

Abstract
Culturing and genomic sequencing of Mycobacterium tuberculosis (MTB) from tuberculosis (TB) cases is the basis for many research and clinical applications. The alternative, culture-free sequencing from diagnostic samples, is promising but poses challenges to obtain and analyse the MTB genome. Paradoxically, culture is assumed to impose a diversity bottleneck, which, if true, would entail unexplored consequences. To unravel this paradox we generate high-quality genomes of sputum-culture pairs from two different settings after developing a workflow for sequencing from sputum and a tailored bioinformatics analysis. Careful downstream comparisons reveal sources of sputum-culture incongruences due to false positive/negative variation associated with factors like low input MTB DNA or variable genomic depths. After accounting for these factors, contrary to the bottleneck dogma, we identify a 97% variant agreement within sputum-culture pairs, with a high correlation also in the variants’ frequency (0.98). The combined analysis from five different settings and more than 100 available samples shows that our results can be extrapolated to different TB epidemic scenarios, demonstrating that for the cases tested culture accurately mirrors clinical samples.
Similar content being viewed by others

Introduction
Mycobacterium tuberculosis (MTB) research from clinical samples usually involves a culturing step to obtain sufficient bacteria for downstream applications, including MTB whole-genome sequencing (WGS) studies. As a consequence, our current knowledge of MTB characteristics, including its biology during infection, evolution, epidemiology, and diagnostics, is largely based on cultured samples1,2. It has been hypothesised that the cultivation procedure may constrain the genetic diversity of MTB, either by selecting for specific variants more suited to in vitro growth as happens for some drug resistance mutations or lineages3,4,5, or simply due to the bottleneck imposed by the culture inoculum6. If true, this could distort our understanding of bacterial diversity, particularly at the within host level and even affect epidemiological and drug resistance inferences that rely on the presence or absence of a few single nucleotide polymorphisms (SNPs). Direct sequencing from clinical samples is an alternative as it could bypass the potential disadvantages of culturing7. However, the implementation of culture-free sequencing techniques is challenging due to the complexity of the sample matrix with low amounts of mycobacterial DNA and a mix of contaminants including host genetic material8,9.
Previous efforts on culture-free genome sequencing have focused on developing new and affordable protocols for MTB culture-free WGS and assessing their reliability for clinical applications, mainly for AMR diagnosis and transmission inference10,11,12,13,14,15. Those studies show contradictory results regarding overall genetic diversity comparison between culture-based and culture-free WGS. Some publications have identified no significant differences11,12, while others have reported a reduction in genetic diversity when culturing13,16. These contradictory results probably reflect limitations of the studies, often focused in one single setting and the limited quality of culture-free sequences prevent a proper comparison of genetic diversity between settings. This is specially true to identify low frequency variants which are more likely to suffer from any technical (i.e. sample processing) and analytical limitations17. Therefore, the question of whether culture does actually impose such a bottleneck remains largely unsolved despite its importance. The main goal of this work is to determine, in different TB settings, whether culture reflects the original MTB variability present in the diagnostic samples, typically sputum, making the culture a suitable sample for clinical research.
Here we have put together our own comprehensive dataset including samples from two settings differing in tuberculosis (TB) incidence, burden of AMR, and HIV co-infection; and achieving sufficient sequencing depth to properly compare genetic diversity, especially regarding low-frequency variants. We successfully sequence 61 high-quality sputum-culture pairs from Georgia and Mozambique, which are middle- and high- burden TB settings. For the sputa, we implemented a culture-free WGS approach based either in direct (dWGS) or bait-enrichment (eWGS) sequencing, depending on the amount of MTB DNA. In addition we carry out experimental benchmarking to detect major sources of artifactual genetic variation in culture-free approaches. We also reanalyse available datasets10,13,16 to generalise our results across settings and sequencing approaches. Importantly, we develop a tailored analysis workflow to address the absence of standardised laboratory protocols and bioinformatic pipelines, carefully considering artefacts beyond the role of potential contaminants in the sample. Our customised workflow is capable of demonstrating that culture accurately reflects sputum diversity in all the evaluated settings, albeit with some individual exceptions. Our results indicate that the current knowledge of M. tuberculosis diversity based on culturing methods is robust, in the scenarios tested, and therefore the genetic diversity observed in culture generally mirrors that present in the diagnostic sample. In addition, we provide in-vitro and in-silico tools to test the correlation in scenarios not contemplated in this work.
Results
Selection of sputum-culture pairs
Out of the initial 95 sputum-culture pairs available, 80/95 (84.2%) were suitable for WGS since 4/95 (4.2%) cultures did not grow and 11/95 (11.6%) sputa were MTB negative according to qPCR results and %MTB (Cq>35 and <1%MTB). Given their quality, in terms of Cq and %MTB DNA, 48/80 (60%) sputa underwent dWGS and 32/80 (40%) were enriched before sequencing (Fig. 1a; see Methods). Percentage of MTB was obtained before and after the enrichment step. We observed that sputa with an initial MTB% within 0.5-10% reached 25.5-98.4% after the enrichment, while those containing initially 10-25% of MTB reached more than 90% of MTB (see Methods).

a qPCR Cq vs %MTB. 95 sputa are each one represented by a point. %MTB was obtained by performing a pre-sequencing run in order to determine if each sputum contained enough MTB DNA to be sequenced directly (light purple) or if it required a previous enrichment step (dark purple). Sputum samples in orange were considered negative (Cq>35 and %MTB < 1%). Shape indicates sample origin: triangles for Mozambique, dots for Georgia. Dashed lines represent thresholds to decide which WGS approach to follow, the horizontal line highlights MTB% = 20%, the vertical line highlights Cq = 25. b Mean depth vs coverage. 86 sputa sequenced with enrichment (eWGS, square) or directly (dWGS, diamond). Colour represents the sequencing quality, good quality samples (the ones used for comparison analysis) are in green and bad quality samples in red. The dashed lines represent the coverage and depth cut-off values to consider a good quality sample, the horizontal line highlights a coverage = 0.95 and the vertical line highlights depth = 30X. Source data is provided as a Source Data file.
A total of 19 sputa sequences did not meet the minimum quality criteria (30X depth and 95% coverage) (Fig. 1b), thus these pairs were excluded from further analysis. The sequencing performance of the remaining 61 pairs was: median depth 48X for eWGS (30-157X), 74X (33-302X) for dWGS and 114X (46-347X) for cultures. Median genome coverage, at a minimum of 20X depth, was above 96% in all sputa and cultures (Table 1). See below the samples’ sequencing workflow in the Methods section. All samples’ information is available at Supplementary Data 1 and Supplementary Fig. 1.
Tailored SNP calling analysis for culture-free WGS
To determine if culture-free sequencing approaches introduced a bias during the variant calling, we compared fixed and low-frequency variants detected in trios of eWGS, dWGS and culture-WGS from the same TB case in three suitable examples. After variant calling, we implemented a “recovery” step in which all variants not passing filters in one sequencing approach, but accurately called in another approach, were re-included or “rescued” in the analysis. Our rationale was that culture-free WGS is performed in samples with low amounts of MTB DNA, hence increasing the chance of variants not passing the filters due to lower sequencing depths. Before applying our customised calling filters, we identified up to 22 variants only detected in eWGS, all of them at low frequencies (median=22%, range=10-54%). Exclusive variants were also detected in dWGS (up to 8) and in culture-WGS (up to 3) but the magnitude of the differences were minor compared to eWGS (Fig. 2a Venn diagrams in blue). We screened manually the discrepant variants in the eWGS approach, and identified that all of them appeared in supplementary alignments (Supplementary Fig. 2). A supplementary alignment is a read segment split from the primary read and aligned to a different region of the genome that can produce false positive calls in the variant calling step18,19. After discarding supplementary alignments, most discrepant SNPs disappeared giving a concordance of 99-100% in the variant calling between the three sequencing approaches (Fig. 2a).

a Venn diagrams of the comparisons between trios of direct sputum (dWGS), enriched sputum (eWGS), culture WGS. Amount and percentage of exclusive and common variants are denoted. Blue and orange Venn diagrams represent comparisons of variant calls from default unfiltered bams (including supplementary alignments and filtered bams, respectively. b Comparison of the amount of supplementary alignments between direct (sputum dWGS and culture WGS, light purple) and enriched sputum samples (eWGS, dark purple) in all 61 paired-samples. Median (M) and the total amount of samples (n) are shown. Asterisk (*) highlights a significant p-value (Wilcox test, p-value = 0.0002049). Data are presented as box-plots: centre line represents the median, upper bound located at 75th percentile, lower bound at 25th percentile, whiskers at minimum and maximum values and the outliers. Each dot represents one sample. c On the left there is the comparison of the discrepant SNPs exclusive in sputum, either dWGS and eWGS, before and after filtering supplementary alignments. Colours stand for variant calls from bams before discarding supplementary alignments (blue) and after discarding them (orange). The x-axis is discontinued. The right part shows the percentage of supplementary alignments in sputum files, either dWGS (light purple) and eWGS (dark purple). Plot c contains 32/61 pairs, the 16 ones containing a higher percentage of supplementary alignments in each eWGS and dWGS. Samples are ordered from the highest to the lowest amount of supplementary alignments. The complete version containing the 61 pairs can be seen in Supplementary Fig. 3. d Correlation between the percentage of supplementary alignments and the amount of SNPs removed when discarding supplementary alignments from sputum bam files (represented as SNP difference and calculated as follows: discrepant SNPs exclusive in sputum in Default Bams—Filtered Bams). Colours represent whether the sputum samples have been sequenced directly (light purple) or previously enriched (dark purple). Regression lines, Pearson correlation coefficients (one-side) (Corr) and p-values are shown in the plot. Source data are provided as a Source Data file.
The extended analysis of all the 61 sputum-culture pairs demonstrated that overall the 88.5% (307/347) of the discrepant variants between sputa (either eWGS or dWGS) and paired cultures were false positives introduced by supplementary alignments, and these false positive variation was accumulated in 11/61 sputa (taking into account the sputum samples with more than 5 SNPs discarded due to supplementary alignments). Particularly, eWGS sputa accumulated significantly higher false positive variants than dWGS sputa and cultures (Wilcox test, p-value < 0.001, Fig. 2b) likely due to a higher amount of chimeric reads causing supplementary alignments. After discarding them from the bam files, we identified a mean of 18.3 false positive SNPs (range 0–106 SNPs corresponding to 1–7.4%) per eWGS- culture pair (Fig. 2c, Supplementary Fig. 3) whereas, dWGS-culture pairs showed a lower mean value of 0.3 (1–5 SNP corresponding to 0.3–5%) false positive SNPs (Fig. 2c, Supplementary Fig. 3). Notably, a high, positive and significant correlation (corr=0.95 [confidence interval: 0.85–0.98], p-value < 0.001) was obtained between supplementary alignments and false positive SNPs for eWGS, whereas, in the case of dWGS, the correlation was lower and with limited impact on variation (Fig. 2d). Supplementary alignments accounted for only 1.2% of the reads on average (range: 0.2–7.5; dWGS: 1.0%, eWGS: 2.3%), therefore not impacting the depth and genome coverage (Supplementary Data 1).
Sputum-culture genetic diversity comparison
After applying our tailored SNP calling pipeline we evaluated the genetic diversity between sputum and culture. Overall, a mean of 913 (range 770–1063), representing 97% of the total SNPs detected, were common in sputum and culture (Supplementary Fig. 4). The SNPs rescued represent approx 3% of total SNPs, with a mean of 24 (range 5–69) (Table 2; Fig. 3a; Supplementary Fig. 5). We noticed that rescued SNPs can have an impact, added to that of/in addition to supplementary alignments, when evaluating the correlation between sputum-culture pairs and can affect both sputum and culture estimated diversity. The effect of both supplementary alignments and the rescue step in the total of discrepancies per sample is detailed in Supplementary Fig. 6. Regarding exclusive variants, we also observed a small proportion of sputum-exclusive SNPs (mean 1, range 0–10) and culture-exclusive (mean 1, range 0–11). In other words, most of the sputum-culture pairs (49/61, 80.3%) did not display any discrepancies in sputum; 9/61 (14.8%) presented 1–5; and only 3/61 (4.9%) had between 5 and 10 sputum-exclusive SNPs (Fig. 3b).

a Comparison of the amount of common and exclusive variants (Venn diagrams on the left) and comparison of frequency of variants in sputum (dWGS or eWGS) and culture (on the right). Colours represent common or exclusive variants. Percentage of MTB reads in the dWGS is shown above each plot. The complete figures containing the 61 pairs can be seen in Supplementary Fig. 4 and Supplementary Fig. 5. b Analysis of sputum-exclusive variants in this dataset and published ones. Percentage of pairs in each dataset with 0, 1-5 or more than 5 sputum-exclusive SNPs (all were not fixed variants). Colours represent the dataset. Abbreviations of countries/regions stand for: MZ-Mozambique, GE-Georgia, UK-United Kingdom, LIT-Lituania, SA-South Africa, VLC-Valencia (Spain). c Histogram of the difference of frequency (freq) between variants obtained in sputum versus culture (frequency in sputum - frequency in culture). Colours represent whether the variants are common or exclusive. Percentages of SNPs are shown on the top of the bars. The plot includes the percentage of SNPs that have a frequency difference equal to 0.Total number of variants is shown in grey boxes (n). d Differences of sputum-exclusive SNPs published in the original paper (in blue) versus the ones found by running our pipeline (in orange) for Goig et al.10 (upper panel) and Nimmo et al.16 (bottom panel) datasets. Source data are provided as a Source Data file.
It could be argued that culture-free WGS will be more relevant for determining the frequency of SNPs rather than just their presence/absence. The correlation between the frequencies of SNPs observed in both sputum, either dWGS and eWGS, and culture (common SNPs) was 0.98 (0.93–0.99, p-value < 0.001) (Fig. 3a; Supplementary Fig. 5). By going deeper, we observed that most of the common SNPs (98%) had a small frequency difference within both paired samples (median = 0, ranging 0–10%) with higher frequencies in culture than in sputum (Fig. 3c). To highlight, 83.4% (46445/55700) presented the same frequency in sputum and culture pairs. As expected, these differences were slightly higher for rescued SNPs with 70% of them ranging 0–10% frequency and a 28% ranging 11–30% frequency (Fig. 3c).
Regarding the exclusive variants, we observed a total of 40 sputum-exclusive SNPs distributed among 12 sputa, with 17 out of 40 (42%) falling within a frequency interval of 30-40%. For those culture-exclusives, which were present in 12 cultures, 73.8% (31/42) were found within a frequency range of 10-20% (Fig. 3c).
The results for discrepant variants suggest that, in some cases, variants with intermediate frequencies (between 30-80%) in sputum can be missed in culture. In any case, the number of exclusive variants present only in sputum or in cultures were similar and low compared to those commonly called. To highlight, no fixed-SNP (>90% frequency) was detected within them.
Generalisation of results in different datasets
Finally, we extended our analysis to publicly available sputum-culture sequencing data from three different clinical settings10,13,16. In those, authors described differences in genetic diversity between sputum and culture pairs. We only analysed those sequences meeting the quality criteria (see Methods) for comparisons. After applying our pipeline, we observed that 88% of pairs differed in less than 5 SNPs (Fig. 3b), all of them below 90% frequency. These results are very similar to the ones obtained for the dataset generated in this study (samples from Georgia and Mozambique; Fig. 3b). For those studies with the number of discrepant SNPs available in the publication (Goig et al.10 and Nimmo et al.16), our pipeline reduces the number of discrepant SNPs from an average of 10.4 SNPs (median 7, range 4–32) to 0 SNPs in all 7/7 sputum-culture pairs of Goig et al.10 dataset; and from an average of 8.7 SNPs (median 5, range 1–45) to 1 SNPs (median: 0, range 0–7) in 28/32 pairs from Nimmo et al.16. dataset (Fig. 3d). Contrary to the Goig et al.10 dataset, in three samples of Nimmo et al.16. dataset, the number of SNPs did not decrease before and after applying our tailored pipeline, and there was even an increase in one of the pairs. For that sample (RF0003, Fig. 3d) the number of discrepant SNPs in sputum is higher than for the rest of comparisons.
Overall, the results from our dataset suggest that almost all the genetic variability present in the original sputum is represented by culture both in terms of presence/absence of SNPs but also in terms of SNP frequency correlation within the samples. Re-analysis of available datasets also points to a high sputum-culture concordance after discarding false-positive variants, mostly due to the bioinformatic analysis implemented.
Phylogenetic classification and drug-resistance profile
We obtained a 100% concordance between sputum and culture lineage (L) prediction. We identified 31/61 L4 strains (20 from Mozambique and 11 from Georgia) and 16/61 L2 (8 strains from each dataset). The remaining strains from Mozambique belonged to L1 (13/61 samples) and L3 (1/61 sample). Regarding the re-analysed datasets the lineage distribution was 1/52 L1, 12/52 L2, 2/52 L3 and 37/52 L4. Concordance at lower taxonomic levels according to Coll et al. classification20 was also 100%. Sublineage classification is shown in Supplementary Data 2.
All the 61 sputum samples matched with their paired cultures when constructing a Maximum likelihood tree (Supplementary Fig. 1) obtaining a 0 SNP in all pairs (fixed-SNP > 90% frequency) pairwise distance. Two samples from Mozambique were in the same transmission cluster (measured at a 5 SNP distance cut-off) which was equally detected when culture and sputum genomes were analysed separately.
Regarding AMR-conferring variants, the agreement between the resistance profile predicted in sputum and culture was 100%. We obtained 10/61 pairs with at least one AMR-conferring mutation (6 from Georgia, 4 from Mozambique): 7/10 mono or poly-resistant, 2 MDR and 1 pre-XDR. All these were fixed-SNPs in both sputum and culture. No low-frequency AMR-conferring variants were found. See Supplementary Data 3 for resistance SNPs information. Regarding the re-analysed datasets, we identified 31/52 strains with at least one AMR-conferring mutation in the sputum and the culture. All of them were fSNPs except one from Shockey’s10,13,16 dataset (P10) which was a heteroresistant variant present at 70% in both sputum and culture.
Discussion
In this study, we explore the role of culturing from sputum diagnostic samples as a bottleneck for MTB genetic diversity. In terms of SNPs’ presence/absence, both fixed and minority variants, after comparing the genomic variability, most culture-sputum pairs (91.8%, 56/61) showed a difference of less than 5 (0–3) SNPs. Strikingly, the correlation of the SNPs’ frequencies was very high (0.98, p-value < 0.001). Our findings show that culture mirrors the genetic diversity present in the sputa analysed, considering both the presence/absence of SNPs and their frequencies. We corroborate our results by reanalyzing previously published datasets, suggesting that any apparent contradictions were likely due to differences in bioinformatic pipelines. Importantly, our findings were consistent across the datasets included, encompassing samples from different clinical settings and laboratories.
One key finding of our study was the detection of false-positive variation driven by chimeric reads most likely produced during sample enrichment and library preparation steps. While this is a common issue, we observed that they were significantly higher in the enriched sputa as compared to direct sputa sequenced. The presence of chimeric reads was potentially due to the low concentration of DNA in sputa. Considering that not all DNA was from MTB, the low quantity of target DNA could lead to non-specific amplifications in both library preparation and PCR reactions carried out during the enrichment. Our hypothesis is that such amplifications likely generate chimeric reads, resulting in supplementary alignments during the mapping and, therefore, false variant calls. Furthermore, a high correlation between supplementary alignments and false-positive SNPs was obtained for eWGS sputa, with special impact on those samples with very low MTB DNA to be captured. Importantly, while removing supplementary alignments reduces by more than 80% the discrepant variants in some cases, it has no impact on depth and coverage. However, a more dedicated study of the factors affecting supplementary alignments is warranted as we identify some non-enriched samples with a high number of alignments.
Another source of discrepancies is driven by differences in depth throughout the genome, which is more relevant in culture-free WGS approaches. To solve this, here we implemented a recovery step to rescue the false negative variants present in the sputum or culture. It was observed that the rescued SNPs only make up less than 3% of the total SNPs analysed in the joint dataset. However, because differences between culture and sputum are subtle, they can make a great impact on the interpretation of sputum-culture differences in specific pairs. This phenomenon is more obvious when looking at specific examples like those presented in Fig. 3a. It is worth noting that the absence of culture could have resulted in the oversight of these variants in the sputum, highlighting the critical role of achieving minimum sequencing depths for diversity comparisons. Overall, these findings unequivocally indicate that achieving an accurate representation of the original genetic diversity depends on sequencing depth, appropriate bioinformatic filters and careful post-processing of results.
The main aim of this study was to assess whether culturing distorts the genetic diversity of the bacterial population present in sputum. By performing an enrichment step, we succeed in increasing the MTB DNA by 26.5-fold on average, allowing high-quality sequences of sputa with as low as 1% of MTB DNA, similar to the values obtained by Mann et al. 21. Since we have not explored the potential of culture-free WGS as a diagnostic tool for TB, we performed as many runs as necessary to achieve sufficient coverage and depth to reach our goal, regardless of the sequencing cost per sample. With this effort we were able to perform culture-free sequencing, with high quality, in 76% (61/80) of diagnostic samples, suggesting that further improvements of the technique will be needed as shown by others15. In fact, while previous studies showed that smear-negative and scanty sputa could be enriched and sequenced occasionally10,11,12,13, a formal testing of the limit of detection is still needed. Meanwhile, intermediate alternatives like targeted next-generation sequencing tools are finding a room in the TB diagnostics pipelines22.
Understanding that the diversity of M. tuberculosis is reflected in both sputum and culture has an impact on TB research, since the overwhelming majority of studies today rely on culture sequencing to assess diversity, even in those cases where surgery23 or post-mortem24 samples are interrogated. Our analysis suggests that diversity of the diagnostic sample is well represented in those studies and that substantial variation is not missed in the matching culture. However, as a limitation, in this study we have not been able to analyse the detection of genotypes carrying AMR mutations (or any other mutation) with a fitness cost in culture. Most of our samples are drug susceptible and our analysis has identified only one case of heteroresistance for quinolones which is equally detected in sputum and culture. Thus, the high correlation observed in our sputum-culture pairs may not apply in situations where a subpopulation has a mutation with associated fitness cost. Mutations such as these are known to cause growth delay and affect culture-based diagnostics, particularly in the case of rifampicin-resistance conferring mutations occurring in rpoB4,25. Similarly, this limitation also applies to cases where lineages with different growth rates coexist, potentially affecting the identification of polyclonal infections, as it has been previously suggested for Lineage 6 and animal strains5. Identification of those scenarios requires specific analysis23 out of the scope of this work. For some cases, particularly in the datasets from Georgia and South Africa16, we identified a higher disagreement between sputum and culture. This may be due to the presence of unknown fitness-cost associated mutations or to polyclonal infections with genetically close strains, which are more likely to happen in those settings, particularly if there are high transmission hotspots26. Finally, a proportion of non-cultivable bacterial populations in the sputum of patients has been described in some studies, which may be relevant at the clinical level27,28. In the case that those populations are genotypically different (currently unknown) it may be reflected in the few SNPs and pairs with sputum exclusive SNPs or it may well be a phenomenon occurring below our limit of detection of 10% frequency. Those specific situations remain to be tested and here we provide tools for an accurate assessment of the sputum-culture diversity. Nevertheless, our dataset reflects the reality of many TB cases, which are from single infections and carry drug-resistance mutations that do not cause growth problems in culture.
In conclusion, our results highlight the importance of evaluating and applying appropriate filtering steps when sequencing complex samples, such as sputum, in order to detect and discard sources of false variation. After developing and applying a tailored bioinformatics analysis, we show that culture accurately captures the genetic diversity present in diagnostic samples and this is true across settings and laboratories tested. More specific scenarios remain to be analysed and here we provide laboratory and bioinformatics tools to do so. On one hand, from a diagnostic point of view our results reflect the long road ahead towards whole genome-based diagnostics, as highlighted in recent publications15. On the other hand, from a research point of view our results support the large body of work based on culture sequencing.
Methods
Ethics
The samples from Mozambique came from a study that was approved by the National Bioethics Committee for Health of Mozambique (CNBS, Ref:369/CNBS/17) and the Internal Bioethics Committee of CISM. Regarding samples coming from Georgia, the ethical approval was obtained from the Institutional Review board (IRB) of the National Center for Tuberculosis and Lung Diseases within the framework of observational clinical study NCT02715271. All methods were performed in accordance with the relevant guidelines and regulations. An informed consent was signed by all participants after providing a verbal explanation and written information about the study. Regarding participants under 18 years of age, the informed consent was obtained from their relatives (parents or guardians). All data were de-identified before the analysis.
Dataset
We received sputum-culture paired samples from the Centro de Investigação em Saúde de Manhiça (Mozambique) and the National centre for Tuberculosis and Lung Diseases located in Tbilisi (Georgia). Mozambique and Georgia are considered high- and medium- burden tuberculosis countries. No statistical method was used to predetermine sample size. To ensure that the bacillary load present in sputum samples was enough for sequencing, we selected 50 sputum samples from Mozambique graded High/Medium according XpertUltra result and 3+/2+ according to smear microscopy result. Regarding Georgia, we processed all 45 sputum samples received due to the lack of bacillary load information. In summary, we processed 95 sputum-culture paired samples.
Sputum samples, collected at diagnosis, were homogenised and decontaminated in origin countries. In Mozambique, they performed the N-acetyl-L-Cysteine-sodium hydroxide (NALC-NaOH) or Kubica method29, while in Georgia they followed the modified Petroff protocol, that uses 4% NaOH, which was validated under supervision of the WHO Supranational TB Reference Laboratory in Antwerp30.
The paired cultures were grown in 7H11 solid media, supplemented with OADC and glycerol to ensure a high amount of bacteria for sequencing.
All samples, cultures and sputa, were received and processed at FISABIO biosafety level 3 (BSL-3) facilities in Valencia, Spain.
Samples’ processing
DNA extraction from sputum samples
DNA extraction from sputum leftovers was based on a differential cell lysis procedure to remove non-MTB contaminant DNA using MolYsis basic5 kit (Molzym, Germany). We followed a modified version of the original protocol that entailed an initial lysis of non-mycobacterial cells to remove contaminant DNA, followed by an inactivation of MTB cells at 95 °C for 15 min and a mechanical cell disruption using FastPrep. DNA precipitation steps were performed using ethanol, sodium acetate and Glycoblue. DNA extraction steps are detailed as follows:
-
1.
Saline wash
-
11.
Centrifuge 1 mL of sample at 13,000 rpm during 15 min
-
12.
Remove supernatant by pipetting without disturbing the pellet (leave 200uL of liquid)
-
13.
Add 1 mL of PBS and resuspend the pellet by pipetting.
-
11.
-
2.
Lysis of human cells with MolYsis Basic5 kit:
-
21.
Add 250 µL of CM buffer, mix by slow pipetting
-
22.
Incubate 5 min at room temperature (Note: CM is a chaotropic buffer)
-
23.
Add 250 µl buffer DB1 and 10 µl MolDNase B (do not premix) to each sample and immediately vortex for 15 s.
-
24.
Incubate at room temperature for 15 min.
-
25.
Centrifuge the tubes at 13,000 rpm for 10 min.
-
26.
Discard the supernatant taking care to not disturb the pellet.
-
27.
Add 1 mL of RS buffer and pipette up and down until the pellet is resuspended.
-
28.
Centrifuge the tubes at 13,000 rpm for 10 min (During this step, set a thermal block to 95 °C for MTB inactivation.
-
29.
Discard the supernatant taking care to not disturb the pellet.
-
30.
Add 700uL of sterile mqH2O and resuspend the pellet by pipetting up and down.
-
3.
MTB Cells Inactivation:
-
31.
Spin down tubes at maximum speed 30 s and incubate at 95 °C for 15 min.
-
4.
Mechanical cell disruption
-
41.
Transfer all the volume 700uL to a 2 mL FastPrep Lysing Matrix B tube.
-
42.
Break the cells using the FastPrep Mycobacterium tuberculosis program using 1 pulse instead of two (45 s at 6.5 m/s). Microscopic glass beads break the mycobacterial cell.
-
43.
Spin the tubes in the centrifuge and transfer 450 uL to a new microcentrifuge tube.
-
5.
DNA ethanol precipitation procedure:
-
51.
Add 1/10 vols (45ul) of sodium acetate 3 M to the supernatant.
-
52.
Add 1.5ul of GlycoBlue.
-
53.
Add 2 vol (1000 μl) of cold EtOH 96% and vortex 10 s.
-
54.
Incubate at −20 °C for 30–60 min.
-
55.
Centrifuge at 13,000 rpm or maximum speed for 15 min and remove the supernatant leaving a small volume of liquid to not disturb the pellet.
-
56.
Add 1 ml of EtOH 70% centrifuge at maximum speed for 5 min and remove the ethanol without disturbing the pellet.
-
57.
Let the ethanol dry (but not overdry), preferably in a vacuum centrifuge machine.
-
58.
Resuspend the pellet in 50 μl of Tris-Hcl 10 mM and dissolve the DNA by pipetting up and down.
DNA extraction from culture samples
Cultures were heat inactivated at 90 °C during 30 min and then MTB DNA was extracted following the standardised CTAB protocol31 based on an overnight cell wall digestion with lysozyme, followed by incubation steps with proteinaseK, SDS, CTAB and NaCl and a final DNA precipitation step with isopropanol. All bacterial cultures and DNA extraction steps were performed in a BSL-3 laboratory.
qPCR Conditions
We assess the concentration of MTB in sputum samples to decide the sequencing approach. The qPCR was performed in a total volume of 20 μL including 10 μL of Kapa Fast Probe Master Mix 2X, 2 μL of forward and reverse primers mix 2.5 μM, 0.6 μL of probe 10 μM; and 1 ng of DNA. We used DNA normalised to 0.5 ng/μL. The qPCR assay consisted on the amplification of a 65 bp region within the Rv2341 gene using the following primers: Forward-GCCGCTCATGCTCCTTGGAT, Reverse-AGGTCGGTTCGCTGGTCTTG, Probe-TGAGTGCCTGCGGCCGCAGCGC32.
Sequencing selection
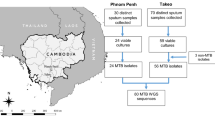
We prepared NexteraXT (Illumina) libraries for all samples. Libraries from cultures were sequenced directly. Regarding sputum samples, we classified libraries for dWGS (direct) or eWGS (enriched) depending on Cq and %TB obtained in pre-sequencing runs as described in ref. 10. Therefore, we performed a qPCR targeting Rv2341 gene10,32 to quantify the amount of MTB DNA. Sputa that obtained a Cq value above 35 (equivalent to less than 2 genome copies) were considered negative (not suitable for sequencing) (See qPCR conditions section). Libraries were prepared for all MTB positive samples (Cq<35) and a pre-sequencing run was performed to estimate the percentage of MTB. According to qPCR and pre-sequencing %MTB result we performed dWGS in sputa obtaining a Cq<25 and more than 20% of MTB and the rest sputum samples underwent an enrichment step before sequencing. The enrichment step consisted of a MTB DNA capture using RNA biotinylated baits (See Enrichment step section). See the flowchart in Fig. 4.

Diagram summarising the sequencing steps and the amount of samples that have passed or have been discarded (in red). Abbreviations: C-Culture, S-Sputum, P-Pair, dWGS-sputum samples not enriched, eWGS-sputum samples enriched.
WGS was performed in Illumina Miseq (2x300bp) or NextSeq (2x150bp) platforms. Samples were sequenced until a minimum of 30X median depth was reached.
Enrichment step
We used myBaits kit (Arbor Biosciences) to perform the hybridisation MTB DNA capture step following the myBaits protocol Version 4.01. This protocol is based on a hybridization of already prepared whole genome sequencing libraries (prepared using NexteraXT kit, Illumina) with RNA biotinylated baits at 65 °C overnight (at least 24 h). During this step, baits hybridised to denatured MTB DNA. Then, most non-captured DNA is discarded by different cleaning steps with streptavidin-coated magnetic beads. Finally, the MTB library is amplified by a post-capture PCR in a 50uL reaction containing 25uL of KAPA HiFi HotStart ReadyMix PCR Kit (Roche), 200 nM of Illumina sequencing primers P5 and P7; and 15uL of captured library. Post-capture PRC conditions are the following: 15 cycles of 20 s to 98 °C, 30 s to 65 °C and 1 min to 72 °C.
The RNA-biotinylated baits panel was designed and developed by Arbor Biosciences using as reference the inferred ancestor genome of the MTB (NC_000962.3), genetically equidistant to all the MTB lineages. This panel is available upon request.
We evaluated the enrichment step by obtaining the fold-change of the percentage of MTB reads between the 32 sputum samples before and after the enrichment. The median fold-change was 10.96 (Fig. 5). In other words, sputa containing an initial MTB% within 0.5–10% got a 64.8% (average) while those with an initial MTB% of 10–25% reached 94.3% after undergoing the enrichment step (Fig. 5).

Comparison of %MTB DNA before and after the enrichment step of the 32 sputum samples that have been enriched. Each point represents a sputum sample. Colours represent samples’ origin orange for samples from Georgia and purple for samples for Mozambique. Lines link each sputum sample before and after the enrichment. Source data are provided as a Source Data file.
Bioinformatics analysis
Core analysis
Analysis was performed using our routine pipeline available at https://gitlab.com/tbgenomicsunit/ThePipeline for both culture and sputum samples. First, reads were trimmed by quality with FastP33; non-MTB reads were discarded using Kraken (v0.10.5)8,34. MTB reads were mapped to the reference ancestor genome (NC_000962.3)35, which is genetically equidistant to all lineages, using BWA (version 0.7.10-r789)18.
Samples with a median depth below 30X and less than 95% of the genome covered were discarded for downstream variant comparison analysis. For the remaining good quality samples, variants were called with VarScan (version 2.3.7)36 and Samtools v1.1537 by applying stricter coverage and frequency filters for culture samples than for sputum samples (variants called in at least 3 reads, in both strands for sputum; variants appearing in 6 reads, in at 10X depth and in both strands, for culture) (see parameters for VarScan in Fig. 6). For the genetic diversity comparison analysis, we discarded SNPs appearing in high density regions with GATK (version 4.0.2.1)38 or genomic regions that are known to be challenging for short-read mapping such as repetitive genes or mobile elements (PE/PPE gene families)39 by using a customised Python2 (version 2.7.5) script. Highly conserved genes such as rrs and rrl were also discarded for the comparisons to avoid false positive variability coming from non-MTB reads not discarded by Kraken. We also obtained Pearson’s correlation of the variant frequency obtained in each sputum-culture pair (the mean, median and the range).

Fixed SNPs (fSNPs) are the ones called at a frequency above 90%. After obtaining SNP files additional filters were applied as described in the Core analysis section.
Capture technical validation
To investigate if the capture step introduced bias in variant calling, we analysed sputum samples sequenced by direct (dWGS) and enrichment (eWGS) methods and compared to the respective culture (hereafter, trios-analysis). All discrepant positions within trios, but particularly between eWGS and dGWS samples, were manually checked on the reads alignment to the reference genome in Tablet viewer (v1.17.08.17, see Supplementary Fig. 2) to identify the causes of the inconsistency. All discrepant variants were identified in supplementary alignments, which are part of a read that is split and aligned to a different part of the genome than the primary alignment18,19, were identified as the main source of false positive variants. Therefore, we discarded them from bam files running samtools v1.15 (command: samtools view -bh -f0 -F256 -F2048 $BAM_FILE), before variant calling step. Afterwards, we evaluated the impact of discarding supplementary alignments in diversity comparison in all the 61 pairs (sputum-culture) analysed.
After applying all filters described above, a pairwise comparison of variants was conducted within sputum samples and their corresponding cultures. At this point we included a recovery step based on searching for the discrepant SNPs, in the files of the paired sample obtained from a less restrictive variant calling. The reason was that in samples with suboptimal depth the missing SNP may still exist but may have been lost during the filtering steps. Parameters used in VarScan pileup2snp for generating rescued SNP files for comparison analysis were: --min-coverage 2, --min-reads2 1, --min-freq-for-hom 0.9, --min-var-freq 0.01.
The average of rescued SNP shared within sputum and culture pairs was 24 (5–69) representing the 2.5% (0.5–7.9%) of the total variants, highlighting the importance of the step when dealing with samples of heterogeneous coverage.
To verify if rescue was biassing the comparison analysis, we conducted a concordance analysis to corroborate that our rescue approach was not artificially removing true discrepant positions. We analysed 35 sputum-culture pairs with depth of coverage >50x and >95% of the genome covered by applying the same calling pipeline (see the variant calling parameters for cultures in Fig. 6) and skipping the rescue step. Results showed a median of 886 (range: 828–1059) of common variants, 2 (range: 0–6) sputum-exclusive variants and 2 (range: 0–16) culture-exclusive variants. This corresponded to a median Cohen’s Kappa coefficient40 of 0.998 (0.992-1) which represented an almost perfect agreement. In this case, the median Pearson’s correlation of variants frequencies was 0.939 (0.454–0.991).
Finally, variants appearing in both samples were classified as “common”, variants rescued were classified as “common rescued”, sputum-exclusive variants were called “only sputum” and those culture-exclusive were classified as “only culture”.
Validation using available datasets
We applied our pipeline described to analyse genetic variability on available sputum-culture paired data sets. We downloaded the sequences under the following project accession numbers from ENA: PRJEB920612, PRJNA48671316 and PRJEB3760910. Sequences were analysed and only pairs passing the quality filters were compared as explained above (see Bioinformatic Analysis from Methods).
Resistance profiling and phylogenetic classification
The resistance profile was obtained by looking for DR-conferring mutations (>10% frequency) listed in the WHO catalogue (version from 03/09/2021)41. Lineage and sublineage classification of the strains was determined by looking for phylogenetic variants reported in bibliography20,42.
Overall, even though the sequencing quality for some sputum was not enough for the comparison analysis, we were able to classify phylogenetically 74/80 (92.5%) sputum samples sequenced and 72/80 (90.0%) at a sublineage level according Coll et al. classification20.
Pairwise distance between each sputum-culture pair was obtained using the R package ape43 based on a multiple alignment of fixed SNPs (fSNPs, >90% frequency). In addition, we also obtained the pairwise distance to see whether paired samples clustered together. Neighbour joining trees were constructed using MEGA version X44.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequences generated in this paper have been deposited in the European Nucleotide Archive (ENA) under project accession number: PRJEB64897. Detailed information about the samples can be found at Supplementary Data 1. We also downloaded the sequences under the following project accession numbers from ENA: PRJEB9206 (https://doi.org/10.1128/jcm.00486-15)12, PRJNA486713 (https://doi.org/10.1186/s12864-019-5782-2)16 and PRJEB37609 (https://doi.org/10.1016/S2666-5247(20)30060-4)10. Source data are provided with this paper.
Code availability
The pipeline used for analysing sputum and culture sequences has been developed and validated in our laboratory and it is available at: https://gitlab.com/tbgenomicsunit/ThePipeline.
References
Meehan, C. J. et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat. Rev. Microbiol. 17, 533–545 (2019).
Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16, 202–213 (2018).
Gagneux, S. et al. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312, 1944–1946 (2006).
Miotto, P., Cabibbe, A. M., Borroni, E., Degano, M. & Cirillo, D. M. Role of disputed mutations in the rpob gene in interpretation of automated liquid MGIT culture results for rifampin susceptibility testing of Mycobacterium tuberculosis. J. Clin. Microbiol. 56, e01599–17 (2018).
Gehre, F. et al. Deciphering the growth behaviour of Mycobacterium africanum. PLoS Negl. Trop. Dis. 7, e2220 (2013).
Dhillon, J., Fourie, P. B. & Mitchison, D. A. Persister populations of Mycobacterium tuberculosis in sputum that grow in liquid but not on solid culture media. J. Antimicrob. Chemother. 69, 437–440 (2014).
Mohamed, S., Köser, C. U., Salfinger, M., Sougakoff, W. & Heysell, S. K. Targeted next-generation sequencing: a Swiss army knife for mycobacterial diagnostics? Eur. Respir. J. 57, 2002132 (2021).
Goig, G. A., Blanco, S., Garcia-Basteiro, A. L. & Comas, I. Contaminant DNA in bacterial sequencing experiments is a major source of false genetic variability. BMC Biol. 18, 1–15 (2020).
Eshetie, S. & van Soolingen, D. The respiratory microbiota: new insights into pulmonary tuberculosis. BMC Infect. Dis. 19, 92 (2019).
Goig, G. A. et al. Whole-genome sequencing of Mycobacterium tuberculosis directly from clinical samples for high-resolution genomic epidemiology and drug resistance surveillance: an observational study. Lancet Microbe 1, e175–e183 (2020).
Votintseva, A. A. et al. Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J. Clin. Microbiol. 55, 1285–1298 (2017).
Brown, A. C. et al. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J. Clin. Microbiol. 53, 2230–2237 (2015).
Shockey, A. C., Dabney, J. & Pepperell, C. S. Effects of host, sample, and in vitro culture on genomic diversity of pathogenic mycobacteria. Front. Genet. 10, 477 (2019).
Doyle, R. M. et al. Direct whole-genome sequencing of sputum accurately identifies drug-resistant mycobacterium tuberculosis faster than MGIT culture sequencing. J. Clin. Microbiol. 56, e00666–18 (2018).
Nilgiriwala, K. et al. Genomic sequencing from sputum for tuberculosis disease diagnosis, lineage determination, and drug susceptibility. Prediction. J. Clin. Microbiol. 61, e0157822 (2023).
Nimmo, C. et al. Whole genome sequencing Mycobacterium tuberculosis directly from sputum identifies more genetic diversity than sequencing from culture. BMC Genomics 20, 389 (2019).
Goossens, S. N. et al. Detection of minor variants in Mycobacterium tuberculosis whole genome sequencing data. Brief. Bioinform. 23, bbab541 (2022).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Structural variants and the SAM format—the long (reads) and short (reads) of it. https://cmdcolin.github.io/posts/2022-02-06-sv-sam.
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 1–5 (2014).
Mann, B. C., Jacobson, K. R., Ghebrekristos, Y., Warren, R. M. & Farhat, M. R. Assessment and validation of enrichment and target capture approaches to improve Mycobacterium tuberculosis WGS direct patient samples. J. Clin. Microbiol. 61, e0038223 (2023).
Use of targeted next-generation sequencing to detect drug-resistant tuberculosis. https://www.who.int/publications/i/item/9789240076372.
Moreno-Molina, M. et al. Genomic analyses of Mycobacterium tuberculosis from human lung resections reveal a high frequency of polyclonal infections. Nat. Commun. 12, 2716 (2021).
Lieberman, T. D. et al. Genomic diversity in autopsy samples reveals within-host dissemination of HIV-associated Mycobacterium tuberculosis. Nat. Med. 22, 1470–1474 (2016).
Van Deun, A. et al. Mycobacterium tuberculosis borderline rpoB mutations: emerging from the unknown. Eur. Respir. J. 58, 2100783 (2021).
Pandey, P. et al. Mycobacterium tuberculosis polyclonal infections through treatment and recurrence. PLoS ONE 15, e0237345 (2020).
Mukamolova, G. V., Turapov, O., Malkin, J., Woltmann, G. & Barer, M. R. Resuscitation-promoting factors reveal an occult population of tubercle Bacilli in Sputum. Am. J. Respir. Crit. Care Med. 181, 174–180 (2010).
Chengalroyen, M. D. et al. Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med. 194, 1532–1540 (2016).
Kubica, G. P., Dye, W. E., Cohn, M. L. & Middlebrook, G. Sputum digestion and decontamination with N-acetyl-L-cysteine—sodium hydroxide for culture of mycobacteria. Am. Rev. Respir. Dis. 87, 775–779 (1963).
Tripathi, K. et al. Modified Petroff’s method: an excellent simplified decontamination technique in comparison with Petroff’s method. Int J. Recent Trends Sci. Technol. 10, 461–464 (2014).
Somerville, W., Thibert, L., Schwartzman, K. & Behr, M. A. Extraction of Mycobacterium tuberculosis DNA: a question of containment. J. Clin. Microbiol. 43, 2996–2997 (2005).
Goig, G. A. et al. Towards next-generation diagnostics for tuberculosis: identification of novel molecular targets by large-scale comparative genomics. Bioinformatics 36, 985–989 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Comas, I. et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42, 498–503 (2010).
Koboldt, D. C. et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Marin, M. et al. Benchmarking the empirical accuracy of short-read sequencing across the M. tuberculosis genome. Bioinformatics 38, 1781–1787 (2022).
McHugh, M. L. Interrater reliability: the kappa statistic. Biochem. Med. 22, 276–282 (2012).
Walker, T. M. et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe 3, e265–e273 (2022).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 48, 1535–1543 (2016).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Acknowledgements
This work has been supported by the following: European Research Council (ERC): H2020-ERC-COG/0800; Ministerio Español de Ciencia e Innovación: PID2022-137607OB-I00 and Fundació La Caixa: HR21-00415 received by I.C; Stop TB partnership (TB REACH): STBP/TBREACH/GSA/W5-30 received by A.G.B; and International Science and Technology Center (ISTC): Project #G-2143; National Institute of Allergy and Infectious Diseases (NIH) received by N.S. We would like to thank Katharine Walter for reviewing the manuscript and providing her feedback.
Author information
Authors and Affiliations
Contributions
A.G.B., B.S.C. and E.M. were responsible for data and samples collection in Mozambique. S.V., I.K., Z.A., A.R., A.G., M.S. and N.S. were responsible for data and samples collection in Georgia. C.M.L., M.T.P., L.V. processed and sequenced the samples. M.T.P., M.G. and I.C. conceived and designed the experiments. C.M.L., M.G. and I.C. were involved in data interpretation. C.M.L., G.G., M.G. and I.C. wrote the paper. All the authors reviewed and approved the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Maha Farhat who co-reviewed with Brendon Mann; Conor Meehan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mariner-Llicer, C., Goig, G.A., Torres-Puente, M. et al. Genetic diversity within diagnostic sputum samples is mirrored in the culture of Mycobacterium tuberculosis across different settings. Nat Commun 15, 7114 (2024). https://doi.org/10.1038/s41467-024-51266-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51266-0
- Springer Nature Limited