

Abstract
Preclinical studies suggest that simultaneous HER2/VEGF blockade may have cooperative effects in gastroesophageal adenocarcinomas. In a single-arm investigator initiated clinical trial for patients with untreated advanced HER2+ gastroesophageal adenocarcinoma, bevacizumab was added to standard of care capecitabine, oxaliplatin, and trastuzumab in 36 patients (NCT01191697). Primary endpoint was objective response rate and secondary endpoints included safety, duration of response, progression free survival, and overall survival. The study met its primary endpoint with an objective response rate of 81% (95% CI 65–92%). Median progression free and overall survival were 14.0 (95% CI, 11.3–36.4) and 23.2 months (95% CI, 16.6–36.4), respectively. The median duration of response was 14.9 months. The regimen was well tolerated without unexpected or severe toxicities. In post-hoc ctDNA analysis, baseline ctDNA features were prognostic: Higher tumor fraction and alternative MAPK drivers portended worse outcomes. ctDNA at resistance identified oncogenic mutations and these were detectable 2–8 cycles prior to radiographic progression. Capecitabine, oxaliplatin, trastuzumab and bevacizumab shows robust clinical activity in HER2+ gastroesophageal adenocarcinoma. Combination of VEGF inhibitors with chemoimmunotherapy and anti-PD1 regimens is warranted.
Similar content being viewed by others

Introduction
Gastroesophageal adenocarcinomas (GEAs) are the 2nd leading cause of cancer-related mortality worldwide1 and often present with incurable advanced disease. Treatment is focused on systemic therapy, which until recently has consisted of a fluoropyrimidine and platinum chemotherapy doublet2. Incorporation of PD1 checkpoint inhibitors with first line therapy have shown an improvement in objective response rate (ORR), progression free survival (PFS) and overall survival (OS), creating a new standard of care, especially for patients with PD-L1 positive tumors3.
Amplification of the HER2 is found in 15–20% of gastroesophageal adenocarcinomas4, comparable to the rates of HER2 amplification seen in breast cancers5. The ToGA trial established the efficacy of adding trastuzumab to doublet chemotherapy for HER2+ advanced gastroesophageal adenocarcinomas, showing an improvement in survival, thus becoming the standard of care for these patients over a decade ago6. More recently, the KEYNOTE-811 trial demonstrated that addition of the checkpoint inhibitor pembrolizumab to the fluoropyrimidine, platinum and trastuzumab triplet significantly improved progression free survival in PD-L1 CPS positive patients7,8.
Targeting tumor angiogenesis in advanced gastroesophageal adenocarcinomas has delivered mixed results. Although the VEGFR-2 blocking monoclonal antibody, ramucirumab, has demonstrated an overall survival benefit in two randomized clinical trials, REGARD9 and RAINBOW10, the effect of anti-VEGF agents in the 1st line setting for gastroesophageal cancers has been underwhelming. The addition of the VEGF-A inhibitor, bevacizumab, to a fluoropyrimidine-platinum doublet in the AVAGAST trial led to a statistically significant improvement in response rate and PFS, but did not improve the primary end point of overall survival11. Similarly, addition of ramucirumab (RAINFALL trial) or the VEGF-Trap, ziv-aflibercept (ZAMEGA trial), to 1st line chemotherapy in unselected gastroesophageal adenocarcinomas did not show any significant clinical benefit12,13.
HER2 overexpression increases VEGF levels via increased HIF1α protein synthesis in human breast cancer cell lines14 and HER2 and VEGF expression are positively correlated in primary human breast cancers15. Furthermore, dual inhibition of HER2 and VEGF was associated with improved anti-tumor activity in pre-clinical models of gastric cancer16,17.
Based on these promising pre-clinical data, we sought to establish the safety and efficacy of bevacizumab in combination with capecitabine, oxaliplatin (CAPOX) and trastuzumab in patients with previously untreated HER2 positive locally advanced or metastatic gastroesophageal adenocarcinomas in an investigator-initiated phase 2 clinical trial. Since our study preceded KEYNOTE-811, checkpoint inhibition with a PD1-directed agent was not included as part of our standard therapy backbone. Here, we show encouraging efficacy of bevacizumab in combination with CAPOX and trastuzumab in patients with previously untreated HER2 + GEA supporting further investigation of this regimen in larger randomized studies. In additional correlative ctDNA analysis we identify molecular biomarkers of response and resistance.
Results
Patient characteristics
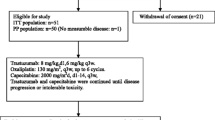
Between March 2011 and February 2015, 61 patients were screened for the study of whom 24 were ineligible. Thirty-seven patients were enrolled on the study, of which one withdrew consent prior to starting. A total of 36 patients received at least one cycle of treatment and were included in the analysis at the time of data lock on June 08, 2022 (Fig. 1). Patients received a median of 19 cycles of therapy (Interquartile range (IQR): 8–34.5 cycles) and have a median duration of follow-up of 23.2 months (IQR: 11.0–46.9 months).

Patient flowchart showing selection of cohort included in the trial.
Patient characteristics are reported in Table 1. The median age for the cohort was 55.5 years (Range 32–79) and consisted predominantly of male patients (n = 32, 89%). Esophageal (47%) or GE junction (28%) tumors were most common, and the most frequent sites of metastatic disease were the lymph nodes (72%), liver (56%) and lungs (25%). Nine patients (25%) had received prior chemotherapy for non-metastatic disease. Thirty five of thirty six tumor samples were tested for HER2 immunohistochemistry; 27 (75%) were IHC 3 + , 6 (17%) were 2 + , and 2 (6%) were 0 + . All 8 tumors that were IHC < 3 + , were FISH positive with HER2/CEP17 ratio of 2.0 or higher (median 2.5; range 2.0–3.0). Since our study was conducted prior to NCCN guidelines for universal microsatellite instability and CPS testing in gastroesophageal adenocarcinomas, we do not have that information available for this cohort.
Efficacy
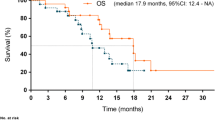
Of 36 patients, 26 patients (72.2%) had a partial response (PR), and 3 patients (8.3%) had a complete response (CR) for an objective response rate of 81% (95% CI 65–92%), meeting the primary endpoint of the study (Fig. 2A). Five additional patients had stable disease (SD), leading to a disease control rate (DCR = CR or PR or SD) of 94.4%. The median time to response was 1.9 months (range 1.4–6.2 months) and the median duration of response was 14.9 months (range: 2.4–95.9 months, Fig. 2B). Median PFS was 14 months (95% CI, 11.3–36.4) and the 6-month progression free survival was 77.1% (95% CI, 64.3–92.4) (Fig. 2C). Median OS was 23.2 months (95% CI, 16.6–36.4) (Fig. 2D). One-year OS was 72.2% (95% CI, 59.0–88.4).

A Waterfall Plot showing Best Response per RECIST 1.1. B Spider plot demonstrates evolution of radiographic response over time as measured by the change in sum of target lesions measured by RECIST 1.1 (N = 36). Progression free survival (PFS, C) and Overall survival (OS, D), respectively, for the entire cohort. Progression free survival E and Overall survival F, respectively, stratified by subjects with tumors that were IHC HER2 3+ vs. IHC HER2 2+ (N = 33). P-value was calculated using two-tailed log rank test E, F.
In a planned exploratory analysis, we stratified response and survival by patients with tumors that were HER2 IHC 3+ vs. patients with tumors that were HER2 IHC 2+ and FISH +. Twenty-four of 27 patients (89%) with HER2 IHC 3+ tumors had an objective response compared to 3 of 6 patients (50%) with HER2 IHC 2+ tumors. Patients with HER2 IHC 3 + , had a statistically significant longer median PFS of 22.4 months (95% CI, 14.0-NA) (Fig. 2E) compared to patients with HER2 IHC 2+ and FISH + , who had a median PFS of 7.4 months (95% CI, 3.3-NA, p = 0.0045). This longer PFS translated into an improved median OS of 27.6 months (95% CI, 19.3–49.1) (Fig. 2F) for patients with HER2 IHC3+ compared to median OS of 11.7 months (95% CI, 4.7-NA) in patients with HER2 IHC 2+ and FISH+ disease (p = 0.0047, Supplementary Data 1). There was no difference in PFS or OS based on location of the primary tumor (Supplementary Fig. 1A, B).
At the time of data lock, no patients remained on study. Twenty-four patients discontinued treatment due to progression, 4 patients due to toxicity or physician decision, 3 patients to take a chemotherapy holiday, 3 patients transitioned to definitive chemoradiation, and 1 patient died on treatment from an unrelated cardiac illness and the last patient was lost to follow up. Nineteen of the twenty-four patients who discontinued trial therapy due to disease progression had evidence of radiographic progression in metastatic lesions, two were diagnosed with clinical progression of the primary tumor, two additional patients had disease progression in locoregional lymph nodes and information regarding site of disease progression is unavailable for one patient. Five patients (14%) were alive at the point of last contact. Two of these patients had a complete response on trial and are now undergoing surveillance. One patient, who progressed on therapy, later had a complete response with a checkpoint inhibitor. The last two patients moved out of state after more than 5 years on trial therapy and were lost to follow up.
Safety
All 36 patients are evaluable for toxicity. Severe toxicity in the first five patients (Part 1) was primarily grade 3 diarrhea (3 of 5 patients) and, per protocol, effected a 30% dose reduction of capecitabine from 1700 mg/m2/day to 1200 mg/m2/day in all subsequent patients. In Parts 2 and 3, the most common grade 3-4 toxicities related to CAPOX included peripheral sensory neuropathy (11%), lymphopenia (8%) and diarrhea (6%). Grade 3-4 toxicity related to bevacizumab included thromboembolic events and hypertension, each occurring in three patients (8%). Trastuzumab-related heart failure was documented in two patients (6%) (Supplementary Data 2).
Except for oxaliplatin, treatment dose was maintained for prolonged periods of time in most patients. Predictably, oxaliplatin required dose attenuation and discontinuation for most patients after 6–16 cycles of therapy. By contrast, dose attenuation of capecitabine was unusual in the 31 patients treated at 1200 mg/m2/day. Bevacizumab was held in 13 patients and trastuzumab was held in 9 patients.
Second Line Therapy
Following study therapy, twelve patients (33%) received no further treatment, six patients (17%) received one treatment regimen, nine patients (25%) received two treatment regimens, seven patients (19%) received three treatment regimens, and two patients (6%) received four treatment regimens. The most common regimens were chemoradiation, paclitaxel/ramucirumab, irinotecan and pembrolizumab. 13 patients received one further line of HER2-directed therapy for at least one cycle.
HER2 2 + IHC cases show significantly higher HER2 heterogeneity
HER2 amplifications have been noted to display significant heterogeneity in gastroesophageal tumors18,19 and this may impact response to therapy. To understand HER2 heterogeneity better, we performed single cell DNA HER2 FISH and quantified the frequency of HER2 amplified cells on diagnostic tumor biopsies for a subset of patients in our trial (n = 14). We found varying percentages of HER2 amplified cells among our cohort (Fig. 3A, B) and HER2 2 + IHC samples showed significantly higher heterogeneity with lower fraction of HER2 amplified cells compared to HER2 3 + IHC samples (Fig. 3C, Frequency of HER2 amplified cells: 2 + IHC vs. 3 + IHC, 0.28 vs. 0.93, p = 0.0099). We were unable to assess the impact of heterogeneity within HER2 3+ cases due to its rare occurrence in our cohort.

A Fraction of cells displaying HER2 amplifications as determined by single cell HER2 FISH shows significant heterogeneity. A minimum of 50 tumor nuclei were measured to determine HER2 amplified fraction (y-axis) (N = 14) B Representative images from HER2 FISH showing absent (left), intermediate (middle) and high levels (right) of HER2 heterogeneity. Red probe binds HER2 locus, and green probe binds centromeric region of chromosome 17 (CEP17). Ratio of HER2/CEP17 is used to identify HER2 amplification and polysomy chromosome 17. Middle panel: Normal tissue provides contrast with no evidence of HER2 amplification. Blue dashed circles: HER2 negative cells, Red dashed circle: HER2 positive cells. C. HER2 2 + IHC (n = 3) tumors show significantly higher HER2 heterogeneity or lower HER2 amplified fraction compared to HER2 3 + IHC (n = 10). Dots represent individual patients, box extends from 25th to 75th percentile, horizontal line within box represents median, and whiskers extend a maximum of 1.5 x interquartile range (IQR). A, C. Groups compared using two-tailed Wilcoxon rank-sum test C.
Baseline ctDNA profiling identifies additional MAPK alterations and high ctDNA tumor fraction as important predictors of patient outcomes
To understand biomarkers of response and resistance we performed post-hoc ctDNA based genomic profiling on baseline or pre-treatment plasma samples using low pass whole genome sequencing (LP-WGS, PredicineSCORETM) and a 152-gene next generation sequencing (NGS) panel (PredicineCareTM) for 30 patients treated on our trial. Of 30 cases, 29 (96.7%) had at least one pathogenic alteration (mutation, rearrangement, or amplification) identified and the median number of pathogenic alterations per sample were 6 (Range: 2–45, Supplementary Fig. 2A). HER2 amplifications were observed in the plasma ctDNA in 76.7% of the cohort (23/30, Fig. 4A). Interestingly, and in contrast to prior published reports20, neither the presence nor degree of HER2 amplification noted on ctDNA had any prognostic impact on outcomes in our cohort (Supplementary Fig. 2B–E). Review of the genomic profile of these tumors revealed a high prevalence of TP53 alterations (83%) and lower rates of mutations in PIK3CA and CDKN2A (Fig. 4A). These results are consistent with the genomic profile of the chromosomal instability (CIN) subtype in gastroesophageal adenocarcinomas which harbor HER2 amplifications4.

A A CLIA-approved 152-gene next generation sequencing (NGS) assay (Predicine CARETM) was used to perform ctDNA based genomic profiling on diagnostic &/or pre-treatment plasma samples from 30 cases in our clinical trial. Genomic profiling showed high rates of TP53 alterations and HER2 amplifications were identified in 76.7% of the cohort (23/30). B 56.7% of the cases in our cohort (17/30) have evidence of additional MAPK driver alteration in addition to HER2. Amplifications in the receptor tyrosine kinases (RTKs) including in EGFR, KRAS, MET, and FGFR1 are the most common additional MAPK alterations. Cases with additional MAPK alterations (MAPK Alt) have a significantly worse progression free C and overall survival D compared to those who’s tumors without additional MAPK alterations (MAPK Wild Type, WT). Patients with high tumor fraction (TF) at diagnosis, estimated using a combination of low pass whole genome sequencing (LP-WGS) using ichorCNA39 and somatic mutation allele frequencies in ctDNA NGS assay, display a significantly worse progression free E and overall survival F. P-value was calculated using two-tailed log rank test C–F. BRR Best radiographic response, CR Complete Response, PR Partial Response, SD Stable disease, PD Progressive disease.
Next we studied whether genomic findings on ctDNA could provide further prognostic information. For this we first studied the baseline genomic profiles of three patients who showed signs of intrinsic resistance to this regimen (Fig. 2B, ID20, 28 with PD, and ID29 with SD with 6% target lesion increase). Interestingly, both cases with PD at first restaging imaging had evidence of amplifications in alternative genes in the MAPK pathway in the pre-treatment specimen including a high level MET amplification in ID28, and amplification of multiple MAPK genes including FGFR1/2, and NRAS in ID20 (Supplementary Data 3–5). The latter also had evidence of CCNE1 amplification, which has been described a mechanism of resistance to HER2 therapies in breast and gastroesophageal cancers21,22. ID29 had slight increase in target lesions but failed to meet threshold of PD also showed evidence of concomitant activating KRAS mutation and amplification. To assess the impact of pre-existing MAPK alterations which might mediate resistance to HER2 directed therapies in gastroesophageal cancers more systematically we assayed for their presence in our entire cohorts baseline ctDNA testing. Indeed, we identified several cases (17/30, 56.7%) where ctDNA revealed amplifications in other members of the MAPK pathway, and several receptor tyrosine kinases (RTK) such as EGFR, FGFR1, and MET, which might mediate resistance to HER2 directed therapy (Fig. 4B). Response rates (CR + PR) were numerically lower in cases with MAPK alterations (70.6% [12/17] vs. 92.3% [12/13], p = 0.51, Supplementary Data 6). Impressively, patients with additional MAPK alterations showed a significantly worsened PFS (median 12.5 months, 95% CI 5.2-NA vs. median 22.7 months, 95% CI 11.2-NA, p = 0.0067; Fig. 4C) and OS (median 16.5 months, 95% CI 8.0–27.6 vs. median 32.3 months, 95% CI 17.8-NA, p = 0.015; Fig. 4D) compared to those without additional MAPK alterations (i.e., MAPK wild-type, WT). In a similar analysis using a previously described AMNESIA gene panel (EGFR/MET/KRAS/PI3K/PTEN mutations, and EGFR/MET/KRAS amplifications), which predict worse outcomes in HER2+ gastroesophageal adenocarcinoma23, yielded similar results in our cohort with AMNESIA+ patients having worse clinical outcomes (Supplementary Fig. 3A–C). Presence of concomitant CCNE1 amplification had no prognostic impact (Supplementary Fig. 3D, E).
Next, we estimated the impact of ctDNA abundance or level at diagnosis on survival. Patients with high TF ( > 50th percentile) had a statistically significant worse PFS (median 11.3 months, 95% CI 5.2–18 vs. median 22.7 months, 95% CI 18.1-NA, p = 0.0013; Fig. 4E) and OS (median 15.4 months, 95% CI 8.0–27.6 vs. median 28 months, 95% CI 17.8-NA, p = 0.022; Fig. 4F) compared to patients with low TF ( ≤ 50th percentile).
Dynamic ctDNA changes at early time points after trastuzumab monotherapy
We next asked whether dynamic changes in ctDNA early in treatment could be predictive of response, potentially antedating the radiographic response. All patients in this trial received trastuzumab monotherapy on C1D1 and received the complete regimen with chemotherapy, trastuzumab and bevacizumab one week later as C2D1. We compared ctDNA based TF at both time points to assess for changes in ctDNA levels (Fig. 5A). Interestingly, cases which displayed higher ctDNA % decrease ( > median) displayed a trend towards improvement in PFS (median PFS 18.1 months, 95% CI 11.33-NA) compared to tumors with either lesser ctDNA % decrease ( < median) or increase in ctDNA levels (Median PFS 5.2, 95% CI 2.96-NA, p = 0.06, Fig. 5B). Similarly, overall survival for cases with higher ctDNA decrease was numerically higher but did not reach statistical significance (Median OS 26.6 months, 95% CI 18.1-NA vs. Median OS 10.2 months, 95% CI 6.9-NA, p = 0.17, Supplementary Fig. 4A). Change in ctDNA levels at one week was not dependent of degree of HER2 positivity on IHC or amplification levels on ctDNA (Supplementary Fig. 4B, C), likely due to low number of patients in the analysis.

A Initial therapeutic strategy on this trial included trastuzumab monotherapy which was followed by the complete regimen being given 7 days later. Tumor fraction at time of trastuzumab monotherapy and one week post are shown. Cases with % ctDNA decrease > median are in blue, and those with % ctDNA decrease <median or ctDNA increase are in red (n = 11 each). Cases with tumor fraction <0.05 (horizontal dashed line) were excluded from the analysis. B Cases with higher drop in ctDNA (blue) after one week of trastuzumab monotherapy have trend towards improved PFS. P-value was calculated using two-tailed log rank test B.
Serial ctDNA reveals temporal response dynamics and precedes disease progression
To understand the putative genetic mechanisms of resistance to therapy, we performed ctDNA based mutational profiling using the same NGS panel in 19 patients who had plasma available at the time of disease progression and compared mutational profiles from time of progression to that at diagnosis. Ten of the nineteen profiled patients had evidence of new genetic alterations on ctDNA (Fig. 6A). Several cases had acquired known oncogenic alterations in previously characterized resistance mechanisms to HER2 signaling including in genes in the MAPK pathway such as KRAS (n = 1), NRAS (n = 1), HRAS (n = 1), BRAF (n = 1) or PI3K pathway including biallelic PTEN alterations (n = 1) and PIK3CA mutations (n = 1). One case acquired 4 unique oncogenic ERBB2 alterations. We also identified emergent mutations in TP53, ATM, and CDKN2A, which were identified in 2 patients each (Supplementary Data 7). The circulating abundance of these newly acquired mutations, as measured by their variant allele fraction (VAF), suggested that these alterations were likely subclonal nature given these were significantly lower than overall ctDNA tumor fraction or than VAF of truncal alterations in TP53 in the majority of cases (Supplementary Data 8).

A Comparison of ctDNA NGS profiles from diagnosis and at the time of progression on therapy in 19 cases are shown. 10 cases show evidence of novel alterations (orange boxes) including in several putative resistance alleles such as KRAS, NRAS, PIK3CA, and PTEN. Interestingly, Pt 9 shows evidence of multiple new ERBB2 alterations. B Longitudinal monitoring of dynamics of resistance (red) and truncal alleles (black) in select four cases using ultra-deep sequencing for higher sensitivity. In general, low level resistance alleles are identified at diagnosis, are effective suppressed with therapy re-emerging several cycles prior to radiographic progression.
To understand the temporal dynamics of the emergence of putative resistance alleles we conducted personalized blood-informed MRD mutational profiling based on cancer variants identified using plasma samples collected at baseline and/or disease progression. Specifically, we profiled longitudinally banked plasma spanning the entire treatment course in four select patients from the cohort of ten patients who had newly diagnosed resistance alleles noted above. With a priori knowledge of the exact alteration being detected, ultra-deep sequencing at 100,000X depth using PredicineCARE ULTRA panel identified these acquired resistance alleles at extremely low abundance that would otherwise below the regular detection threshold at the standard 20,000X sequencing depth (Supplementary Figs. 5). Interestingly, we were now able to identify several of the acquired putative resistance alleles at low abundance in the initial diagnostic samples. These putative resistance alleles typically became undetectable with initiation of therapy. With continued therapy, all four profiled cases showed re-emergence of resistance (and recurrent elevation of truncal mutations) up to 2–8 cycles prior to radiographic progression (Fig. 6B).
ctDNA methylation profiling provides tumor agonistic approach to track changes in disease burden
In cases profiled with longitudinal mutation based ctDNA MRD monitoring (noted above in Fig. 6B) we noted that TP53 and other truncal alterations present at baseline ctDNA testing also up-trended, showing very similar dynamic trends as putative resistance alleles, suggesting that molecular progression precedes radiographic progression. We hypothesized that serial tumor burden monitoring with mutation based ctDNA panels may present practical challenges due to subclonal evolution of the tumor with new mutations emerging at low allele frequencies and that a tumor agonistic approach may be complementary in this effort. Hence, we employed PredicineALERT, a tumor agnostic cfDNA based methylation panel to generate circulating tumor methylation signature against a panel of normal healthy donor plasma methylation signatures. We profiled two cases, which had remaining cfDNA at all serial timepoints, who also underwent longitudinal ctDNA based MRD testing (Pt ID 9 & 15). Strikingly, cfDNA based tumor burden based on differential methylation fragments (DMFs) showed nearly identical dynamics as changes in average mutation allele frequency derived from the MRD panel, confirming that we were capturing the true cancer related methylome and suggesting its potential utility in tracking global tumor burden (Fig. 7A–C). More focused analysis on regions known to be hypermethylated in gastric cancer24,25 (Fig. 7D) confirmed robust methylation present in ctDNA at those loci at treatment naïve timepoints, their disappearance with therapy and re-emergence prior to radiographic progression consistent with evolution of disease burden with therapy.

Comparison of ctDNA derived tumor burden over course of therapy in index cases ID9 A and ID15 B using average mutation allele frequency (MAF) derived from ctDNA based MRD assay (black), and ctDNA based tumor agonistic methylation panel using differential methylation fragment burden (DMFs, red) shows similar dynamics and confirms molecular ctDNA progression precedes radiographic progression. C Heatmap of changes in signal over 2396 cancer associated methylation regions over the course of therapy with re-emergence of signal starting cycle 16 of chemotherapy in ID15. D Fifteen loci known to be hypermethylated in gastric cancer showing high methylation signature in ctDNA at baseline and changes of response and re-emergence around cycle 16 in ID15.
Discussion
We report the clinical outcomes of the CAPOX BETR trial, which tested the addition of bevacizumab to the standard triplet combination of capecitabine, oxaliplatin and trastuzumab in HER2+ advanced gastroesophageal adenocarcinoma. Long-term results from this single arm study demonstrate a favorable ORR of 81%, median PFS of 14.0 months and median OS of 23.2 months. Although comparison with global randomized phase 3 trials is confounded in multiple ways, we note that the results of this regimen seem superior to those seen in the experimental arm of the ToGA trial6 and the standard arm of the JACOB trial, which used a similar chemotherapy doublet with trastuzumab.
The treatment paradigm for advanced HER2+ gastroesophageal adenocarcinomas has evolved since the initiation of our trial. The addition of the PD1 inhibitor, pembrolizumab, has improved the ORR and improves PFS in patients with PD-L1 CPS positive tumors compared to the ToGA and JACOB regimens7,8. Although long-term survival results of the KEYNOTE-811 study are not yet mature, we note that the CAPOX BETR regimen shows numerically similar outcomes as seen in two single arm phase 2 trials which tested the addition of PD-1 inhibitors to 5FU/platinum + HER2 therapy26,27. In addition to the preclinical evidence which supported the design of our, this new study provides contextual clinical evidence for the rationale of combining anti-VEGF and anti-HER2 therapies in gastroesophageal adenocarcinomas. Recent data from the HER-RAM study which tested continuation of trastuzumab in 2nd line advanced HER2+ gastroesophageal adenocarcinoma in combination with paclitaxel and ramucirumab showed an encouraging 54% ORR and mPFS of 7.1 months28, also supports consideration of combining anti-HER2 and anti-VEGF therapies in future trials. Furthermore, given the complementary mechanism of action, and potential for synergy it is tempting to consider quadruplet regimens with 5FU/platinum + HER2 and VEGF with anti-PD-1 blockade. Ongoing strategies combining chemoimmunotherapy (fluoropyrimidines + platinum + PD1) with alternative anti-VEGF tyrosine kinase inhibitors, regorafenib29 and lenvatinib30, in treatment naïve unselected gastroesophageal cancer patients have shown good tolerance and encouraging clinical efficacy in early phase 2 trials with planned or ongoing larger randomized phase 3 studies. Our data suggests that a similar approach focused on targeting VEGF in HER2+ gastroesophageal cancers may further improve outcomes.
ctDNA is an emerging tool with increased utilization in clinical practice. Prior studies have revealed that ctDNA can identify the genomic landscape of gastroesophageal adenocarcinomas and may better capture genomic heterogeneity which is common in this tumor18,31. Similar to these findings our study confirms robust recovery of tumor mutational profiles using panel based ctDNA NGS assays. Using this approach we found that more than half of HER2+ gastroesophageal adenocarcinomas had an additional pathogenic alteration in the MAPK pathway which portended worse outcomes. Similar findings were recently shown on tumor based NGS profiling of cases enrolled on the JACOB trial where presence of alterations in AMNESIA genes (KRAS, MET, PIK3CA, and EGFR) predicted worse clinical outcomes32. In addition, higher ctDNA tumor fraction was associated with significantly worse outcomes, a finding previously suggested on ctDNA testing in gastroesophageal cancers by Maron et al.31, albeit their results did not reach statistical significance. This is interesting since clinicians have long known that patients with higher tumor burden tend to have clinically worse outcomes. However, quantification of disease burden radiographically is challenging, especially outside of clinical trial scenarios. Our data suggest that ctDNA profiling may allow comprehensive prognostication of HER2+ gastroesophageal adenocarcinomas based on the presence (or absence) of additional MAPK driver alterations and may provide an objective metric for tumor burden as molecular stratification metrics for future trials.
Serial changes in ctDNA levels has been used to predict response to therapy in several tumors including gastroesophageal cancers33, as well as to identify mechanisms of HER2 resistance, especially those which might occur subclonally in spatially disparate sites making them harder to detect using tumor NGS18,21,33. In an effort to better understand dynamics of ctDNA change we performed longitudinal profiling of banked samples. Our trial design with single agent trastuzumab dose exposure for one week prior to initiation of the entire systemic therapy regimen provided us a unique opportunity to evaluate very-short term changes in ctDNA levels after one week of targeted therapy alone, a time-point which hasn’t been evaluated for response prediction. Surprisingly, despite the short exposure to targeted therapy alone we found that changes in ctDNA TF after only 1 week of trastuzumab monotherapy were associated with numerically improved PFS and OS in patients with more effective suppression of ctDNA. Even though these results did not reach statistical significance, likely due to the low number of samples available for serial ctDNA testing, the trend seen is biologically interesting and hypothesis generating. We believe these data suggest significant HER2 addiction in a subset of HER2+ gastroesophageal cancers some of which show rapid and dramatic reductions in ctDNA with HER2 inhibition. Further exploration of these concepts could pave the way for adaptive treatment strategies, including early chemotherapy discontinuation or targeted therapy only approaches, which are guided by serial ctDNA changes. In line with these observations we also note that longitudinal ctDNA based monitoring of putative resistance alleles revealed their emergence several weeks to months prior to evidence of radiographic progression, and many of these putative resistance alleles were even present a low abundance at disease diagnosis. This suggests that pre-existing subclonal genetic diversity of tumors is an important predictor of resistance and hence benefit from anti-HER2 therapies, a finding also suggested by single cell RNA sequencing analysis of HER2+ gastroesophageal tumors33. Lastly, we utilize a tumor agnostic cfDNA based methylation panel to identify cancer related methylation signature which robustly tracks changes in disease burden and offers a complementary approach to mutation based tracking which may be more prone to variations in assessing overall tumor burden due to emergence of subclonal mutations. Interestingly, the ctDNA findings in our study are consistent with mechanisms of action and resistance to trastuzumab and it is difficult to appreciate any clear signature of addition of an anti-VEGF agent on the ctDNA analysis. We think this is in part related to the mechanism of action of bevacizumab which likely mediates its effects through its impact on the tumor microenvironment especially tumor angiogenesis and effectiveness of an anti-tumor immune response. The clinical outcomes seen in our study do seem to indicate added clinical benefit over chemotherapy-trastuzumab containing regimens and hence further work in understanding predictors of response to anti-VEGF agents in HER2+ GEAs is necessary.
Our work has several limitations. First, our sample size is relatively small, is biased towards younger, predominantly male, and white patient population. This is reflective of higher rates of gastroesophageal junction, and esophageal cancers in the US which have higher incidence in men (at a 4:1 ratio), and also a lack of a diverse patient population at the treating centers in our trial. Future studies must include appropriate measures to ensure inclusion of a more representative patient cohort potentially by inclusion of centers with a more diverse patient population. In addition, the majority of patients responded to therapy limiting response/non-response comparisons. Second, we had limited number of HER2 2 + IHC cases analyzed by FISH and hence our finding of higher HER2 heterogeneity in these patients needs to be validated in larger cohorts. Third, we did not have adequate remaining archival tissue from cases to perform PD-L1 IHC or tumor based NGS profiling. The latter could provide further insight into spatial heterogeneity being captured by ctDNA based approaches. Lastly, even though we identify putative genetic resistance alterations in a number of cases, they appear subclonal in nature, were often present even at diagnosis on deeper analysis, and hence their functional significance and potential benefit from their targeting is unclear.
In summary, results of this study support further clinical evaluation of VEGF pathway inhibitors in combination with chemotherapy, trastuzumab, and PD1 inhibitors in gastroesophageal adenocarcinoma. ctDNA based tumor profiling is an important tool that can help molecularly stratify patients and longitudinal monitoring is an appealing strategy for response and resistance assessments.
Methods
Study design
The CAPOX-BETR trial (NCT01191697) is an investigator initiated, open-label, single arm multi-center phase 2 trial (https://clinicaltrials.gov/study/NCT01191697?intr=NCT01191697&rank=1). The study protocol was approved by the institutional review board (IRB) at all participating institutions (Dana-Farber/Harvard Cancer Center IRB, and Johns Hopkins University IRB) and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guideline. All patients signed an IRB-approved consent prior to enrollment. Three medical centers in the United States participated in the study. The study is designed in two parts. Part I would evaluate the incidence of Grade 3–5 toxicity during the first three cycles of combination chemotherapy in the first five patients. Based on pre-specified toxicity parameters, part II enrollment would then continue at the same dose or at attenuated dose using a Simon two-stage rules. In the first stage 20 patients will be treated if ≤6 major responses were observed, the trial would be terminated early (early termination probability 0.61). Otherwise the study would proceed to recruit another 16 patients to a total of 36 patients.
Study population
Eligible participants were adults who had histologically confirmed newly diagnosed metastatic or locally advanced unresectable, HER2-positive adenocarcinoma of the esophagus, gastroesophageal junction, or stomach. HER-2 positivity was defined as 3+ staining by immunohistochemistry or was FISH positive (HER2/CEP ratio ≥ 2.0), similar to criteria used in the ToGA trial6. Participants were required to have measurable disease, defined by RECIST 1.1, an ECOG performance status <2 and adequate organ and bone marrow function. Prior therapy with capecitabine, oxaliplatin, bevacizumab or trastuzumab was not permitted. Patients may have received either (neo) adjuvant therapy, as long as it was completed at least 6 months prior to study entry. Patients were required to have LVEF ≥ 50% as determined by MUGA scan or echocardiogram. Patients were not permitted to have active brain or CNS metastases, known dihydropyrimidine dehydrogenase deficiency, uncontrolled hypertension, history of CVA, TIA, MI or unstable angina in the past 6 months, evidence of bleeding diathesis or coagulopathy. Any chemotherapy or radiotherapy to greater than 25% of the bone marrow or major surgery had to be completed at least 4 weeks prior to entering the study. Minor surgery, including placement of a vascular access device, was permitted 7 or more days prior to the first dose of bevacizumab. No other investigational agents or warfarin were permitted but low molecular weight heparin and ASA ≤ 325 mg/day were deemed acceptable. Patients with serious, unhealed wounds, bone fractures, skin ulcers, > grade 1 peripheral neuropathy, or known to be seropositive for HIV, Hepatitis B or C, lacking physical integrity of the upper gastrointestinal tract or having a malabsorption syndrome, were excluded. History of other malignancy was permitted, as long as the subject was cancer-free for at least 3 years and deemed by the investigator to be at low risk for recurrence. Further details of study are provided in trial protocol (Supplementary Note 1).
Treatment Protocols
On day 1 of cycle 1, patients received a loading dose of trastuzumab 4 mg/kg. Cycle 1 was 1 week in duration. On day 1 of cycle 2 and all subsequent cycles (every 3 weeks), patients received non-commercial bevacizumab 7.5 mg/kg, followed by trastuzumab 6 mg/kg, followed by oxaliplatin 130 mg/m2, followed by capecitabine 1200 mg/m2/day (1700 mg/m2/day in the first 5 patients), rounded to the nearest 500 mg increment, given as in two daily divided doses on days 1–14. To receive treatment on day 1 of cycle 2 and all subsequent cycles, patients had to have an absolute neutrophil count ≥ 1.0 K/uL, a platelet count ≥ 75 K/uL, an ECOG Performance Status ≤ 2 and recovery of all clinically significant study drug related non-hematological toxicity to grade 1 or less. There were no dose attenuations of trastuzumab. Trastuzumab was held for LVEF < 50%. Trastuzumab was discontinued if held for more than 12 weeks. There were no dose attenuations of bevacizumab. Bevacizumab was held for venous thrombosis, grade 2 proteinuria (UPCR > 1.9), or grade 3 congestive heart failure. Bevacizumab was discontinued for fistula, GI perforation, nephrotic syndrome, arterial thromboembolic event, wound dehiscence, reversible posterior leukoencephalopathy, grade 4 congestive heart failure, uncontrolled hypertension, recurrent grade 3 hemorrhage, grade 4 hemorrhage, or if held for more than 9 weeks.
Oxaliplatin could be attenuated in 3 dose levels, each representing a 25% dose reduction of the previous dose level. Capecitabine could be attenuated in 3 dose levels, each reducing the dose by one 500 mg tablet/day compared to the previous dose level. Investigators were permitted to lower the dose by two dose levels for any grade 4 toxicity. Oxaliplatin and capecitabine were held for any grade 2–4 toxicity. Treatment resumed once toxicity recovered to grade 1 or better. Investigators were required to attenuate the drug that had most likely caused the toxicity. For ANC < 1.0/K/uL, plts <75 K/uL, or febrile neutropenia, both oxaliplatin and capecitabine were held and attenuated. Both agents were held but not attenuated for grade 2 or higher renal insufficiency. Oxaliplatin was held and attenuated for grade 2 neuropathy and permanently discontinued for severe neuropathy. Capecitabine was held and attenuated for recurrent grade 2 or severe diarrhea, hyperbilirubinemia, or palmar/plantar erythrodysesthesia.
Treatment was continued until evidence of clinical, biochemical, or radiographic progression, unacceptable toxicity, participant withdrawal from the study or physician assessment based on change in health of the patient. Further details of dose adjustments, and treatment criteria are provided in the trial protocol.
Assessments
An interval history, physical exam, complete blood count with differential and comprehensive metabolic panel were performed prior to the start of each cycle. Computed tomography of the chest, abdomen and pelvis were performed within 28 days of starting treatment, after completing cycle 3 therapy, and then every 2 cycles until cycle 24 and then every 3 cycles thereafter. MUGA or echocardiogram was performed prior to starting treatment, after completing cycle 3 therapy and then every 4th cycle while on study. Plasma and mononuclear cells were drawn pretreatment on cycle 1 day 1, cycle 2 day 1, and day 1 of every even cycle thereafter and upon discontinuation of treatment.
HER2 heterogeneity testing
The analysis of HER2 gene amplification status and intra-tumoral heterogeneity was performed using fluorescence in situ hybridization (FISH) on formalin-fixed paraffin-embedded tumor specimens as previously described34. Briefly, a board-certified molecular pathologist performed microscopic review of HER2-stained immunohistochemical sections and/or H&E-stained routine sections using validated digital pathology or traditional microscopic workflows, and selected tumor-rich region(s) for hybridization. Hybridization was performed on 4-micron thick sections of formalin-fixed paraffin-embedded tumor material. A dual-color FISH assay was performed using the PathVysion assay (Abbott Molecular) using a probe specific to the chromosome 17q HER2 locus and a copy number control probe recognizing centromere 17 (CEP17 located at 17p11.1-q11.1). The inclusion of CEP17 probe allows for relative copy number of the HER2 gene to be determined as a ratio and detecting any polyploidy ( > 2 copies) of chromosome 17. Signal quantification was used to generate a HER2/centromere 17 ratio and HER2/cell ratio calculated from ≥ 50 tumor nuclei. The PathVysion Kit is FDA approved as an in vitro test, indicated as an aid in the assessment of patients for whom trastuzumab treatment is being considered. Estimate of HER2 intra-tumoral heterogeneity was determined using fraction of tumor cells showing evidence of HER2 amplification within a single tumor section in individual patients.
Circulating tumor (ct)DNA analysis
Plasma ctDNA analysis was performed using PredicineCARETM35, and PredicineCARETM Ultra, CLIA certified next generation sequencing (NGS) panel assays; PredicineSCORETM36, a low-pass whole genome sequencing assay; PredicineEPICTM, and PredicineALERTTM, a whole genome and panel based methylation assay. PredicineCARE assay detects variants in patients with tumor fraction lower than 0.5% (VAF 0.25%) whereas PredicineCARE Ultra assay detects variants in patients with tumor fraction as low as 0.1% (VAF 0.05%). For PredicineSCORETM, library is used for low pass whole genome sequencing (LP-WGS) as previously mentioned. Specifically, plasma samples are sequenced at 1-3x sequencing depth under the platform name PredicineCNB™36.
Cell-free DNA (cfDNA) processing, sequencing and data analysis were performed as previously described37. Briefly, cfDNA is extracted, quantified prior to library construction. Library undergoes selection with NGS panel prior to sequencing, and sequenced data is analysised using Predicine DeepSea pipeline. A variant identified in cfDNA is considered a somatic mutation only when (i) at least three distinct fragments (at least one of them should be double-stranded) contained the mutation; and (ii) the mutation allele frequency is higher than 0.25% or 0.1% for hotspot mutations. Candidate somatic mutations are further filtered on the basis of gene annotation to identify those occurring in protein-coding regions. Intronic and silent changes are excluded, while mutations resulting in missense mutations, nonsense mutations, frameshifts, or splice site alterations are retained. Mutations annotated as benign or likely benign are also filtered out based on ClinVar database, or common germline variants including 1000 genomes, ExAC, gnomAD and KAVIAR with population allele frequency >0.5%. Finally, hematopoietic expansion-related variants that have been previously described, including those in DNMT3A, ASXL1, TET2, and specific alterations within ATM (residue 3008), GNAS (residue 201, 202), or JAK2 (residue 617).
Copy number variation is estimated at gene level. The pipeline calculates the on-target unique fragment coverage based on consensus bam files, which is first corrected for GC bias, and is then adjusted for probe level bias (estimated from a pooled reference). Each adjusted coverage profile is self-normalized (assuming diploid of each sample) and then compared against correspondingly adjusted coverages from a group of normal reference samples to estimate the significance of the copy number variant. To call an amplification or deletion of gene, it requires the absolute z-score and copy number change pass minimum thresholds.
ctDNA fractions are estimated based using previously described methods37,38. Mutant allele fraction (MAF) and ctDNA fraction are related as MAF = (ctDNA * 1) / [(1 - ctDNA) * 2 + ctDNA *1], and so ctDNA = 2 / ((1 / MAF) + 1).
Copy number burden analysis using low-pass whole genome sequencing
Low-pass whole genome sequencing (LP-WGS) with an overall average coverage of 3x was performed on patient samples. ichorCNA algorithm39 was applied to GC and mappability-normalized reads to estimate plasma copy number variations using hidden Markov model (HMM). The pipeline first estimated the segment level (1 Mb genomic regions) copy number deviation as log2 ratio of the normalized reads between the test sample and a pool of normal plasma samples, then the sample level copy number burden (CNB score) was calculated as the logarithm of the sum of absolute CNV z-scores, where higher/lower CNB score indicates higher/lower CNV abnormality compared with normal background. The CNB score cutoff of 5.6 was defined as three standard deviations away from the population mean of normal plasma CNB scores.
Combined tumor fraction estimation
LPWGS-derived tumor fractions were estimated by ichorCNA software39 and mutation-derived were estimated based on the methods described above. Mutation-derived tumor fractions provide higher sensitivity than CNV or LPWGS-derived tumor fractions. However, mutation-derived tumor fractions have limitations, which include: (1) difficult to accurately call somatic mutations without normal control; (2) inaccurate MAF estimation caused by local copy number changes; (3) the mutation-derived tumor fractions are often under-estimated due to the limitation of the target panel size. On the other hand, LPWGS-derived tumor fractions do not have such limitation due to its genome-wide coverage. In addition, LPWGS-derived tumor fraction has advantages of its robustness and without the need of paired normal control. The limitations of LPWGS-derived mutations include low sensitivity (tumor fraction estimation is unreliable when it is less than 10%) and many samples may not have CNV changes. Considering the strength and limitations of both tumor fraction estimation methods, here we used a combination tumor fraction estimation method. The combination tumor fraction equals LPWGS-derived tumor fraction when LPWGS-derived tumor fraction is higher than 10%, otherwise the combination tumor fraction equals to the mutation-derived tumor fraction.
PredicineCARETM Ultra analysis
We tracked MRD status of mutations identified at time of resistance on PredicineCARE using PredicineCARE Ultra. To analyze this data we leveraged the workflow from PredicineBEACON that is a personalized MRD assay and tracks up to 50 personalized variants plus 500 hot spot mutations. We followed the same BEACON MRD workflow and leveraged the baseline variants detected using PredicineCARE and tracked its MRD status using the MRD sequencing depth of 100,000x in PredicineCARE Ultra. To detect a known MRD variant in the follow-up time points, the variant should have at least one confident variant support fragment. To call a sample as MRD positive, two or more MRD variants are detected, and one of them should have double-stranded variant support.
The tumor fraction of the MRD sample was estimated based on the allele fractions of autosomal somatic mutations:
Where \({{TF}}_{b}\) is the tumor fraction of the matched baseline sample,\(\,i\) is the selected mutation site for MRD tracking, \(n\) is the total number of selected mutation sites for MRD tracking, \(m\) is the number of mutated fragments at the mutation site, and \(t\) is the total number of fragments at the mutation site; \({m}_{b}\) and \({t}_{b}\) are mutated and total fragments at the mutation site at the baseline level.
cfDNA methylation analyses
The PredicineEPICTM assay generates whole-genome DNA methylation profiles and provides an orthogonal approach for assessing tumor burden using abnormally methylated fragment scores. Approximately 5 ng cfDNA was used for library construction and methylation treatment using proprietary PredicineEPICTM reagents. A small portion of methylation treated libraries were used for whole-genome sequencing in 25x paired-end 2x150bp sequencing, and the rest of libraries were used for PredicineALERTTM methylome panel sequencing in 300x paired-end 2x150bp sequencing. Paired-end sequences were first aligned to the hg19 reference assembly, and DNA fragments were then built by combining reads from the same molecules based on mapping locations and UMIs. A horizontal beta-value (ratio of methylated CpG-sites) was calculated for each fragment covering four or more CpG-sites. A background model was generated using plasma samples from healthy donors allowing for the identification of differentially methylated DNA fragments (empirical p-value < 0.001). A set of 34 hold-out normal plasma samples (not used in the generation of the background model) were tested in the same way as negative controls. The number of abnormal fragments were normalized by sequencing depth. Differentially methylated fragment regions (DMFRs) were determined from clusters of co-localized CpGs covered by more than one abnormal differentially methylated fragments. The total number of differentially methylated fragment (DMFs) was calculated within the DMFRs. Baseline-informed analysis was conducted by only considering DMFRs in baseline (C1D1) samples, excluding DMFRs found in more than 1 healthy donor sample. Loci known to be hypermethylated in gastric cancer were identified based on prior reports24,25.
Statistical analyses
The primary endpoint for the trial was objective response rate (complete response + partial response) as measured by RECIST 1.1 on central independent blinded review. Secondary endpoints included safety and toxicity of the combination regimen, duration of response (DOR), median progression free survival (PFS), and median overall survival (OS). The regimen would be considered promising if 15 or more of 36 evaluable patients had an objective response in the final analysis. This was based on a historical response rate of ≤30% for the CAPOX combination versus the new response rate of at least 50% in this combination plus bevacizumab and trastuzumab, using a type I error of 10% (one-sided) and power of 86%. The first patient was enrolled on study on March 7, 2011, and the last patient was enrolled on the study on February 18, 2015. PFS and OS, along with the associated confidence intervals, were calculated using R (v4.0.3) and the survminer (v0.4.9) and survival v(3.3-1) packages. Survival was compared between two groups using two-tailed log rank test using survival v(3.3-1) package. Progression free survival was defined as time until either progression or death, and patients were censored at date when therapy was discontinued for any reason other than progression or death. Duration of response was calculated for all patients who achieved an objective response defined as the time from best response until date of progression or death. Group comparisons between continuous variables were performed using the Mann-Whitney test using the wilcox.test function in R (v4.0.3). Visualizations were generated in R using ComplexHeatmap (v2.6.2), ggplot2 (v3.3.6), and ggpubr (v0.4.0).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Deidentified patient data are available to academic investigators for research purposes upon email request to Harshabad Singh (Harshabad_singh@dfci.harvard.edu). These requests are subject to approval by the institutional review board and would require data use agreement after which data will be available for a duration of 1 year. Cohort details, outcomes, clinical trial protocol with formal statistical plan is provided along with the paper as well as on clinicaltrials.gov (https://clinicaltrials.gov/study/NCT01191697?term=peter%20enzinger&rank=2&tab=results). The clinical trial protocol is available in the supplementary information file as Supplementary Note 1. Low-pass whole genome sequencing data generated in this study have been deposited to dbGAP with controlled access under accession code phs003706.v1.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003706.v1.p1]”. These data are available under restricted access for patient privacy. Permanent employees of an institution at a level equivalent to a tenure-track professor or senior scientist with laboratory administration and oversight responsibilities may request access through dbGAP. Requests are managed by the NCI Data Access Committee, take less than 2 days for approval and access is permitted for 12 months. Source data are provided with this paper.
Code availability
Clinical data from the trial were analyzed in R using survminer (v0.4.9) and survival v(3.3-1) packages. Visualizations were generated in R using ComplexHeatmap (v2.6.2), ggplot2 (v3.3.6), and ggpubr (v0.4.0). No custom code was generated for this part of the analysis. Genomic data from ctDNA assays were analyzed using proprietary software and custom code which may be made available for research purposes after obtaining a non-exclusive limited revocable license from Predicine (pdu@predicine.com). No right will be granted to sublicense, or distribute the software to a third party.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Cunningham, D. et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N. Engl. J. Med. 358, 36–46 (2008).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398, 27–40 (2021).
Cancer Genome Atlas Research, N. et al. Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017).
Andrulis, I. L. et al. neu/erbB-2 amplification identifies a poor-prognosis group of women with node-negative breast cancer. Toronto Breast Cancer Study Group. J. Clin. Oncol. 16, 1340–1349 (1998).
Bang, Y. J. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697 (2010).
Janjigian, Y. Y. et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 600, 727–730 (2021).
Merck. KEYTRUDA® (pembrolizumab) Plus Trastuzumab and Chemotherapy Met Primary Endpoint of Progression-Free Survival as First-Line Treatment in Patients With HER2-Positive Advanced Gastric or Gastroesophageal Junction (GEJ) Adenocarcinoma. (June 16, 2023).
Fuchs, C. S. et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 383, 31–39 (2014).
Wilke, H. et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 15, 1224–1235 (2014).
Ohtsu, A. et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J. Clin. Oncol. 29, 3968–3976 (2011).
Cleary, J. M. et al. FOLFOX plus ziv-aflibercept or placebo in first-line metastatic esophagogastric adenocarcinoma: A double-blind, randomized, multicenter phase 2 trial. Cancer 125, 2213–2221 (2019).
Fuchs, C. S. et al. Ramucirumab with cisplatin and fluoropyrimidine as first-line therapy in patients with metastatic gastric or junctional adenocarcinoma (RAINFALL): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 420–435 (2019).
Laughner, E., Taghavi, P., Chiles, K., Mahon, P. C. & Semenza, G. L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 21, 3995–4004 (2001).
Konecny, G. E. et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin. Cancer Res. 10, 1706–1716 (2004).
Singh, R., Kim, W. J., Kim, P. H. & Hong, H. J. Combined blockade of HER2 and VEGF exerts greater growth inhibition of HER2-overexpressing gastric cancer xenografts than individual blockade. Exp. Mol. Med. 45, e52 (2013).
Liu, K., Chen, H., You, Q., Shi, H. & Wang, Z. The siRNA cocktail targeting VEGF and HER2 inhibition on the proliferation and induced apoptosis of gastric cancer cell. Mol. Cell Biochem. 386, 117–124 (2014).
Pectasides, E. et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 8, 37–48 (2018).
Grillo, F., Fassan, M., Sarocchi, F., Fiocca, R. & Mastracci, L. HER2 heterogeneity in gastric/gastroesophageal cancers: From benchside to practice. World J. Gastroenterol. 22, 5879–5887 (2016).
Zhang, L. et al. ERBB2 Copy number as a quantitative biomarker for real-world outcomes to anti-human epidermal growth factor receptor 2 therapy in advanced gastroesophageal adenocarcinoma. JCO Precis Oncol. 6, e2100330 (2022).
Kim, S. T. et al. Impact of genomic alterations on lapatinib treatment outcome and cell-free genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann. Oncol. 29, 1037–1048 (2018).
Scaltriti, M. et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl Acad. Sci. USA 108, 3761–3766 (2011).
Pietrantonio, F. et al. Biomarkers of primary resistance to trastuzumab in HER2-positive metastatic gastric cancer patients: the AMNESIA case-control study. Clin. Cancer Res. 24, 1082–1089 (2018).
Watanabe, Y. et al. Sensitive and specific detection of early gastric cancer with DNA methylation analysis of gastric washes. Gastroenterology 136, 2149–2158 (2009).
Zeng, Y. et al. DNA methylation: an important biomarker and therapeutic target for gastric cancer. Front Genet. 13, 823905 (2022).
Janjigian, Y. Y. et al. First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: an open-label, single-arm, phase 2 trial. Lancet Oncol. 21, 821–831 (2020).
Lee, C. K. et al. A single arm phase Ib/II trial of first-line pembrolizumab, trastuzumab and chemotherapy for advanced HER2-positive gastric cancer. Nat. Commun. 13, 6002 (2022).
Kim, C. G. et al. Trastuzumab combined with ramucirumab and paclitaxel in patients with previously treated human epidermal growth factor receptor 2-positive advanced gastric or gastroesophageal junction cancer. J Clin Oncol. 20, 394–440 (2023).
Cytryn, S. L. et al. First-line regorafenib with nivolumab and chemotherapy in advanced oesophageal, gastric, or gastro-oesophageal junction cancer in the USA: a single-arm, single-centre, phase 2 trial. Lancet Oncol. 24, 1073–1082 (2023).
Yanez, P. E. et al. First-line lenvatinib plus pembrolizumab plus chemotherapy versus chemotherapy in advanced/metastatic gastroesophageal adenocarcinoma (LEAP-015): Safety run-in results. J. Clin. Oncol. 41, 411–411 (2023).
Maron, S. B. et al. Circulating tumor DNA sequencing analysis of gastroesophageal adenocarcinoma. Clin. Cancer Res. 25, 7098–7112 (2019).
Pietrantonio, F. et al. HER2 copy number and resistance mechanisms in patients with her2-positive advanced gastric cancer receiving initial trastuzumab-based therapy in JACOB trial. Clin. Cancer Res. 29, 571–580 (2023).
Maron, S. B. et al. Determinants of survival with combined HER2 and PD-1 blockade in metastatic esophagogastric cancer. Clin. Cancer Res. 29, 3633–3640 (2023).
Mitra, D. et al. Enrichment of HER2 amplification in brain metastases from primary gastrointestinal malignancies. Oncologist 24, 193–201 (2019).
Kwan, E. M. et al. Plasma cell-free DNA profiling of PTEN-PI3K-AKT pathway aberrations in metastatic castration-resistant prostate cancer. JCO Precis Oncol. 5, PO.20.00424 (2021).
Davis, A. A. et al. Genomic complexity predicts resistance to endocrine therapy and CDK4/6 inhibition in hormone receptor-positive (HR+)/HER2-negative metastatic breast cancer. Clin. Cancer Res. 29, 1719–1729 (2023).
Renouf, D. J. et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 13, 5020 (2022).
Vandekerkhove, G. et al. Circulating tumor DNA reveals clinically actionable somatic genome of metastatic bladder cancer. Clin. Cancer Res. 23, 6487–6497 (2017).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1324 (2017).
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P50CA127003. Research was also supported in part by NIH Grants 5U10CA031946-31and 5U10CA021115-38, as well as by Genentech, South San Francisco CA. Predicine, Inc. Hayward CA supported ctDNA analysis.
Author information
Authors and Affiliations
Contributions
H.S. and P.C.E. performed primary data analysis, interpretation, designed correlative analysis and formulation of conclusion of results. K.E.L. was responsible for clinical data collection for clinical trial. K.K. and H.Z. performed statistical analysis. R.J.K., N.J.M., T.A.A., J.A.C., E.M.R., S.J.K., M.Y., J.M.C., D.A.R., J.A.M., A.J.A., B.M.C., P.C.E. were responsible for patient care. A.M.H., J.R., L.K.B., E.A. were responsible for collection of biospecimens on clinical trial for correlative analysis. L.K.B. supervised clinical data collection. L.L.R., G.M., E.G., J.K.L. performed HER2 FISH analysis for HER2 heterogeneity. P.C.E. conceived, and designed the clinical trial and wrote the protocol. All authors reviewed the manuscript, provided edits, and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
HS receives research funding from AstraZeneca and has consulted for Dewpoint Therapeutics, Inc. and Merck Sharp & Dohme, LLC. RJK reports receiving advisory board/consulting fees from Astellas, AstraZeneca, Bristol Myers Squibb, Daiichi Sankyo, Eisai, Eli Lilly, EMD Serono, Exact Sciences, Grail, Ipsen, Merck, Novartis, Novocure, Phillips, Takeda, Toray and grant support paid to Johns Hopkins University and Baylor University Medical Center from Bristol Myers Squibb and Eli Lilly. TAA receives research funding to his institution from Lilly, Sanofi; receives consulting fees from Eisai and AstraZeneca; stock ownership in Merck. JAC receives consulting fees from Ipsen, Novartis, Pfizer. MY receives research funding from Janssen Pharmaceuticals and fees for peer review services from UpToDate. SJK reports consulting/advisory role for Merck, BMS, Astellas, Eli Lilly, AstraZeneca, Amgen, Daiichi-Sankyo, Novartis, Pfizer, Mersana, and Exact Sciences. SJK reports stock/equity for Turning Point Therapeutics, Nuvalent Therapeutics. JMC receives research funding to his institution from Merus, Roche, Servier, and Bristol Myers Squibb. He receives research support from Merck, AstraZeneca, Esperas Pharma, Bayer, Tesaro, Arcus Biosciences, and Apexigen; he additionally received honoraria for serving on the advisory board of Incyte and Blueprint Medicines. DAR has served as a consultant for Boston Scientific, Celgene, Instylla, Taiho Pharmaceuticals and serves on the scientific advisory board of AxialTx. LLR has received honoraria from PeerView, Medscape, Clinical Care Options, consulting honoraria from Abbvie, Personal Genome Diagnostics, Bristol Myers Squibb, Loxo Oncology at Lilly, Amgen, Meck, AstraZeneca, Sanofi-Genzume, and EMD Serono. LLR is currently an employee of Foundation Medicine, Inc. with stock in Roche Holding AG. AJA has consulted for Anji Pharmaceuticals, Affini-T Therapeutics, Arrakis Therapeutics, AstraZeneca, Boehringer Ingelheim, Oncorus, Inc., Merck & Co., Inc., Mirati Therapeutics, Nimbus Therapeutics, Plexium, Revolution Medicines, Reactive Biosciences, Riva Therapeutics, Servier Pharmaceuticals, Syros Pharmaceuticals, T-knife Therapeutics, Third Rock Ventures, and Ventus Therapeutics. A.J.A. holds equity in Riva Therapeutics. A.J.A. has research funding from Bristol Myers Squibb, Deerfield, Inc., Eli Lilly, Mirati Therapeutics, Novartis, Novo Ventures, Revolution Medicines, and Syros Pharmaceuticals. JAM served on a scientific advisory board for Merck Pharmaceutical. BMW is a consultant for Agenus, Celgene/BMS, EcoR1 Capital, GRAIL, Ipsen, Mirati, Revolution Medicines, and Third Rock Ventures; research support from AstraZeneca, Celgene/BMS, Eli Lilly, Harbinger Health, Novartis, and Revolution Medicines, outside the described work. PCE has served as a consultant and received honoraria from ALX Oncology, Arcus Bioscience, Astellas, Astra-Zeneca, Blueprint Medicines, Bristol-Myers Squibb, Chimeric Therapeutics, Celgene, Coherus, Daiichi-Sankyo, Five Prime, Ideaya, Istari, Legend, Lilly, Loxo, Merck, Novartis, Ono, Servier, Taiho, Takeda, Turning Point Therapeutics, Xencor, Zymeworks. The remaining authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Qianxing Mo, Wencheng Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, H., Lowder, K.E., Kapner, K. et al. Clinical outcomes and ctDNA correlates for CAPOX BETR: a phase II trial of capecitabine, oxaliplatin, bevacizumab, trastuzumab in previously untreated advanced HER2+ gastroesophageal adenocarcinoma. Nat Commun 15, 6833 (2024). https://doi.org/10.1038/s41467-024-51271-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51271-3
- Springer Nature Limited