data16. As shown by the Gene Ontology (GO, Fig. 4A) and KEGG analysis (Fig. 4B), macrophage ILF3 was significantly associated with immune response and pathways related to immune response (such as NF-κB, TNF, and HIF1 signaling pathway were apparently enriched with macrophage ILF3). Macrophages play a pivotal role in AAA pathogenesis, where they mainly accumulate in the adventitial layer, serving as essential producers and responsive entities to inflammatory mediators, thereby orchestrating the intricate inflammatory dynamics within the aortic wall17.

A Representative Gene Ontology terms of differentially expressed genes (DEGs) enriched in ILF3+ macrophage clusters based on functional enrichment analysis. B Representative Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway data for macrophage DEGs enriched in ILF3+ vs ILF3-. C Immuno-staining of macrophage-derived MCP-1, TNF-α, IL-6, and IL-10 levels in AAA lesions of ILF3M-WT ApoE−/− and ILF3M-KO ApoE−/− mice treated with Ang II for four weeks (n = 6 per group; Scale bar=25 μm). D Plasma IL-6, TNF-α, MCP-1, IL10 levels in ILF3M-WT ApoE−/− and ILF3M-KO ApoE−/− mice treated with Ang II for four weeks (n = 10 per group). E Western blot analysis and quantification of ILF3, MCP1, TNF-α, IL-6, and IL-10 expression in peritoneal macrophages from the ILF3 deficiency and control groups with or without Ang II treatment (1 μM; n = 6 per group). Data are represented as mean ± SD. P values were calculated by one-sided Fisher’s exact test in (4A and 4B). For comparisons between two groups, an unpaired Student’s t-test with two-tailed analysis was performed if the data met the assumptions of normality and equal variances; otherwise, the Wilcoxon rank-sum test was used (4D). For comparisons among four groups, one-way ANOVA followed by Tukey’s post hoc multiple comparisons test was used (4E).
Utilizing immunofluorescent double staining techniques, we found the number of CD68+ macrophages was reduced in ILF3M-KOApoE−/− mice (Fig. 4C) and increased in ILF3M-TgApoE−/− mice compared with control mice (Supplementary Fig. 5A). Meanwhile, the results revealed the expression of several inflammatory cytokines and chemokines, including monocyte chemoattractant protein-1 (MCP-1), IL-6, TNF-α, and IL-10 exhibited a pronounced decrement in the ILF3M-KOApoE−/− mice (Fig. 4C). Conversely, these were significantly upregulated in the ILF3M-TgApoE−/− group (Supplementary Fig. 5A). Notably, the macrophage-produced immunosuppressive cytokine IL-10 expression was diametrically opposed to the aforementioned pro-inflammatory mediators (Fig. 4C; Supplementary Fig. 5A). Moreover, similar results were further validated in the AAA mouse model by assessing serum samples through Elisa (Fig. 4D; Supplementary Fig. 5B).
In vitro, we treated the isolated peritoneal macrophages (Fig. 4E) and bone marrow-derived macrophages (Supplementary Fig. 5C) from the macrophage ILF3 deficiency and wild-type mice with Ang II. Immunoblotting analysis demonstrated that macrophages deficient in ILF3 showed decreased expression of MCP-1 and inflammatory cytokines (IL-6 and TNF-α). IL-10 levels were, however, intensified due to the lack of ILF3. The above results showed that deficiency of macrophage-derived ILF3 attenuates Ang-II-induced inflammatory cell infiltration and concomitant inflammation, while an elevation in ILF3 expression exerts the opposite effect.
Macrophage ILF3 mediates NF-κB and Keap1-Nrf2 pathways via p105 and Keap1
To investigate the machinery between ILF3 and subsequent physio-biologic features of macrophages, we conducted proteomics using peritoneal macrophages from mice of ILF3M-WT and ILF3M-KO. Our findings revealed 1233 uniquely differentially expressed proteins attributable to ILF3 deficiency (Supplementary Fig. 6A; and Supplementary Data 1), although a small amount of residual expression of ILF3 was observed after its knockout. GO enrichment analysis, aligning with our scRNA-seq results, demonstrated ILF3’s strong association with inflammatory response (Supplementary Fig. 6B). KEGG pathway analysis highlighted ILF3’s role in key pathways such as Fluid Shear Stress and Atherosclerosis and NF-κB pathway (Fig. 5A), with significant alterations in p105 (nfkb1) and Keap1 (Fig. 5B). These factors are known to regulate various inflammatory cytokines and chemokines via NF-κB and Keap1-Nrf2 signaling pathway18,19,20. Western blotting validated these findings, showing decreased Keap1 and increased p105 and p50 in ILF3M-KO mice across both peritoneal and bone marrow-derived macrophages (Supplementary Figs. 6C–6F). Additionally, ILF3 deficiency could inhibit the Ang II-induced p65 nuclear translocation and increased cytoplasmic expression of p105 and p50 (Fig. 5C–E; Supplementary Figs. 6G, H), and moderated impaired Nrf2 nuclear translocation while lowering Keap1 expression (Figs. 5C, D, F; and Supplementary Fig. 6G, H). Furthermore, Co-IP analysis showed more prevalence of p50:p105 and p50:p50 complexes, while less p50:p65 complex in ILF3M-KO peritoneal macrophages (Figs. 5G, H). Given that p65 and Nrf2 are well-established transcription factors in the inflammatory response, Electrophoretic Mobility Shift Assay (EMSA) showed reduced NF-κB DNA binding in ILF3-deficient macrophages (Fig. 5I). On the contrary, ILF3 deficiency increased Nrf2-ARE DNA binding, especially, the Nrf2 binding to the ARE sequence of key inflammatory (e.g., IL-6, MCP1; Fig. 5J, Supplementary Figs. 6I, J). Collectively, the above results indicate that ILF3 mediates the inflammatory response via the NF-κB and Keap1-Nrf2 signaling.

A Bubble plot showing Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of differentially expressed genes (DEGs) in macrophages of ILF3M-WT vs. ILF3M-KO mice (one-sided Fisher’s exact test). The top 10 significantly enriched KEGG pathways. B KEGG pathway analysis for DEGs in significant KEGG pathway terms was visualized as a chord dendrogram composed of ribbons. C–D Western blot analysis and quantification of cytoplasmic and nuclear p105, p50, p65, Keap1, and Nrf2 expression in peritoneal macrophages of ILF3 deficiency and control groups with or without Ang II treatment (1 μM; n = 6 per group). E–F Immunofluorescence staining for p65 (red, E) and Nrf2 (green, F) in peritoneal macrophages from ILF3 deficiency and control mice with or without Ang II treatment (1 μM); nuclei were stained with DAPI (blue, Scale bar, 20 μm; n = 6 per group). G–H Co-immunoprecipitation (Co-IP) assay and visualization of interactions between p50, p105, and p65 with an anti-p50 monoclonal antibody in peritoneal macrophages from ILF3 deficiency and control mice after Ang II stimulus (1μm, 24 h, n = 3 per group). I–J, EMSA analysis showing the DNA binding activity of both NF-κ B (I) and Nrf2 (J) in nuclear extracts of peritoneal macrophages from ILF3 deficiency and control mice after Ang II stimulus (1 μm, 24 h, n = 6 per group). K–L Western blot analysis and quantification of MCP-1, TNF-α, IL-6, and IL-10 expression in peritoneal macrophages from ILF3M-WT and ILF3M-Tg mice treated with si-Keap1 and AdV-p105 inhibitor under the stimulus without or with Ang II (1 μM; n = 5 per group). Data are represented as mean ± SD. For comparisons between more than two groups, one-way ANOVA followed by Tukey’s post hoc multiple comparisons test was used (5D, 5L). For comparisons between two groups, an unpaired Student’s t-test with two-tailed analysis (5H).
To elucidate whether ILF3’s regulation of NF-κB and Keap1-Nrf2 signaling depends on p105 and Keap1 expression, we used p105-overexpressing adenovirus (AdV-p105, Supplementary Figs. 7A–B) and Keap1 small interfering RNA (si-Keap1, Supplementary Figs. 7C–D). Western blotting and immunofluorescence revealed that p105 overexpression limited p65 translocation in ILF3M-Tg mice, without altering Keap1/Nrf2 expression (Supplementary Fig. 8A–D). EMSA comfirmed that ectopic p105 expression reduced NF-κB activation, leaving the Nrf2 pathway unaffected caused by overexpression of macrophage ILF3 (Supplementary Figs. 8E, F). Additionally, Keap1 knockdown restored nuclear Nrf2 expression (Supplementary Figs. 8C, D; Supplementary Figs. 8G, H) and Nrf2 activation (Supplementary Fig. 8I-K) under ILF3 overexpression, without inhibiting NF-κB pathway (Supplementary Fig. 8L). And both si-Keap1 and AdV-p105 significantly reduced ILF3 overexpression-induced inflammation in macrophages (Figs. 5K, L; Supplementary Fig. 9). In summary, our data demonstrate that ILF3 deficiency modulates inflammatory balance through dual mechanisms: inhibiting NF-κB via p105 upregulation and activating Keap1-Nrf2 pathway due to reduced Keap1 expression.
Macrophage ILF3 promotes p105 mRNA instability and Keap1 translation efficiency
To unravel the underlying mechanisms behind the altered expression of Keap1 and p105 in ILF3-deficient macrophages, we initially assessed protein degradation pathways as contributing factors. This was achieved by employing MG132, a proteasome inhibitor, and hydroxychloroquine (HCQ), a classical lysosomal inhibitor. Neither of these inhibitors altered the changes observed in peritoneal macrophages (Supplementary Fig. 10A) and bone marrow-derived macrophages (Supplementary Fig. 10B) from ILF3M-KO mice. Considering ILF3’s established role in RNA metabolism, a fact substantiated by both our enrichment analysis and previous studies10, we next conducted RNA Immunoprecipitation sequencing (RIP-seq). The results revealed that 83.2% of ILF3-bound RNAs were involved in protein coding, highlighting ILF3’s critical role in biological functions via RNA metabolism (Fig. 6A). Furthermore, RIP-seq showed ILF3 was able to bind to both p105 and Keap1 RNA (Figs. 6B, C; Supplementary Fig. 10C, D). RT-PCR analysis indicated that ILF3 deficiency increased p105 mRNA expression without affecting Keap1 RNA levels in peritoneal and bone marrow-derived macrophages (Fig. 6D; Supplementary Fig. 10E). Assessment of mRNA stability showed that deficiency of ILF3 slowed the degradation of p105 mRNA but had no impact on Keap1 mRNA (Fig. 6E; Supplementary Figs. 10F–H). To further explore ILF3’s influence on Keap1 protein expression, we performed IP-MS and found no interaction between ILF3 and Keap1. However, eIF4A1 emerged as a potential mediator, given its role in initiating cap-dependent eukaryotic protein translation (Fig. 6F). Subsequent Co-IP confirmed the interaction between ILF3 and eIF4A1 (Fig. 6G, H). Moreover, we employed the catRAPID method to estimate the binding potential of eIF4A1 protein to Keap1 mRNA21. The catRAPID identified a direct interaction between eIF4A1 protein and Keap1 mRNA, with a Global Score of 1.00 (scaling from 0 to 1, where 0 indicates no RNA-binding ability and 1 indicates strong affinity). And it revealed that the predominant binding site for eIF4A1 is located in the 5’ UTR of Keap1 mRNA (Supplementary Fig. 10I), which was further validated by an in vitro RIP assay (Fig. 6I). These findings propose the assembly of an ILF3/eIF4A1/Keap1 mRNA complex, which might account for the observed reduction in Keap1 protein levels. Ribosomal profiling confirmed our suspicions, and similar results were acquired using RT-PCR, which showed that macrophage ILF3 deficiency inhibits the translation efficiency of Keap1 (Figs. 6J, K; and Supplementary Data 2). Besides, results showed knockdown of eIF4A1 via si-eIF4A1 suppressed the Keap1 translation efficiency (Supplementary Fig. 10J) and inhibited the elevated translation of Keap1 due to ILF3 overexpression (Supplementary Fig. 10K). In summary, our findings prove that ILF3 deficiency in macrophages enhances p105 mRNA stability and inhibits Keap1 translation, thereby modulating the inflammatory response through the NF-κB and Keap1-Nrf2 pathways.

A Distribution of ILF3-binding regions on the genome relative to RIP-Sequence. B–C Representative ILF3 RIP-seq peaks were shown as track signals in an integrative genomic viewer of p105 (Nfkb1) (B) and Keap1 (C). Peak area is highlighted in light blue and peak region is denoted as red rectangle. D Relative mRNA levels of Keap1 and p105 in peritoneal macrophages from ILF3M-WT and ILF3M-KO mice (n = 6 per group). E Relative p105 remaining RNA levels at different time points after Actinomycin-D (Act-D) treatment in peritoneal macrophages from ILF3M-WT and ILF3M-KO mice (n = 6 per group). F, Venn diagrams showing the proteins immunoprecipitated with anti-IgG and anti-ILF3 antibodies in wild-type peritoneal macrophages. Blue represents proteins immunoprecipitated with anti-ILF3 under basal conditions (NC). Pink represents proteins immunoprecipitated with anti-IgG. G–H, Co-IP assay showing interactions between ILF3 and eIF4A1 with an anti-ILF3 antibody (G) and anti-eIF4A1 (H) antibody in peritoneal macrophages from control mice (n = 3 per group). I RIP assay for enrichment of Keap1 mRNA with eIF4A1 in wild-type peritoneal macrophages; IgG served as an internal control (n = 3). J Translation profiles for Keap1 computed from Ribo-seq data without induction. Positions of the genetic parts and gene are shown above the profiles. The Y axis indicates sequence depth. K, Box plot showing the translation efficiency (TE) of Keap1 mRNAs in peritoneal macrophages from ILF3M-WT and ILF3M-KO mice (n = 3 per group). Data are represented as mean ± SD. For comparisons between two groups, an unpaired Student’s t-test with two-tailed analysis (6D and 6 K). For 6E, statistical analysis was performed using Two-way ANOVA and Sidak’s multiple comparison post-hoc test (***p < 0.001).
Pharmacological therapy with BM alleviates macrophage ILF3 high expression induced AAA progression
Bardoxolone methyl (BM, CDDO-Me) is an oral oleanolic acid-derived semi-synthetic triterpenoids and has been proven as an effective inducer of Keap1-Nrf2 (induce Keap1-Nrf2 disassociation, promotes Nrf2 protein stabilization and nuclear translocation22) as well as an inhibitor of the NF-κB pathway(inhibit NF-κB p65 to the nucleus23), which is undergoing clinical trials to explore the potential to treat chronic kidney diseases24,25. Given the shared targets and mechanisms between BM and ILF3, we aim to explore the potential of BM in treating AAA with high ILF3 expression, while simultaneously further validating the underlying mechanisms of ILF3’s action. Initially, we administered BM to ILF3M-WT ApoE−/− mice and observed that BM treatment resulted in a reduction of AAA incidence, suggesting a protective effect of BM against the development of AAA (Supplementary Fig. 11). Furthermore, the in vitro research indicated that BM could restrain the inflammatory response induced by ILF3 overexpression via hindering NF-κB pathway and enhancing the Keap1-Nrf2 pathway (Supplementary Figs. 12A–12F). While no significant changes in blood pressure were observed across different treatment groups (Supplementary Table 4), BM potently attenuated exacerbated AAA progression in Ang II-treated ILF3M-Tg ApoE−/− mice (Fig. 7A). This was evidenced by reduced incidence of aneurysms (Fig. 7B), rupture rate (Fig. 7C), and mortality (Fig. 7D) attributed to ectopic ILF3 expression. Vascular ultrasound imaging and maximal external aortic diameter measurement further corroborated BM’s inhibitory effects on vessel dilation in ILF3M-Tg ApoE−/− mice (Figs. 7E, F). Furthermore, BM treatment significantly ameliorates Ang II-induced elastin degradation (Figs. 7G, H). The CaCl2-induced AAA model also revealed that BM treatment ameliorates ILF3 overexpression-caused abdominal aorta dilation (Figs. 7I, J) and elastin disruption (Supplementary Figs. 12G and 12H). In sum, the above results suggest that pharmacological intervention with BM may counteract the effects of macrophage ILF3 overexpression on NF-κB and Keap1-Nrf2 pathway and mitigate the detrimental effects of ILF3 overexpression in macrophages on AAA by restoring inflammatory homeostasis inflammatory homeostasis (Fig. 8).

A Schematic representation of study design and representative images showing the morphology of the whole aorta from all groups showing the macroscopic characteristics of aneurysms. B–D The AAA incidence (B) rupture rate (C) and Kaplan-Meir survival curve (D) across different groups (ILF3M-WTApoE−/−, n = 20; ILF3M-TgApoE−/− and BM-treated ILF3M-TgApoE−/− mice, n = 19). Statistical analyzes employed Fisher’s exact test with a two-tailed analysis for (B, C) and the Log-rank test for (D). E and F Representative ultrasound images (E) and aorta external diameter measurements (F) of Ang II-infused ILF3M-WT ApoE−/−, ILF3M-Tg ApoE−/−, and BM-treated ILF3M-Tg ApoE−/− mice (n = 9 per group). G and H Representative hematoxylin and eosin (HE) and Verhoeff-Van Gieson (VVG) staining (G) and elastin degradation grading (H) of the mouse aorta obtained from ILF3M-WTApoE−/−, ILF3M-TgApoE−/−, and BM-treated ILF3M-TgApoE−/− mice after AngII stimulus (n = 10 per group; Scale bar=100 μm). I and J Schematic representation of study design and representative photographs (I) of saline-treated and CaCl2-induced aneurysm and maximal abdominal aortic diameter (J) n = 9 per group). Data are represented as mean ± SD. For comparisons between the three groups, one-way ANOVA followed by Tukey’s post hoc multiple comparisons test was used (7F, 7H, 7J). Boxplots were created using the first and third quartiles to define the bounds of the box, with the median shown in a circle. The minima are defined as the first quartile minus 1.5 times the inter-quartile range (IQR), and the maxima are defined as the third quartile plus 1.5 times the IQR. Violin plots display the density distribution of the data points through smoothed histograms.

Under the AAA pathological environment, ILF3 was upregulated in aneurysmal macrophages. Macrophage ILF3 activation accelerates the degradation of p105 mRNA and increases nuclear p65 translocation, intensifying pro-inflammatory responses via activation of the NF-κB pathway. On the other hand, macrophage ILF3 may bind to Keap1 mRNA via the protein complex ILF3-eIF4A1 and amplifies Keap1 translation, which contributes to the suppression of anti-inflammatory responses via restraining Keap1-Nrf2 activity. That destroys inflammatory homeostasis in focal AAA lesions, resulting in the development of AAA. Pharmacological therapy with bardoxolone methyl (BM) ameliorates ILF3 overexpression-induced AAA development via crosstalk with the NF-κB and Keap1-Nrf2 pathways.
Discussion
The overarching findings of our study demonstrated that ILF3 is upregulated in aneurysmal macrophages. This upregulation of macrophage ILF3 appears to accelerate the formation of AAA, as corroborated by human and mouse AAA tissue data. Importantly, we found that the activation of macrophage ILF3 disrupts the inflammatory balance within localized AAA lesions. This disruption occurs through two primary mechanisms: enhancing the NF-κB pathway by destabilizing p105 mRNA and inhibiting the Keap1-Nrf2 pathway by increasing Keap1 translational efficiency via the ILF3/eIF4A1 complex. Moreover, our study highlights the therapeutic potential of BM in mitigating the adverse effects of AAA exacerbated by macrophage ILF3 activation.
Prior studies have shown that ILF3 is involved in the progression of cardiovascular diseases such as myocardial infarction26,27. Our previous work demonstrated that elevated ILF3 expression could promote arteriosclerotic calcification by acting on the promoter regions of BMP2 and STAT128. A recent study revealed that ILF3 participates in the process of circACTA2 reducing CDK4 mRNA stability and protein expression via competing for association with CDK4 mRNA, thus leading to Ang II-induced VSMC death15. However, the specific role of ILF3 in AAA development is still unclear. Here, using bioinformatic and immunological methods, we have identified markedly elevated levels of ILF3 in AAA tissues of humans and mice. Notably, this increase was closely associated with a heightened accumulation of vascular macrophages, denoting the potential role of macrophage ILF3 in AAA progression.
The adventitial remodeling and inflammation are the main pathological features of AAA9,29. Macrophages are key phenotypes of immune cells in the adventitial, where they secrete pro-inflammatory mediators, including TNF-α, IL-6, and MCP-1 to initiate a positive feedback loop to extensive remodeling of the aortic wall and eventual aortic dissection30,31. Mellak et al. found that macrophage depletion inhibits the development of AAA using bone marrow transplantation in ApoE−/− mice32. However, the underlying molecular mechanisms that regulate macrophagic inflammatory homeostasis in situ are still to be comprehensively understood. Our study addresses this gap through a comprehensive approach involving bioinformatic and proteomic analyzes. We discovered a robust association between ILF3 and inflammatory responses, specifically implicating the NF-κB and Keap1-Nrf2 pathways. Our gene silencing experiments showed that inhibiting macrophage ILF3 led to a marked reduction in macrophage infiltration. The dampened inflammatory response was directly associated with a pronounced reduction in aortic remodeling and dissection in mouse models of AAA, thereby unequivocally establishing a causal link between ILF3 activity and AAA pathology. Furthermore, our in vitro and in vivo analyzes added another layer of understanding. We found that ILF3 deficiency mitigates the release of the chemokine MCP-1 and reduces the levels of pro-inflammatory cytokines such as IL-6 and TNF-α. Given that MCP-1 is a potent chemotactic factor for macrophage recruitment33 and that IL-6 and TNF-α are critical players in vascular inflammation34,35, our findings have broad implications. It suggests that targeting ILF3 could disrupt multiple facets of the inflammatory cascade that contribute to AAA development, and further pharmacological targeting of ILF3 could disrupt multiple components of the inflammatory cascade, thereby offering a comprehensive approach to AAA treatment.
Compelling evidence underscores the pivotal role of NF-κB p65-mediated transactivation in the etiology of various chronic inflammatory diseases. Achieving targeted inhibition of p65, tailored to specific tissues and contexts, remains a significant therapeutic objective. In our research, the absence of macrophage ILF3 did not alter p65 levels, yet we observed an upregulation of p105 expression as confirmed by proteomic and immunoblotting analyzes. Previous studies have reported that p105 acts as an essential suppressor of inflammation, with p105 deficiency leading to spontaneous lymphocytic inflammation in the liver and lungs. Additionally, macrophages deficient in p50 have been shown to secrete increased pro-inflammatory cytokines36,37,38. However, the specific mechanisms by which p50/p105 regulates inflammatory responses in AAA lesions remain unclear. In our current study, we found that macrophage ILF3 deficiency caused an evident increase in cytoplasmic p105 and p50, as well as nuclear p50. Additionally, the results of Co-IP assays revealed elevated levels of p50:p105 and p50:p50 dimers, along with a decrease in p65:p50 dimers in ILF3-deficient macrophages, which accounted for the retention of cytoplasmic p65 and subsequent inhibition of inflammation. Furthermore, using RIP-seq and RT-PCR, we proved that ILF3 can bind to p105 mRNA, thus enhancing its degradation. As well as this, we also found that macrophage ILF3 enhances Keap1 protein expression, leading to decreased nuclear Nrf2. Mechanistically, RIP-seq, IP-MS and ribosome profile analysis unveiled that ILF3 augmented Keap1 translational efficiency by forming the ILF3/eIF4A1/Keap1 mRNA complex39. These results demonstrate the role of ILF3 in modulating inflammatory homeostasis and suggest that ILF3 can affect the translation efficiency of specific proteins via the formation of protein complexes.
The NF-κB and Keap1-Nrf2 pathways are vital in regulating inflammatory responses’ physiological homeostasis. Currently, there is an urgent need for therapeutics capable of targeting these two pathways. Herein, we revealed that ILF3 deficiency could inhibit NF-κB-DNA binding activity and increase Nrf2-DNA binding capacity, thus leading to a decreased inflammatory response. BM has been proven as an effective inducer of Keap1-Nrf2 and an inhibitor of the NF-κB pathway40,41. Growing evidence from the Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes (BEACON) trial and the Phase 2 Study of Bardoxolone Methyl in Patients with Chronic Kidney Disease and Type 2 Diabetes (TSUBAKI) study shows that BM not only can effectively increase eGFR and improve kidney function, but also can prevent cancer development and mitigate radiation-induced damage24,42,43. Our current investigation unveiled that BM counteracts the alterations in NF-κB and Keap1-Nrf2 DNA activity induced by ILF3 overexpression in vitro. This observation further substantiates the role of ILF3 in modulating the inflammatory response through its impact on the NF-κB and Keap1-Nrf2 signaling pathways. Notably, our in vivo assessments demonstrated that BM effectively attenuates the rapid progression of AAA associated with ectopic ILF3 expression, which offers a promising therapeutic direction for treating patients with AAA characterized by elevated ILF3 levels.
In conclusion, our study revealed the pivotal role of macrophage ILF3 on AAA development. We observed that in the pathological context of AAA, there is an upregulation of macrophage ILF3, which correlates with a worsening inflammatory response. Our mechanistic insights reveal that silencing macrophage ILF3 could offer arterial protection by mitigating inflammatory dilation. Specifically, macrophage ILF3 accelerates the degradation of p105 mRNA, enhancing the nuclear translocation of p65. This process activates the NF-κB pathway, leading to amplified pro-inflammatory responses. Moreover, ILF3’s interaction with Keap1, through the ILF3-eIF4A1 complex, augments Keap1 translation, which in turn dampens anti-inflammatory activities by restraining the Keap1-Nrf2 pathway. These mechanistic insights suggest that targeting macrophage ILF3, either through gene silencing or specific pharmacological treatments, presents a promising avenue for AAA therapy.
Methods
Microarray data collection and analysis
The NCBI-GEO database (https://www.ncbi.nlm.nih.gov/geo/) was searched for microarray or RNA-sequencing data related to abdominal aortic aneurysm. We evaluated two studies, GSE57691 and GSE12591. Only normal aorta and abdominal aortic aneurysm samples were selected in each dataset. GSE57691 (Platform GPL570) included 10 control aortic specimen of organ donors or aortic specimen from 29 patients with AAA (mean maximum aortic diameter=68.4 ± 14.3 mm). GSE12591 (Platform GPL7215) comprised 3 whole aortas of 17 week old male ApoE−/− mice exposed to angiotensin II (1.44 µg/kg/min) for 4 weeks where there was clear evidence of aortic aneurysm formation and those exposed to saline infusion (n = 3). We used the GEO2R online tool to analyze the raw data of microarrays and identify DEGs between the control tissues and AAA. GEO2R is an interactive web toll that allows users to compare different groups of samples in a GEO series to examine differentially expressed genes according to experimental conditions. Fold Change > 1.2 and P < 0.05 were used as the cut-off standards to obtain DEGs.
Single cell RNA-seq data processing
Sequencing data were acquired from the Gene Expression Omnibus (accession no. GSE166676). The Seurat package (Version 3.2.0) in R software (Version 4.2.1) was used for cross-sample adjustment, processing, and quality control. After removing low-quality cells, gene expression matrices were normalized using the Normalize Data function, and 2000 features with high cell-to-cell variation were identified using the Find Variable Features function. The Run PCA, Find Neighbors and Find Clusters functions were used to reduce dimensionality and cluster the cells, followed by dimensional reduction with the Run Uniform Manifold Approximation and Projection (UMAP) and Runtsne functions. By matching the cell cluster gene signature with the assumed cell type specific marker, the aggregated cells are mapped to the corresponding cell type.
The average gene expression level was calculated and the log-two-fold change between the specific cell cluster and other cells was applied as the test statistic. Enrichment analysis for the functions of differentially expressed genes (DEGs) was conducted using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functions in the Cluster Profiler in R; enrichment in GO terms and KEGG pathways were calculated based on hypergeometric distribution. The functions with p-value < 0.05 were considered significantly different.
Collection of human tissue samples
Human AAA tissues from ten male patients undergoing surgical repair for AAA in the Beijing Anzhen Hospital (N = 10) were donated by Dr. Wen-jian Jiang. The detailed information on the ten patients is listed in Extended Data Table S1. The protocol was approved by local Ethics Committee of Beijing anzhen hospital capital medical university (#2018004). Patient consent was obtained for the use of this tissue. Age-matched non-AAA segments acquired from organ donors were harvested during explantation (N = 10) of Shandong provincial hospital. The protocol was endorsed by the Ethics Committee of Shandong First Medical University (#2022-303).
Animals
Animal models and pharmacological therapy
ILF3Flox/Flox mice, which were generated by inserting the mouse ILF3-201 transcript between the first and second exons of the mouse ROSA26 locus. Specifically, the targeting vector used for this purpose was engineered to include a splice acceptor (SA), a loxP-flanked STOP cassette containing three polyadenylation signals, and the ILF3 cDNA, followed by an additional polyadenylation signal. This vector was introduced into C57BL/6 J zygotes via microinjection, along with Cas9 mRNA and sgRNA targeting the first intron of the Gt(ROSA)26Sor locus (ROSA26). The procedure was conducted by Viewsolid Biotech Co, Ltd, Beijing, China. These ILF3Flox/Flox mice were hybridized with C57BL/6 LysMCre mice to generate macrophage-ILF3 conditional overexpression mice (ILF3Flox/FloxLysMCre+; hereafter referred to as ILF3M-Tg) and its littermates (ILF3Flox/FloxLysMCre-; hereafter referred to as ILF3M-WT). ILF3flox/flox mice, possessing two loxP sites flanking exon 3 of the ILF3 gene (Viewsolid Biotech Co, Ltd; Beijing, China), were crossed with C57BL/6 LysMCre mice to generate macrophage-ILF3 knockout mice (ILF3flox/floxLysMCre+; hereafter referred to as ILF3M-KO) and controls (ILF3flox/floxLysMCre-). ApoE−/− mice were purchased from Beijing Viewsolid Biotechnology (Beijing, China) and crossed with ILF3M-KO, ILF3M-Tg, and their littermates to generate macrophage ILF3 genetic-engineering mice in ApoE−/− background. To avoid potential confounding factors related to sex-specific differences, male mice aged eight weeks old were selected in this study44,45. The mice were housed at 40 ± 5% humidity and 24 ± 2 °C under a 12 h light/dark cycle with access to water and food ad libitum.
To establish an angiotensin II (Ang II)-induced AAA mouse model, a mini osmotic pump (Alzet Model 2004, Alza Corp, Palo Alto, CA, USA) loaded with Ang II (1000 ng/kg/min, Sigma-Aldrich, Saint Louis, MO, USA) or saline (0.9% NaCl) was implanted subcutaneously at the dorsum of the neck for Ang-II or saline perfusion, and with concurrent high-fat feeding. Four weeks later, the abdominal aorta was collected for further analysis. Mouse aortic diameter was measured after cleaning the perivascular tissue using the microscope eyepiece reticle micrometer ruler, as mentioned in previous studies46.
To establish a CaCl2-induced AAA mouse model, infrarenal abdominal aortas were isolated and wrapped with fertile cotton balls presoaked in 0.5 mol/L calcium chloride for 15 min under general anesthesia using isoflurane. After four weeks, the mice were sacrificed for sample collection.
BM (CDDO-Me, Selleck, Shanghai, China) was dissolved in dimethyl sulfoxide (DMSO) and further diluted in PEG300 and PBS. Ang II-infused and CaCl2-treated mice were intraperitoneally injected with BM (1.25 mg/kg/day) or saline on the day after Ang II or CaCl2 treatment47. Four weeks later, the mice were sacrificed for sample collection.
This study was conducted in strict compliance with the proposals outlined in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments have been reviewed and approved by the Institutional Animal Care and Use Committee of Shandong University (KYLL2020-454).
Blood pressure and weight measurement
Blood pressures were measured before and four weeks after Ang-II or saline perfusion using the pulse-based tail-cuff method with a photoelectric device (Softron, BP-2010). Mice were sacrificed four weeks after Ang-II or saline perfusion, and body weights were obtained.
Vascular ultrasound and quantification of AAA
Pulse wave velocity and aortic diameter were assessed using an ultrasound machine (Visualsonics, Vevo2100). Before positioning the collection probe, the mice anesthetized by inhalation of isoflurane were placed on the heated 37 °C plastic pillow, scratched with water ultrasonic transmission gel and coated on the mouse abdomen. Basal temperature and heart rate were observed throughout the ultrasound imaging procedure. Vascular ultrasonography was conducted to measure the maximal luminal diameter, with assessments independently carried out by two observers blinded to the experimental groups. Measurements were taken both the day before Ang II infusion and on day 28 post-infusion, as previously described48,49. B-mode imaging was used to determine the diameter of the suprarenal aorta and quantify the incidence of aneurysms according to the definition of AAA, which is defined by an enlargement of the aortic diameter by ≥50% compared to its baseline50,51. Furthermore, any dissection resulting in an intramural hematoma in the AAA mouse model should be considered, even if the dilation is just 110%50.
Macrophage isolation culture
Peritoneal macrophages were isolated through peritoneal lavage three days after the intraperitoneal injection of sterile suspension of 6% starch. Peritoneal macrophages were harvested, washed in phosphate-buffered saline (PBS), and cultured in DMEM medium with 1% penicillin/streptomycin and 10% bovine serum.
Bone marrow-derived macrophages were isolated from mouse femurs and tibiae, as described previously52. PBS was used to flush out the bone marrows of femurs and tibiae with a syringe with a 27 G needle. Cell suspension under 4 °C to 1500 rpm centrifuge for 5 min. Cells were then cultured with complete DMEM containing 30 ng/ml recombinant macrophage colony-stimulating factor (M-CSF) (R&D, Minnesota, USA) for macrophage differentiation. After 7 days, bone marrow-derived macrophages were acquired and used for further study.
Immunoblot analysis
Protein samples were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels and then transferred to a PVDF membrane. The membranes were blocked with 5% nonfat milk at room temperature for 1 h and then incubated with a specific diluted primary antibody overnight. After being washed 3 times with TBST, membranes were incubated with secondary antibodies at room temperature for 1 h. Chemiluminescence (Millipore, Billerica, MA, USA) identified the bound primary antibodies and measured using Image J (NIH, Bethesda, MD, USA). Antibodies used are as follows: β-actin, RRID: AB_2223172, Cell Signaling Technology, Catalog number: 4970; NF-κB1, RRID: AB_2282895, Cell Signaling Technology, Catalog number: 4717; NF-kappa B, RRID: AB_10859369, Cell Signaling Technology, Catalog number: 8242; NRF2, RRID: AB_2715528, Cell Signaling Technology, Catalog number: 12721; eIF4A1, RRID: AB_, Abcam, Catalog number: ab185946; IL-6, RRID: AB_2927381, Abcam, Catalog number: ab259341; MCP1, RRID: AB_448636, Abcam, Catalog number: ab25124; TNF-α, RRID: AB_2935774, Abcam, Catalog number: ab215188; Keap1, RRID: AB_, Abcam, Catalog number: ab227828; IL-10, RRID: AB_2847946, Abcam, Catalog number: ab189392; ILF3, RRID: AB_2049804, Abcam, Catalog number: ab92355; Lamin B1, RRID: AB_443298, Abcam, Catalog number: ab16048. Expression was normalized relative to the control.
Electrophoretic mobility shift assay (EMSA)
An EMSA was conducted using Light Shift Chemiluminescence EMSA kit (Thermo Fisher Scientific, Rockford, USA) to assess Ang II-induced NF-κB and Nrf2 activation in peritoneal macrophages. Nuclear extracts isolated from cultured peritoneal macrophages (5 × 106/ml) using the NE-PER Nuclear and Cytoplasmic Extract Reagents (Thermo Fisher Scientific, Rockford, USA). The NF-κB and Nrf2 binding assay to predicted sites was conducted using annealed biotin labeled oligonucleotide probes. The labeled oligonucleotide probe designed for NF-κB is 5′(Biotin)-AGTTGAGGGGACTTTCCCAGGC-3′, and the probe for Nrf2 is 5′(Biotin)-TGGGGAACCTGTGCTGAGTCACTGGAG-3′.
RNA immunoprecipitation (RIP)
The RIP assay involved using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) in strict accordance with the manufacturer’s recommendations. Anti-ILF3 (19887-1-AP, Proteintech) and anti-IgG antibodies were used to immunoprecipitation RNAs. The final analysis was performed with qRT-PCR, showing fold enrichment of the target RNAs between the immunoprecipitated and input fractions.
Co-immunoprecipitation (Co-IP)
The cell lysates of macrophages were collected and centrifuged, followed by incubation with anti-P50 (SC-8414, Santa), anti-eIF4A1 (ab185946, Abcam), and anti-ILF3 (19887-1-AP, Proteintech) antibodies at 4 °C overnight. A lysate immunoprecipitated with anti-IgG (ab172730, Abcam) antibody was used as a negative control. The immunocomplexes were then purified using 30 µl of protein G magnetic beads (Bimake, Houston, USA) for 2 hours at 4 °C, centrifuged, and washed with PierceTM IP Lysis Buffer (Thermo Fisher Scientific, Waltham, MA, USA) four to five times. The immunoprecipitated protein was further analyzed by western blot with target antibodies.
Histological analysis
Abdominal aortas were harvested, perfused with saline, and then fixed with 4% paraformaldehyde, followed by embedding in paraffin and serial sectioning. Paraffin-embedded sections (5 µm thick) were prepared for hematoxylin and eosin (HE) staining to observe general morphology and Verhoff-Van Gieson (VVG) staining to evaluate elastin degradation. Elastin degradation was graded using a quantification method by counting the number of breaks per vessel3.
Immunohistochemical and immunofluorescence staining
The paraffin embedded arterial sections were dewaxed and stained with primary antibodies, followed by a biotin-conjugated secondary antibody (1:500) and horseradish peroxidase-conjugated streptavidin (Dianova, Rodeo, CA).
For immunofluorescence staining, sections were stained overnight with specific antibodies at 4 °C and then added with fluorescence-labeled secondary antibodies. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). Specific fluorescence imaging was obtained by laser-scanning confocal microscopy (LSM710, Carl Zeiss, Germany).
Immunoprecipitation mass spectrometry (IP-MS)
IP-MS was conducted as previously described53. Briefly, peritoneal macrophages were obtained from C57BL/6 mice and separated. IP was performed using an immunoprecipitation kit (Abcam, Cambridge, UK). In brief, cells were collected, resuspended, and lysed in lysis buffer containing the protease inhibitor cocktail. The resulting cell lysates and anti-ILF3 antibody (19887-1-AP, Proteintech) were mixed overnight in a rotary mixer at 4 °C. Next, 40 µL protein A sepharose bead slurry was added into each tube and incubated for 1 h at 4 °C, followed by 2000 g centrifugation for 2 min. After washing three times with 1 mL 1× wash buffer, 40 µL 2× SDS-PAGE loading buffer added to the beads and boiled for 5 min to elute the complex.
Western blot analysis was used for the validation of IP samples. The gels were then excised and subjected to MS. UPLC-MS/MS involved using nano ACQUITY UPLC Columns (Waters) with Orbitrap Elite Mass Spectrometers (Thermo Fisher Scientific, Waltham, MA, USA). The resulting MS/MS data were handled with Proteome Discoverer 1.3 (high peptide confidence, peptide ion score > 20).
Flow cytometry
To identify subsets of inflammatory cells in murine models, single-cell suspensions were isolated from mouse aortas following established protocols54. The enzymatically digested suspensions were filtered through a 70 µm sterile mesh and centrifuged at 300 g for 5 minutes at 4 °C. The resulting pellet was resuspended in a buffer containing 0.5% BSA. Cell viability and count were determined using trypan blue exclusion and an automated cell counter (Countess, Life Technologies). Anti-CD16/CD32 (101302, biolgend) was added and incubated for 10 minutes at room temperature to minimize nonspecific binding. Subsequently, cells were stained with specific marker antibodies APC anti-mouse CD83(121509, biolgend), Percpcy5.5 anti-mouse CD45(103132, biolgend), FITC anti-mouse CD11b(101206, biolgend), PE anti-mouse F4/80(12-4801-82, ebioscience), Pacific Blue anti-mouse Ly-6G/Ly-6C(Gr-1)(108429, biolgend), PE anti-mouse CD19(115507, biolgend), APC anti-mouse NK1.1(108709, biolgend) and FITC anti-mouse CD3 (100203, biolgend). Data were further analyzed by NovoExpress® Software.
The enzyme-linked immunosorbent assay (Elisa)
After modeling, blood samples were collected and stored at −80 °C until further analysis. Following the manufacturers’ guidelines, plasma levels of IL-6 (Cat#88-7064, Invitrogen), IL-10 (Cat#88-7105, Invitrogen), TNF-α (Cat#88-7324, Invitrogen), and MCP-1 (Cat#BMS6005, Invitrogen) were quantified using Elisa kits.
Proteomics
Total cellular protein was extracted from isolated peritoneal macrophages derived from both ILF3M-KO mice (n = 3) and wild-type counterparts (n = 3) for analysis through 4D label-free quantification (4D-LFQ) proteomics, conducted by PTM Biolabs (Hangzhou, China). The analysis employed a false discovery rate (FDR) of 5% for both protein identification and peptide spectral matching (PSM), establishing a threshold for differential expression: proteins with fold changes exceeding 1.2 and adjusted P values below 0.05 were classified as differentially expressed proteins (DEPs). Functional enrichment analysis of these DEPs was carried out using a two-tailed Fisher’s exact test. Significance was attributed to Gene Ontology (GO) terms, KEGG pathways, and protein domains that achieved a p-value of less than 0.05.
Ribosome sequencing (Ribo-seq) and RNA Sequencing
Ribosome profiling was performed on isolated peritoneal macrophages from both ILF3M-KO (n = 6) and ILF3M-WT (n = 6) tissues by Gene Denovo (Guangzhou, China). In brief, cycloheximide (Sigma, St Louis, MO, USA; 100 µg/ml) was added to the peritoneal macrophage medium for 2 min to block translational elongation. The supernatant was collected after trituration and centrifuged at 20000 g for 10 min at 4 °C. To prepare RFs, 10 µL of RNase I (NEB, Ipswich, MA, USA) and 6 µL of DNase I (NEB, Ipswich, MA, USA) were added to 400 µL of lysate, mixed and incubated for 45 min at room temperature. Nuclease digestion was halted via adding 10 µL of SUPERase, an RNase Inhibitor (Ambion, Austin, TX, USA). Then 100 μL of digested RFs was added to the prepared Size exclusion columns (Illustra MicroSpin S-400 HR Columns; GE Healthcare; catalog no. 27- 5140-01) and centrifuged (600 g, 2 min). Subsequently, 10 μL of 10% (w/v) SDS was added up to the elution, and RFs (size > 17nt) were separated via RNA Clean and Concentrator-25 kit (Zymo Research; R1017). Further, rRNA and residual DNA probes were removed via RNase H (NEB, Ipswich, MA, USA) and DNase I (NEB, Ipswich, MA, USA). Lastly, the RFs were refined by magnet beads (Vazyme, Nanjing, Jiangsu, China).
Ribo-seq libraries were framed by the NEBNext® Multiple Small RNA Library Prep Set for Illumina® (catalog no. E7300S, E7300L). In brief, adapters were added up to RFs, succeeded by reverse transcription and PCR amplification. The PCR products (140-160 bp size) were enriched to construct cDNA library, sequenced using Illumina HiSeqTM X10. Moreover, we used FASTP to filter the raw data and remove low-quality bases (over 50%) or N bases (over 10%). After trimming the adapter sequences, reads with length between 20–40 bp were kept for analysis. Raw reads were filtered and mapped to the mouse ribosome RNA database using bowtie2. The reads mapped to rRNAs, transfer RNAs (tRNA), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and miRNAs were eliminated. Processed RNA-seq reads were mapped to the rn6 genome using whole-genome alignment by STAR with a 2-pass setting. Ribo-seq data analysis was performed as previously described.[23] Differential translated genes between groups were defined as |log2(FoldChange)| > 0.3 and p-value < 0.05.
For RNA-seq, total RNA of peritoneal macrophages from both ILF3M-KO (n = 6) and ILF3M-WT (n = 6) tissues was isolated utilizing the RNeasy Mini Kit (Qiagen, Germany). Subsequently, strand-specific libraries were constructed employing the TruSeq® Stranded Total RNA Sample Preparation kit (Illumina, USA) following the manufacturer’s instructions. Library quantification was conducted using a Qubit® 2.0 Fluorometer (Life Technologies, USA) and validated by Agilent 2100 bioanalyzer (Agilent Technologies, USA) to confirm the insert size and calculate the mole concentration. Library clustering was performed on a cBot system at a concentration of 10 pM, followed by sequencing on the HiSeq X Ten platform (Illumina, USA) (Illumina, USA). The sequencing raw reads were preprocessed to remove rRNA sequences, sequencing adapters, short fragments, and other low-quality reads. We employed Tophat v2.0.955 for mapping the filtered reads to the mouse mm10 reference genome, allowing for a maximum of two mismatches. Following genomic mapping, Cufflinks v2.1.156 was utilized with a reference annotation to calculate FPKM values for known gene models. Differential expression of genes between the groups was ascertained using the DESeq R package, with stringent criteria set at FoldChange > 2 and p-value < 0.05. Translational efficiency was quantified by the ratio of translating mRNAs to total mRNAs, defined as: TE = (FPKM in Ribo-seq) / (FPKM in RNA-seq). Furthermore, to elucidate the functional similarity among identified genes, we employed the Friends analysis, a method executed using the GOSemSim package57. The raw data from the ribosome sequencing and associated mRNA sequencing conducted in this study are publicly available, which have been deposited in the NCBI Gene Expression Omnibus (GEO) and can be accessed through the GEO Series accession numbers GSE241911, GSE242024, GSE242088, and GSE242185.
mRNA stability analysis and qRT-PCR
Macrophages were suspended in TRIzol Reagent (Invitrogen, Waltham, MA, USA) after treatment with actinomycin D (Act-D, 10 mg/mL) for 0, 2, 4, 8, and 12 h. Total RNA was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany) based on the manufacturer’s recommendations. The total RNA was reverse transcribed by using the PrimeScript RT Reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, Japan), and qRT-PCR was conducted using the LightCycler 480 SYBR Green I Master (Roche Life Science, NSW, Australia). The level of each transcript was normalized to the control gene β-actin and shown relative to the average expression of the control sample (n = 5 under each experimental condition).
Statistical analysis
The ggplot2 package of the interesting plots in the finding of the R foundation and statistic in R v4.2.1 (R Foundation for Statistical Computing, Vienna, Austria)58,59, and Prism 9.0 (GraphPad Inc) were used for statistical analyzes. Continuous parameters are presented as (mean ± SD), and categorical variables are shown as percentages. After verification of the normal distribution of the data via the Shapiro-Wilk test and variance equality via the Levene test, the Student’s t test was used when the variance of the two groups was equal, and the unequal variance t test was used for data with unequal variances. Otherwise, the Mann-Whitney U test is for data sets that are not normally distributed. Brown Forsythe test was used to evaluate the homogeneity of variance in multiple groups ( > 2 groups). After One-way ANOVA analysis, a Tukey post-hoc analysis was used if the data passed the equal variance test; otherwise, a Welch ANOVA test was performed, followed by a post hoc analysis. Two-way ANOVA with a Bonferroni post hoc test was used to test the difference among groups with multiple factors. The survival curve analysis was analyzed using the Kaplan–Meier product-limit approach and compared by the log-rank test. We downloaded publicly available data from the Gene Expression Synthesis (GEO) database (GSE57691 and GSE12591) and obtained DEGs by GEO2R analysis.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD047695. IP-MS datasets are available via ProteomeXchange with identifiers PXD047694. All sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under the BioProject ID PRJNA1050581 for Ribo-seq, PRJNA1050631 for RIP-seq. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
References
Sakalihasan, N. et al. Abdominal aortic aneurysms. Nat. Rev. Dis. Prim. 4, 34 (2018).
Shimizu, K., Mitchell, R. N. & Libby, P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb. Vasc. Biol. 26, 987–994 (2006).
Liu, C. L. et al. Adipocytes promote interleukin-18 binding to its receptors during abdominal aortic aneurysm formation in mice. Eur. Heart J. 41, 2456–2468 (2020).
Filiberto, A. C. et al. Endothelial pannexin-1 channels modulate macrophage and smooth muscle cell activation in abdominal aortic aneurysm formation. Nat. Commun. 13, 1521 (2022).
Wang, S. K. & Murphy, M. P. Immune modulation as a treatment for abdominal aortic aneurysms. Circ. Res 122, 925–927 (2018).
Wallinder, J., Bergqvist, D. & Henriksson, A. E. Proinflammatory and anti-inflammatory cytokine balance in patients with abdominal aortic aneurysm and the impact of aneurysm size. Vasc. Endovasc. Surg. 43, 258–261 (2009).
Vucevic, D. et al. Inverse production of IL-6 and IL-10 by abdominal aortic aneurysm explant tissues in culture. Cardiovasc Pathol. 21, 482–489 (2012).
Gavrila, D. et al. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb. Vasc. Biol. 25, 1671–1677 (2005).
Maiellaro, K. & Taylor, W. R. The role of the adventitia in vascular inflammation. Cardiovasc Res. 75, 640–648 (2007).
Castella, S., Bernard, R., Corno, M., Fradin, A. & Larcher, J. C. Ilf3 and NF90 functions in RNA biology. Wiley Interdiscip. Rev. RNA 6, 243–256 (2015).
Nazitto, R. et al. ILF3 is a negative transcriptional regulator of innate immune responses and myeloid dendritic cell maturation. J. Immunol. 206, 2949–2965 (2021).
Li, K. et al. ILF3 is a substrate of SPOP for regulating serine biosynthesis in colorectal cancer. Cell Res. 30, 163–178 (2020).
Yamada, Y., Matsui, K., Takeuchi, I. & Fujimaki, T. Association of genetic variants with dyslipidemia and chronic kidney disease in a longitudinal population-based genetic epidemiological study. Int J. Mol. Med. 35, 1290–1300 (2015).
Yoshida, T. et al. Association of polymorphisms of BTN2A1 and ILF3 with myocardial infarction in Japanese individuals with or without hypertension, diabetes mellitus or chronic kidney disease. Int J. Mol. Med 27, 745–752 (2011).
Ma, Y. et al. circACTA2 mediates Ang II-induced VSMC senescence by modulation of the interaction of ILF3 with CDK4 mRNA. Aging (Albany NY) 13, 11610–11628 (2021).
Davis, F. M. et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J. Exp. Med. 218, e20201839 (2021).
Raffort, J. et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat. Rev. Cardiol. 14, 457–471 (2017).
Dorrington, M. G. & Fraser, I. D. C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol. 10, 705 (2019).
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J. & Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys. Acta Mol. Basis Dis. 1863, 585–597 (2017).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2, 17023 (2017).
Armaos, A. et al. catRAPID omics v2.0: going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Res. 49, W72–w79 (2021).
Yang, J. L. et al. Keap1-Nrf2 signaling activation by bardoxolone-methyl ameliorates high glucose-induced oxidative injury in human umbilical vein endothelial cells. Aging (Albany NY) 12, 10370–10380 (2020).
Wang, Y. Y., Yang, Y. X., Zhe, H., He, Z. X. & Zhou, S. F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel Ther. 8, 2075–2088 (2014).
de Zeeuw, D. et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 369, 2492–2503 (2013).
Shelton, L. M., Park, B. K. & Copple, I. M. Role of Nrf2 in protection against acute kidney injury. Kidney Int 84, 1090–1095 (2013).
Yamada, Y. et al. Association of a polymorphism of BTN2A1 with myocardial infarction in East Asian populations. Atherosclerosis 215, 145–152 (2011).
Yoshida, T. et al. Association of polymorphisms of BTN2A1 and ILF3 with myocardial infarction in Japanese individuals with different lipid profiles. Mol. Med. Rep. 4, 511–518 (2011).
Xie, F. et al. ILF3 is responsible for hyperlipidemia-induced arteriosclerotic calcification by mediating BMP2 and STAT1 transcription. J. Mol. Cell Cardiol. 161, 39–52 (2021).
Li, H. et al. Lysyl hydroxylase 1 (LH1) deficiency promotes angiotensin II (Ang II)-induced dissecting abdominal aortic aneurysm. Theranostics 11, 9587–9604 (2021).
Sakaue, T. et al. Perivascular adipose tissue angiotensin ii type 1 receptor promotes vascular inflammation and aneurysm formation. Hypertension 70, 780–789 (2017).
Wu, M. Y., Li, C. J., Hou, M. F. & Chu, P. Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int J. Mol. Sci. 18, 2034 (2017).
Mellak, S. et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe-/- mice. Arterioscler Thromb. Vasc. Biol. 35, 378–388 (2015).
Harada, T. et al. Focal adhesion kinase promotes the progression of aortic aneurysm by modulating macrophage behavior. Arterioscler Thromb. Vasc. Biol. 37, 156–165 (2017).
Hiromi, T. et al. Excessive EP4 signaling in smooth muscle cells induces abdominal aortic aneurysm by amplifying inflammation. Arterioscler Thromb. Vasc. Biol. 40, 1559–1573 (2020).
Dale, M. A., Ruhlman, M. K. & Baxter, B. T. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb. Vasc. Biol. 35, 1746–1755 (2015).
Yu, Y., Wan, Y. & Huang, C. The biological functions of NF-kappaB1 (p50) and its potential as an anti-cancer target. Curr. Cancer Drug Targets 9, 566–571 (2009).
Saccani, A. et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 66, 11432–11440 (2006).
Cao, S., Zhang, X., Edwards, J. P. & Mosser, D. M. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 281, 26041–26050 (2006).
Li, Z. et al. USP9X controls translation efficiency via deubiquitination of eukaryotic translation initiation factor 4A1. Nucleic Acids Res 46, 823–839 (2018).
Chien, J. Y., Chou, Y. Y., Ciou, J. W., Liu, F. Y. & Huang, S. P. The effects of two nrf2 activators, bardoxolone methyl and omaveloxolone, on retinal ganglion cell survival during ischemic optic neuropathy. Antioxid. (Basel) 10, 1466 (2021).
Yang, C. C., Lin, C. C., Jou, M. J., Hsiao, L. D. & Yang, C. M. RTA 408 inhibits interleukin-1β-induced MMP-9 expression via suppressing protein kinase-dependent NF-κB and AP-1 activation in rat brain astrocytes. Int J. Mol. Sci. 20, 2826 (2019).
Sun, Q. et al. Bardoxolone and bardoxolone methyl, two Nrf2 activators in clinical trials, inhibit SARS-CoV-2 replication and its 3C-like protease. Signal Transduct. Target Ther. 6, 212 (2021).
Nangaku, M. et al. Randomized clinical trial on the effect of bardoxolone methyl on GFR in diabetic kidney disease patients (TSUBAKI study). Kidney Int Rep. 5, 879–890 (2020).
Golledge, J., Thanigaimani, S., Powell, J. T. & Tsao, P. S. Pathogenesis and management of abdominal aortic aneurysm. Eur. Heart J. 44, 2682–2697 (2023).
Ailawadi, G. et al. Gender differences in experimental aortic aneurysm formation. Arterioscler Thromb. Vasc. Biol. 24, 2116–2122 (2004).
Ding, Y. N. et al. SIRT6 is an epigenetic repressor of thoracic aortic aneurysms via inhibiting inflammation and senescence. Signal Transduct. Target Ther. 8, 255 (2023).
Hisamichi, M. et al. Role of bardoxolone methyl, a nuclear factor erythroid 2-related factor 2 activator, in aldosterone- and salt-induced renal injury. Hypertens. Res. 41, 8–17 (2018).
Barisione, C. et al. Rapid dilation of the abdominal aorta during infusion of angiotensin II detected by noninvasive high-frequency ultrasonography. J. Vasc. Surg. 44, 372–376 (2006).
Shridas, P. et al. Adipocyte-derived serum amyloid a promotes angiotensin II-induced abdominal aortic aneurysms in obese C57BL/6J mice. Arterioscler Thromb. Vasc. Biol. 42, 632–643 (2022).
Daugherty, A. & Cassis, L. A. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb. Vasc. Biol. 24, 429–434 (2004).
Trachet, B., Fraga-Silva, R. A., Jacquet, P. A., Stergiopulos, N. & Segers, P. Incidence, severity, mortality, and confounding factors for dissecting AAA detection in angiotensin II-infused mice: a meta-analysis. Cardiovasc Res. 108, 159–170 (2015).
Toda, G., Yamauchi, T., Kadowaki, T. & Ueki, K. Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc. 2, 100246 (2021).
ten Have, S., Boulon, S., Ahmad, Y. & Lamond, A. I. Mass spectrometry-based immuno-precipitation proteomics - the user’s guide. Proteomics 11, 1153–1159 (2011).
Da Ros, F. et al. Targeting interleukin-1β protects from aortic aneurysms induced by disrupted transforming growth factor β signaling. Immunity 47, 959–973 (2017).
Jackson, R. et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 564, 434–438 (2018).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978 (2010).
Allen, M., Poggiali, D., Whitaker, K., Marshall, T. R. & Kievit, R. A. Raincloud plots: a multi-platform tool for robust data visualization. Wellcome Open Res. 4, 63 (2019).
Wickham, H. ggplot2. WIREs Computational Stat. 3, 180–185 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82370455 and 82200466), the National Key R & D Program of China (No.2017YFC0908700, 2017YFC0908703 and 2017YFC1308000), the Taishan Scholar Project of Shandong Province of China (No.ts20190972, No.tsqn202306377), Natural Science Foundation of Shandong Province (No.ZR2022QH296), Key Clinical Research project of Clinical Research Center of Shandong University (No.2020SDUCRCA016), and Academic promotion program of Shandong First Medical University (No.2021QL021).
Author information
Authors and Affiliations
Contributions
Z.W., J.C., Y.W., H.Y., and S.B. contributed equally to this work. Z.W., M.Z., and Y.C. conceived the project. Z.W. and M.Z. wrote the manuscript. Y.Z., W.J., and M.Z. supervised the study. Z.W., J.C., H.Y., S.B., S.W., and Ze.W. performed human studies and carried out analyses. Z.W., J.C., Y.W., B.X., X.Z., and Y.H. performed animal experiments and prepared figures. Z.W., J.C., and M.Z. interpreted the results. All authors reviewed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Zy., Cheng, J., Wang, Y. et al. Macrophage ILF3 promotes abdominal aortic aneurysm by inducing inflammatory imbalance in male mice. Nat Commun 15, 7249 (2024). https://doi.org/10.1038/s41467-024-51030-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51030-4
- Springer Nature Limited