

Abstract
Recent Ebola outbreaks underscore the importance of continuous prevention and disease control efforts. Authorized vaccines include Merck’s Ervebo (rVSV-ZEBOV) and Johnson & Johnson’s two-dose combination (Ad26.ZEBOV/MVA-BN-Filo). Here, in a five-year follow-up of the PREVAC randomized trial (NCT02876328), we report the results of the immunology ancillary study of the trial. The primary endpoint is to evaluate long-term memory T-cell responses induced by three vaccine regimens: Ad26–MVA, rVSV, and rVSV–booster. Polyfunctional EBOV-specific CD4+ T-cell responses increase after Ad26 priming and are further boosted by MVA, whereas minimal responses are observed in the rVSV groups, declining after one year. In-vitro expansion for eight days show sustained EBOV-specific T-cell responses for up to 60 months post-prime vaccination with both Ad26-MVA and rVSV, with no decline. Cytokine production analysis identify shared biomarkers between the Ad26-MVA and rVSV groups. In secondary endpoint, we observed an elevation of pro-inflammatory cytokines at Day 7 in the rVSV group. Finally, we establish a correlation between EBOV-specific T-cell responses and anti-EBOV IgG responses. Our findings can guide booster vaccination recommendations and help identify populations likely to benefit from revaccination.
Similar content being viewed by others

Introduction
Since their discovery in 1976, Ebola viruses have posed a persistent threat to human health. The risk of a resurgence of Zaire Ebola virus disease (EVD) is high, as shown by its 2014–2016 reemergence in West Africa, resulting in over 28,000 cases and 11,325 fatalities. More recently, the 2018–2020 Ebola outbreak in the Democratic Republic of Congo (DRC) caused more than 3400 cases and 2200 deaths1,2. These frequent outbreaks highlight the need for persistent prevention and disease control activities.
To date, two vaccines have been prequalified by the World Health Organization (WHO) and have received marketing authorization by the European Medicines Agency (EMA)3. Merck’s Ervebo® (rVSV-ZEBOV) is a recombinant vesicular stomatitis Indiana virus expressing Zaire Ebola GP. The Johnson & Johnson two-dose combination comprises the adenovirus-based vaccine Zabdeno® (Ad26.ZEBOV), which expresses the Zaire Ebola virus glycoprotein (GP) from the Mayinga strain, and the Mvabea® boost (MVA-BN-Filo), encoding the GP from the Zaire Ebola virus (Mayinga strain), Sudan virus (Gulu strain), and Marburg virus (Musoke strain), along with the nucleoprotein from the Tai Forest virus4.
Both vaccines showed induction of immune responses in non-human primates (NHPs). Ad26.ZEBOV and MVA-BN-Filo two-dose immunization showed full protection in lethal challenge NHP experiments5. One dose of the rVSVΔG-ZEBOV-GP vaccine induced ZEBOV-GP-specific IgG antibodies, which primarily mediate protection against lethal ZEBOV challenge6. CD8+ T-cell responses, but not CD4+ T-cell responses, may play a minor role in protection7.
These vaccines have been tested in numerous clinical studies in European and African adults, adolescents, and children and have shown acceptable safety, tolerability, and robust immunogenicity4,8,9,10,11,12,13,14,15,16. rVSVΔG-ZEBOV-GP has been shown to elicit a strong antibody response that correlates with the early activation of innate immunity, especially of monocytes and type I interferon-induced genes17. This vaccine conferred high protection for the contacts and contacts of contacts of confirmed patients in a Guinean phase 3 ring vaccination study11. Phase 1 and 2 trials of the Zabdeno (Ad26.ZEBOV) and Mvabea (MVA-BN-Filo) two-dose vaccine conducted in Europe, USA, and Africa demonstrated an excellent safety profile and induced robust humoral and cellular immune responses4,9,15,18. These responses persisted for at least three years post-vaccination13,19 and have been predicted to persist for years20. The sporadic nature of EVD outbreaks has limited the ability to conduct large phase 3 efficacy trials of this combination. Therefore, an immune-bridging study from non-human primates of the two-dose Ad26.ZEBOV, MVA-BN-Filo vaccine schedule, with the two vaccines administrated 57 days apart21, led to market authorization. However, despite the availability of authorized vaccines, correlates of protection and the exact level of responses required to maintain lasting protection17 are still uncertain.
The Partnership for Research on Ebola Vaccinations (PREVAC) consortium conducted a large vaccine trial in west Africa aimed to evaluate the safety and immunogenicity of three different vaccine regimens against Zaire Ebola virus disease: Ad26.ZEBOV followed by MVA-BN-Filo 56 days later (referred to as the Ad26–MVA group), rVSVΔG-ZEBOV-GP followed by a placebo 56 days later (the rVSV group), and rVSVΔG-ZEBOV-GP followed by a second dose of the same vaccine 56 days later (the rVSV–booster group)22. The results showed a good safety profile and robust specific IgG responses, as assessed by the magnitude and frequency of responders persisting from 14 days up to 12 months after vaccination23.
Because most studies have focused on humoral responses as a proxy for the correlate of protection, information on the quality and durability of cellular immune responses triggered by these vaccines is scarce. As the current vaccines rely on viral vector platforms, there may be differences in terms of the durability of such responses. These unknowns represent roadblocks to the definition of public health policies in terms of the duration of protection and boosting of anamnestic responses to effectively control the spread of EVD outbreaks. Here, we took the opportunity of the five-year follow-up of the PREVAC participants to investigate the functionality, breadth, and magnitude of vaccine-elicited memory T-cell responses, and to longitudinally monitor these responses over a period of up to 5 years.
Results
Enrollment and baseline characteristics
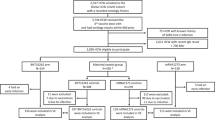
From March 2017 to September 2018, 196 adult participants were included in this PREVAC immunological ancillary study in Guinea from among 4789 in the whole PREVAC trial. In total, 191 participants were included in the per-protocol population after the exclusion of five participants due to an HIV-positive test (n = 4) or discontinuation of the vaccine protocol (n = 1) (Supplementary Fig. 1). The participants were randomized between arms with the following numbers per group: 79 in the Ad26-MVA arm, 27 in the rVSV arm, 9 in the rVSV-booster arm, and 76 in the pooled placebo arm. The baseline demographics of the 191 participants are presented in Table 1. The median age varied from 22 to 27 years between groups, with a small proportion of participants aged over 40. Among the 191 enrolled participants, 31 randomly selected individuals from PREVAC version 4.0 − 11 (35.5%) from the Ad26-MVA, 12 (38.7%) from the rVSV, and 8 (25.8%) from the rVSV-booster groups – were used to assess long term cellular immune responses (Supplementary Table 1). The sampling schedule and immunological assays performed by time point are shown in Supplementary Fig. 2.
Overview of antibody responses and serum biomarkers in vaccinated individuals
We first performed a comprehensive assessment of serum cytokines, chemokines, and inflammatory factors, consisting of 67 markers, in 92 participants randomly assigned to the Ad26-MVA (29), rVSV (28), rVSV-booster (26), or placebo (9) arms among the 191 inclusions in the per-protocol population on day (D)0, D7, and D63 after the prime vaccination. The levels of three pro-inflammatory cytokines, IL-18, IP-10, and TNF-related apoptosis-inducing ligand (TRAIL), were significantly higher in rVSV recipients on D7 post-vaccination than in the placebo group (P = 0.047, P < 0.0001, and P = 0.033, respectively) (Fig. 1).

Quantification of IL-18, IP-10, and TRAIL (pg/mL) in the serum of 92 vaccinated individuals at D0, D7, and D63 post vaccination (n = 29 placebo (grey), n = 28 Ad26-MVA (red), n = 26 rVSV (blue), n = 9 rVSV-booster (green)). The median values ± IQRs are shown. The box plots display the median (central line), the first and third quartiles (boxes), and the whiskers show 1.5 fold the interquartile range (IQR) above and below the boxes. Two-sided Wilcoxon Mann Whitney tests were used for comparisons between the active vaccine arms and the pooled placebo arms. FDR (Benjamini-Hochberg) method was used to adjust for test multiplicity. The exact p-values for IL-18, IP-10, and TRAIL at D7 in the comparison between the placebo group (grey) and the rVSV group (blue) are p = 0.047, p = 1.153e-07, and p = 0.033, respectively. Source data are provided as a Source Data file.
Then, we evaluated the EBOV-IgG specific responses in individuals (n = 93) enrolled in this ancillary study of the PREVAC trial. We confirmed the kinetics of the antibody response in the different arms (Fig. 2) to be similar to those previously described for the entire adult population enrolled in the PREVAC trial23. For both the Ad26-MVA and rVSV–booster groups, the geometric mean concentrations were the highest seven days after booster vaccination (D63) and at month 3 (M3). By M12, the geometric mean concentrations had decreased to the levels observed before receipt of the booster vaccination. At M12, the geometric mean antibody concentrations in EU/mL were 433 [340; 551] in the Ad26–MVA group, 828 [619; 1108] in the rVSV group, 663 [313; 1404] in the rVSV–booster group, and 92 [77; 110] in the placebo group (Table 2).

The geometric mean concentration was based on the log10 concentration with the baseline log10 titer and trial site as covariates. An antibody response was measured by FANG ELISA and defined as an antibody concentration of at least 200 enzyme-linked immunosorbent assay units (EU) per milliliter and an increase from baseline in the antibody concentration by at least a factor of 4. The geometric mean concentrations are shown for placebo (grey) n = 29; Ad26-MVA (red) n = 28; rVSV (blue) n = 27; rVSV-booster group (green) n = 9, respectively. The bars indicate 95% confidence intervals. Source data are provided as a Source Data file.
Assessment of ex-vivo memory T-cell responses in vaccinated individuals
To characterize the T-cell responses elicited by the vaccine strategies, we conducted ex-vivo intracellular cytokine staining (ICS) analyses of the per-protocol population (n = 191) at baseline, two weeks after each vaccination, and at M12. These groups received either rVSV (n = 36), Ad26 (n = 79), or placebo (n = 76) injections on D0 as part of the randomized prime-boost strategy (Supplementary Figs. 1 and 2). On D14, the percentage of EBOV-specific CD4+ T-cell produced cytokines (IFN-γ ± IL-2 ± TNF ± MIP-1β) after stimulation by EBOV peptides was significantly higher in the Ad26-MVA group than the placebo group (median [IQR] of 0.08% [0.01–0.16] vs 0.01% [0–0.05]; p = 0.0267). There were no detectable specific T-cell produced cytokines after a single dose of rVSV (Fig. 3A). On D70 (14 days post-boost), EBOV-specific CD4+ T-cell responses were detectable both in the Ad26-MVA and rVSV-booster groups versus in the placebo group (0.14% [0.08–0.23], p < 0.0001 and 0.05% [0.04–0.06], p = 0.026, respectively). There was no observed significant increase in EBOV-specific CD8+ T-cell responses, regardless of the vaccine strategy, either at D14, D70, or M12 (Fig. 3A). Analysis of the polyfunctionality of the CD4+ EBOV-specific T cells showed a high frequency of cells simultaneously producing IFN-γ, IL-2, MIP-1β, and TNF (Fig. 3B) on D14 and D70 in the Ad26-MVA group and on D70 in the rVSV booster group. At one year post-prime vaccination (M12), no ex-vivo T-cell responses were detectable in any of the groups relative to the placebo group.

A Total cytokine (IFN-γ ± IL-2 ± MIP1β ± TNF) levels (sum of the Boolean gates) produced by EBOV-specific CD4+ or CD8+ T cells from the Placebo (grey, n = 76), Ad26-MVA (red, n = 79), rVSV (blue, n = 27), and rVSV-booster (green, n = 9) groups after in-vitro stimulation on D14, D70, and month 12 post-prime vaccination. Each dot represents an individual value of total cytokine. Results are presented with the background subtracted. The box plots show the median (middle line) and the first and third quartiles (boxes), and the whiskers show 1.5 fold the interquartile range (IQR) above and below the box. Bivariate model was used for inter-arm comparisons of total cytokine levels of active vaccine groups (rVSV group, rVSV-booster group, and Ad26-MVA group) vs placebo group (grey). FDR (Benjamini-Hochberg) method was used to adjust for test multiplicity for each arm comparison separately. The exact p-value for CD4 at D70 in the comparison between the placebo group (grey) and the Ad26-MVA group (red) is p = 5.5e-08. B Functional composition of EBOV-specific CD4+ T-cell responses induced by the Ad26-MVA vaccine on D14 and D70 and by the rVSV-booster vaccine on D70. Responses are color coded according to the combination of cytokines produced. The arcs identify cytokine-producing subsets (IFN-γ, IL-2, MIP-1b and TNF) within the CD4+ and CD8+ T-cell populations. Source data are provided as a Source Data file.
Correlation between cellular vaccine T-cell responses and Ebola-specific IgG responses
We then estimated the correlations between antibody responses from D0 to M12 and EBOV-specific ex-vivo CD4+ T-cell responses from D14 to M12 in all vaccine groups. The Spearman correlation matrix showed that the EBOV-specific CD4+ T-cell responses detected on D14 positively correlated with the antibody titers from D14 to M12, with weak to intermediate correlation coefficients. The total number of IFN-γ+, MIP1β+, and TNF+-CD4+ T cells detected on D70 also positively correlated with the antibody response from M3 to M6 (Fig. 4).

Spearman’s correlation matrix between EBOV-GP antibody and CD4+ T-cell responses from day 0 to month 12 after vaccination in all vaccinated individuals. Correlation analyzes were performed using Spearman’s rank correlation with the two-tailed P-value. A FDR method (Benjamini-Hochberg) was used to adjust for test multiplicity. Colors indicate Spearman’s correlation coefficient. Only significant correlations after adjustment for test multiplicity (p < 0.05) are represented. Source data are provided as a Source Data file.
EBOV-specific memory T-cell responses induced by Ebola vaccines persist up to 60 months after vaccination
We characterized the EBOV-specific T cells long term after vaccination by assessing whether EBOV-specific memory T cells would be detectable after the expansion of antigen-specific T cells in vitro in a subgroup of participants (Supplementary Table 1) at 24, 36, 48, and 60 months after prime vaccination (PREVAC Follow-up study). We detected a high frequency of functional CD4+ T-cell produced cytokines in response to EBOV peptides in all groups. The median frequencies [IQR] of CD4+ cytokine+-specific T cells were 16% [11–21], 15% [12–24], 26% [19–31], and 20% [14-25] for the M24, M36, M48, and M60 follow-up visits, respectively, for Ad26-MVA vaccinated individuals; 11.8% [6.8–16.3], 13% [10–17], 9.8% [7.7–14.5], and 15% [11–19] for the M24, M36, M48 and M60 follow-up visits, respectively, for rVSV vaccinated individuals; and 16% [12–20], 16% [9–21],12% [8–18], and 26% [17–26] for the M24, M36, M48, and M60 follow-up visits, respectively, for rVSV-booster vaccinated individuals (p < 0.05 for all comparisons to non-stimulated conditions, except for the rVSV booster group at M24, p = 0.05) (Fig. 5A–C, left). There was a significant increase in the frequency of EBOV-specific CD8+ T cells from M36 to M60 for the Ad26-MVA group and from M24 to M48 for the rVSV group. We detected no significant levels of specific CD8+ T cells in the rVSV-booster group, except at M48 (p = 0.05) (Fig. 5A–C, right). The frequencies of CD4+ and CD8+ EBOV-specific memory T cells remained elevated from M24 to M60, indicating that vaccine induced T-cell responses are sustained for up to 60 months following the initial vaccination (Supplementary Fig. 3).

Total cytokine (IFN-γ ± IL-2 ± MIP1β ± TNF) levels (sum of the Boolean gates) produced by EBOV-specific CD4+ or CD8+ T cells from individuals in the Ad26-MVA (A), rVSV (B), and rVSV-booster (C) groups following in vitro stimulation on D0 and re-stimulation on D8 with the EBOV peptide pools (1 µg/mL of each) at M24 (n = 11 Ad26-MVA, n = 12 rVSV, n = 6 rVSV-booster), M36 (n = 10 Ad26-MVA, n = 11 rVSV, n = 8 rVSV-booster), M48 (n = 9 Ad26-MVA, n = 11 rVSV, n = 7 rVSV-booster), and M60 (n = 9 Ad26-MVA, n = 10 rVSV, n = 7 rVSV-booster) post-vaccination. The median values ± IQRs are shown. The box plots display the median (central line), the first and third quartiles (boxes), and the whiskers show 1.5 fold the interquartile range (IQR) above and below the boxes. Each dot represents an individual value of total cytokine. Two-sided Wilcoxon signed-rank test was used for comparisons between stimulated and unstimulated responses. FDR method (Benjamini-Hochberg) was used to adjust for test multiplicity for each arm comparison separately. The exact p-values for CD4 T cells in Ad26-MVA group at M24, M36, M48 and M60 in the comparison between the unstimulated condition (grey) and stimulated condition (red) are p = 0.008, for all time points. The exact p-values for CD4 T cells in the rVSV group at M24, M36, M48 and M60 comparing the unstimulated condition (grey) and stimulated condition (blue) are p = 0.002 from M24 to M48 and p = 0.003 for M60. The exact p-value for CD8 T cells at M36 is p = 0.002. Source data are provided as a Source Data file.
Among the groups for which specific T-cell responses persisted for 60 months, a substantial proportion of the EBOV-specific CD4+ and CD8+ T cells showed polyfunctionality, producing up to four cytokines (IFN-γ ± IL-2 ± TNF ± MIP-1β). This was observed across all groups, with most cells producing IFN-γ and MIP-1β (Fig. 6).

Functional composition of EBOV-specific CD4+ (A) and CD8+ (B) T-cell responses induced by the three vaccine strategies at M24, M36, M48, and M60. Responses are color coded according to the combination of cytokines produced. The arcs identify cytokine-producing subsets (IFN-γ, IL-2, MIP-1β, and TNF) within the CD4+ and CD8+ T-cell populations. Source data are provided as a Source Data file.
We then evaluated the secretion of 25 soluble factors produced by PBMCs upon stimulation with EBOV peptide pools from vaccinated individuals long term after prime vaccination (M24, M36, M48, and M60) (Fig. 7).There was a significant difference in the levels of several biomarkers versus the unstimulated condition solely within the Ad26-MVA and rVSV groups until M36. However, starting from M48, only a limited subset of soluble factors showed differences in these two groups relative to the unstimulated condition. We observed no significant differences in biomarker levels between the rVSV-booster group in the stimulated versus unstimulated conditions at any time point.

Heatmap of supernatants from the PBMCs of vaccinated individuals at M24, M36, M48, and M60 after prime vaccination collected on day 2 after stimulation with EBOV peptide pools (sum of EBOV1 and EBOV2 peptide pools). The colors represent standardized expression values centered around the mean, with a variance equal to 1 for each cytokine and time point. The Wilcoxon U test for paired samples was used for comparisons between EBOV peptide pools and unstimulated responses for each vaccine strategy and time point. Significant differences are represented by colors according to the vaccine strategy. Grey columns represent missing data, when participants were not sampled at the specified time point. Source data are provided as a Source Data file.
Specifically within the Ad26-MVA group, we observed significantly higher levels of nine soluble markers in wells stimulated with the EBOV peptide than in the unstimulated condition. There were notably higher levels of the pro-inflammatory factor IP-10, the anti-inflammatory cytokine IL-10, Th1 cytokines (IL-2, and TNF), a marker associated with T-cell activation (RANTES), a marker of cytotoxic activity (Granzyme B), and type I IFN-α, as well as cytokines produced by activated monocytes/macrophages and DCs (MIP-1α, MIP-1β) than in the unstimulated condition. Administration of a single dose of rVSV resulted in significantly higher levels of 16 soluble markers in wells stimulated with the EBOV peptide pools than in the unstimulated condition. These markers included various pro-inflammatory factors (IL-6, IL8, IL1-b), Th1 cytokines (IFN-g, IL-2, TNF), a Th2 cytokine (IL-4), and a Th17 cytokine (IL-17A). In addition, we observed higher levels of a marker of cytotoxic activity (Granzyme B), a marker associated with T-cell activation (CD40L), and cytokines produced by activated monocytes/macrophages and DCs (TNF, MIP-1a, MIP-1b, IL-12p70) than in the unstimulated condition. Neutrophil chemoattractant (CXCL1), growth factor (G-CSF), and the immunoregulatory factor PDL1 were also detected. At M36, we observed the presence of the same cytokines as previously identified in both study arms. By contrast, at M48 and M60, only Th1 cytokines (IFN-g, IL-2) and G-CSF were still observed in the Ad26-MVA arm. Only G-CSF and the pro-inflammatory cytokine IL-1b were detected at M60 in the rVSV group (Fig. 7).
Discussion
We studied T-cell responses to three Ebola vaccine strategies tested in a large phase 2b study performed in West Africa within the international PREVAC consortium22,23. Volunteers were randomized to receive either one injection of the rVSV-ZEBOV vaccine, an injection of the rVSV-ZEBOV vaccine followed by a homologous boost, the Ad26.ZEBOV, MVA-BN-Filo vaccine schedule, or placebo. The long-term follow-up of a subgroup of volunteers allowed us to also evaluate the durability of these responses for up to five years. This is the first study to assess the cellular immune responses of the two EVD vaccines recommended by the WHO Strategic Advisory Group of Experts on Immunization (SAGE) in a high-risk population for EVD.
We demonstrate that the Ad26-MVA and rVSV vaccine strategies elicit long-term memory cellular responses in most vaccinees that persist up to 60 months following first vaccination. We significantly extended the 12-month results on humoral responses of the PREVAC study14,16,22,23. First, we found several differences in the profile of immune responses between the viral vector vaccination strategies. We show an induction of polyfunctional EBOV-specific CD4+ T-cell responses, but not CD8+ T-cell responses, following the Ad26 prime, which were improved by the MVA boost, whereas such responses were barely or not detectable in the rVSV groups. When detectable, ex-vivo CD4+ T-cell responses declined one year following the prime and became significantly no different from those of the placebo group. Our results complete previous studies that have tested the same Ebola vaccines. For example, we found that the frequency of vaccine-elicited T-cell responses in the Ad26, MVA-BN regimen increased following the MVA boost, as previously reported in the phase 2 EBL2001 trial conducted in Europe4. On the contrary, we did not detect an increase in EBOV-specific CD8+ T-cell responses in the PREVAC Guinean population. This discrepancy may be due to differences in host genetic and/or environmental factors24,25. As previously reported26, we observed that ex-vivo T-cell responses following an rVSV prime were low or undetectable. This observation contrasts with the detection of a significant increase in serum IP-10 and TRAIL levels on D7 post-rVSV injection, indicating an activation effect, as already reported27. A difference in the kinetics of the detection of specific T-cell responses cannot be ruled out.
In a subgroup of vaccinees, we evaluated the persistence of long-term T-cell memory responses following vaccination at M24, M36, M48, and M60. We used an enrichment technique consisting of the stimulation of cells in culture with EBOV peptide pools for eight days. This approach previously allowed us to characterize EBOV-specific T-cell responses in long-term EVD survivors28. Our findings show that both the two-dose Ad26-MVA regimen and the single dose rVSV vaccine are able to induce enduring EBOV-specific T-cell responses for up to 60 months after the initial vaccination, with no decline observed over time. Interestingly, CD8+ T-cell memory responses were not detected in the long-term follow-up of the rVSV booster group. This discrepancy could be due to the low number of volunteers in this group resulting in lower statistical power. Notably, analyses of the in-vitro production of cytokines in the supernatant after stimulation of PBMCs with EBOV peptides revealed a common set of biomarkers between the Ad26-MVA and rVSV groups in the long-term follow-up. Beyond the production of Th1 cytokines, granzyme and markers of T-cell activation (PDL1, RANTES, CD40L) were also produced. Although the number of factors produced in vitro decreased over time, G-CSF, a marker of T-cell activation, remained detectable at 60 months in the Ad26-MVA and rVSV groups. One original aspect of our study was the evaluation of the immunogenicity of a homologous prime-boost rVSV administration. To date, data available concerning booster immunizations with rVSV are limited. In the PREVAC trial, administering a second dose of rVSVΔG-ZEBOV-GP 56 days after the initial dose resulted in only a transient increase in antibody concentrations23. In terms of the cellular responses, a booster dose of rVSV induced EBOV-specific CD4+ T-cell responses but failed to stimulate EBOV-specific CD8+ T-cell responses in the long term, unlike the single-dose regimen. Given the high replication capacity of the VSV virus vector, the possibility of an immunodominant response against the rVSV backbone, which might dampen the EBOV T-cell responses in the rVSV booster group, cannot be ruled out. This observation does not preclude the potential benefit of a single delayed rVSV-ZEBOV boost, a strategy under discussion, to evaluate the optimal timing of a rVSV boost in the PREVAC population.
The observation of long-term persistence of specific memory T-cell responses in vaccinees, including functional Th1 memory cells, may provide an additional clue in the quest of correlates of protection and/or markers of vaccine responses. Although correlates of protection against EVD are still unclear, it is now established that antibodies play a crucial role in the protection against EVD. For example, a single dose of rVSVΔG-ZEBOV-GP vaccine induced ZEBOV-GP-specific IgG antibodies, which have been shown to be primarily responsible for protecting against lethal ZEBOV challenges in animals6. Recently, the protective effect of rVSVΔG-ZEBOV-GP vaccination against death among patients with confirmed EVD was reported29. By contrast, CD8+ T cells are of minor importance in providing protection following rVSVΔG-ZEBOV-GP vaccination, whereas they play a crucial role in adenovirus-based and MVA-based ZEBOV vaccines6,30,31.
The above results provide indications of the protective effect of immune responses against infection, but markers of long-term protection are still lacking. Since the 2014-2016 outbreak of EVD in West Africa and the more recent 2018-2020 outbreak in DRC, it is estimated that 500,000 to 1 million individuals have been vaccinated worldwide. However, data on the persistence of long-term humoral responses to these vaccines are limited32,33. In the same vein, the lack of data on the durability of vaccine efficacy hampers a clear definition of the criteria for re-vaccination. To date, there are no immunological indications for a booster vaccination or indications concerning the population that would likely benefit. As CD4+ T cells play a pivotal role in promoting the development and persistence of humoral responses, our demonstration of a correlation between EBOV-specific T-cell responses and anti-EBOV IgG responses may provide a rationale for the need of a long-term boost to maintain T- and B-cell memory responses. Accordingly, a recent clinical trial involving children in Sierra Leone demonstrated that a booster dose of Ad26.ZEBOV administered to those who had undergone the two-dose Ad26.ZEBOV and MVA-BN-Filo vaccine regimen over three years earlier was well tolerated and induced a rapid and robust increase in binding antibodies against Ebola virus34. Our data may also provide information about low responders to the vaccines14 and/or potential breakthrough infections despite EBOV vaccination35,36. There is still a lack of definitive evidence concerning the protective efficacy of the natural immune response following EVD. However, studies have shown substantial immunological disparities between vaccination and natural Ebola virus infection. In EVD survivors, both humoral and cellular immunity are present, but their specific roles in protection are unclear37. Vaccination with VSV- or adenovirus-vectored vaccines demonstrated that both humoral and cellular immunity contributed to protection in NHP38. Vaccination with recombinant serotype 5 adenovirus encoding Ebolavirus GP induced anti-GP specific antibodies, but these antibodies alone did not confer protection, as the transfer of EBOV GP-specific IgG from Ad5-EBOV vaccinated NHPs to naïve animals did not protect them against death following EBOV challenge. However, when CD8 + T cells were depleted, 4 out of 5 vaccinated animals died after challenge, indicating a crucial role for these cells31. These disparities provide compelling evidence that antibody titers alone may not suffice for evaluating the antibody-mediated immunity elicited by vaccination39. We have previously reported that Ebola survivors maintain high levels of EBOV-specific IgG antibodies and robust and polyfunctional EBOV-specific memory T-cell responses for up to two years after acute disease28. Given the large amount of data from both large cohorts of post-Ebola survivors40,41,42,43,44 and vaccinees, a comparative analysis of natural and vaccine responses between populations could provide insights about potential correlates of protection.
Our study had several limitations. Although we were able to analyze ex-vivo T-cell responses and serum biomarker levels in 191 individuals, the number of participants in the five-year follow-up was limited, in particular, in the rVSV booster arm. Further studies are necessary to gain a better understanding of the differences observed between the rVSV and rVSV-booster arms. In addition, the correlation study between EBOV-specific CD4+ T-cell responses and IgG antibody responses was performed up to M12, the primary immunological time point of the PREVAC study23. However, the long-term follow-up of humoral responses until M60 is ongoing under the PREVAC-UP study (EDCTP EU funding program).
Methods
Trial design and participants
The PREVAC trial (NCT02876328) was a randomized, double-blinded, placebo-controlled trial in adults 18 years of age or older and children 1 to 17 years of age at six sites in four West African countries. The general objective was to evaluate the safety and immune responses of three vaccine regimens against Zaire Ebola virus disease: Ad26.ZEBOV (0.5 ml; 5 × 1010 viral particles) followed by MVA-BN-Filo 56 days later (0.5 ml; 1 × 108 infectious units) (the Ad26–MVA group), rVSVΔG-ZEBOV-GP (1.0 ml; 9.4 × 107 plaque-forming units) followed by placebo 56 days later (the rVSV group), and rVSVΔG-ZEBOV-GP followed by rVSVΔG-ZEBOV-GP 56 days later (the rVSV–booster group). As previously published22, participants in the PREVAC trial were enrolled under three distinct protocol versions (2.0, 3.0 and 4.0 (main study)), with a progressive opening of active vaccine arms. In version 3.0, a diluted rVSVΔG-ZEBOV-GP vaccine (approximately 5 × 107 plaque-forming units/mL) was administered to certainin participants randomized in rVSV arms due to safety considerations. Subsequently, the safety alert regarding the rVSVΔG-ZEBOV-GP vaccine was revoked, allowing enrollment under version 4.0, which reintroduced the initially intended vaccine combination (1.0 ml; 9.4 × 107 plaque-forming units). The alternate rVSV vaccine regimens from version 3.0 were excluded from this ancillary study as it deviated from the intended formulation and did not represent the vaccine formulation employed in practical settings. The results of the main study (protocol version 4.0) evaluating the antibody response after up to 12 months of follow-up has been previously published23. The ongoing PREVAC UP project is evaluating long-term immunogenicity with a follow-up of up to five years for participants included in PREVAC.
Participants included in this PREVAC immunological ancillary study (n = 230) were a subset of adult participants at the Landreah site in Guinea of the PREVAC trial using the same inclusion criteria. The primary endpoint of the immunological ancillary study was to assess specific T cell responses induced by the three vaccine strategies and their durability up to five years after the initial vaccination. The secondary endpoints included measuring serum cytokine and chemokine levels before vaccination (Day 0) and after vaccination (Day 7 and Day 63) using Luminex technology, evaluating the ex vivo gene expression profile in whole blood before and three hours after each vaccination (Day 0 and Day 56), as well as on Day 7 and Day 63 (ongoing study), and performing a phenotypic analysis of B and T cell subsets before and after vaccination (not performed due to insufficient cells remaining after T cell response experiments). The sub-study was systematically proposed to adult participants included in the main study at Landreah. Participants included in the immunological ancillary study were asked to provide additional written informed consent for additional blood draws. Participants who tested positive for HIV were excluded. The participants were randomized to one of the three vaccine strategies described above or to a placebo group as part of the PREVAC trial. Participants from different PREVAC protocol versions V2.0, V3.0 and V4.022 were pooled for this ancillary study, except those who received a diluted rVSVΔG-ZEBOV-GP vaccine in V3.0. Consequently, analysis were performed on 196 participants.
After the prime vaccination at entry to the study, follow-up visits occurred on (D7 ( ± 3 days), D14 ( ± 3 days), and D28 ( ± 7 days). The booster dose of vaccine was administered on D56 (53 to 66 days), with further follow-up visits on D63 (7 ± 3 days after the booster vaccination) and D70 (14 ± 3 days after the booster vaccination), and at 3 months ( ± 14 days), 6 months ( ± 1 month), and 12 months ( ± 1 month).
Then, samples from a subset of 31 study participants randomized in active vaccine arms in PREVAC version 4.0 (11 from the Ad26-MVA, 12 from the rVSV, and 8 from the rVSV-booster groups) were randomly selected for the assessment of long-term responses at 24 ( ± 6 months), 36 ( ± 6 months), 48 ( ± 6 months), and 60 ( − 6 months; + 1 month) months within the PREVAC-UP trial. To assess various immunogenicity endpoints, samples were collected at the indicated time points for the immunological assays outlined in Supplementary Fig. 2. PBMC samples were collected and stored at a centralized site in Conakry, Guinea. Immunological assays were conducted at the centralized laboratory of the Vaccine Research Institute in Créteil, France.
Ethics
Regarding the main PREVAC trial, the study protocol and informed consent, along with participants’ informational materials, received approval from the ethics committees of both the sponsor (INSERM IRB 00003888) and the implementing country (Guinea) prior to each version of the protocol being enacted. The study is registered with ClinicalTrials.gov (registration number NCT02876328), EudraCT (2017-001798-18 and 2017-001798-18/3rd), and the Pan African Clinical Trials Registry (PACTR201712002760250).
Antibody responses to Ebola glycoprotein
Serum concentrations of IgG binding antibodies against the Ebola virus surface glycoprotein were measured at baseline and at each follow-up visit until M12 only for participants recruited in the version 4.0 of the PREVAC trial (Supplementary Fig. 2) using the Filovirus Animal Nonclinical Group (FANG) enzyme-linked immunosorbent assay (ELISA) as previously described in ref. 23.
Characterization of EBOV-specific T-cell immune responses
T-cell immunogenicity was evaluated on D14 (14 days post-prime) and D70 (14 days post-boost) and at M12, M24, M36, and M48 post-prime (Supplementary Fig. 2). Cellular responses to EBOV (Ebola virus) peptides were evaluated using two distinct approaches, depending on the timing of the study visit. For visits conducted between D0 and M12, ex vivo intracellular cytokine staining (ICS) was used, with overnight stimulation. For visits occurring between M24 and M60, EpiMax technology45 was used, involving an eight-day amplification period. Briefly, PBMCs were stimulated in vitro with 158 overlapping 15-mer peptides (11-amino acid overlaps) covering the Ebola virus Mayinga variant GP in two pools of 77 (EBOV1) and 81 peptides (EBOV2) (JPT Technologies) (1 µg/mL of each). The total EBOV response was calculated by summing the EBOV1 and EBOV2 responses. Cell functionality was assessed by ICS, with Boolean gating. The percentage of total cytokines was calculated by summing the Boolean gates IFN-γ ± IL-2 ± TNF ± MIP-1β.The flow cytometry panel included a viability marker, antibodies to CD3, CD4, and CD8 to determine T-cell lineage, and IFN-γ, TNF, MIP-1β, and IL-2 antibodies. Gating strategy is shown in supplementary Fig. 4. Data were acquired using a LSRFortessa four-laser (488, 640, 561, and 405 nm) flow cytometer (BD Biosciences) and analyzed using FlowJo software, version 9.9.5 (Tree Star).
Quantification of serum or supernatant analytes
We quantified 67 analytes in serum samples on D7 (7 days post-prime) and D63 (7 days post-boost) using two different commercial kits: the 19-plex Human Luminex Discovery Assay (R&D systems, #LXSAHM-19) and the 48-plex Human Cytokine Screening Panel (Bio-Rad, #12007283). We quantified 25 analytes in supernatants from M24, M36, M48, and M60 PBMCs on D2 after EBOV1 (1 µg/mL) or EBOV2 (1 µg/mL) stimulation using the Human XL Cytokine Premixed Kit (R&D systems, #FCSTM18-25). The total EBOV response was calculated by summing the EBOV1 and EBOV2 responses. The biomarkers measured were as follows: for the 19-plex Human Luminex Discovery Assay: IL-17E/IL-25, IL-23, IL-31, Granzyme A, IL-21, 4-1BB/TNFRSF9/CD137, CCL28, Chemerin, EGF, Fas Ligand/TNFSF6, Midkine, TGFa, Aggrecan, CD40 Ligand/TNFSF5, CXCL14/BRAK, Fas/TNFRSF6/CD95, Flt-3 Ligand, Granzyme B, and PD-L1/B7-H1 and for the 48-plex Human Cytokine Screening Panel: CTACK, Eotaxin, Basic FGF, G-CSF, GM-CSF, GRO-a, HGF, IFN-a2, IFN-g, IL-1a, IL-1b, IL-1ra, IL-2, IL-2Ra, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12(p70), IL-12(p40), IL-13, IL-15, IL-16, IL-17, IL-18, IP-10, LIF, MCP-1, MCP-3, M-CSF, MIF, MIG, MIP-1a, MIP-1b, b-NGF, PDGF-BB, RANTES, SCF, SCGF-b, SDF-1a, TNF-a, TNF-b, TRAIL, VEGF. For the 25-plex Human XL Cytokine Premixed Kit: CCL2/MCP-1, CCL4/MIP-1b, CD40L/TNFSF5, CXCL10/IP-10, G-CSF, GRZ B, IFN-b, IL-7, IL-10, IL-13, IL-17A, B7-H1/PD-L1, TNF-a, CCL3/MIP-1a, IFN-a, IFN-g, IL-1b/IL-1F2, IL-2, IL-4, IL-6, IL-8/CXCL8, IL-12p70, TRAIL, CCL5/RANTES, and CXCL1/GRO-a/KC/CINC-1.
Statistical analysis
According to the methodologist’s calculation, maintaining the allocation ratio of randomization at 2:1:2:1:1, we intended to enroll up to 230 participants in total for this ancillary study across PREVAC versions 2.0, 3.0, and 4.0. A statistical analysis plan was formulated and endorsed prior to conducting the statistical analyses. The immunogenicity endpoints were analyzed per group in the per-protocol population (all randomized participants having received all vaccine administrations initially assigned by the randomization and with available samples for all immunogenicity time points, pooled across protocol versions). The participants from the rVSV and rVSV-booster groups were pooled for analyses of the time points before the boost vaccination at D56 (including Day 14). Total cytokine levels and polyfunctional responses were analyzed using a bivariate model46 for comparisons between the active vaccine groups and the pooled placebo groups. The bivariate model was built to analyze the T-cell responses measured by ICS in vaccine trials taking into account unstimulated control response and stimulated response by antigens, irrespective of the correlation between the non-specific and specific responses. CD4+ and CD8+ T-cell counts were log-transformed to match the conditions of validity of the model. Within-group comparisons of the ICS Epimax data were performed for each time point between the stimulated (EBOV1 and 2 stimulations pooled) and background responses using Wilcoxon signed-rank tests. The concentrations of the 67 measured cytokines assessed at D0, D7, and D63 were compared between the active arms and the pooled placebo groups for each time point using the Wilcoxon Mann Whitney test. Spearman correlations were used to assess the relationship between ICS responses and antibody responses until M12. We used a FDR method (Benjamini-Höchberg method) to adjust for test multiplicity for dependent comparisons47 (adjustment for each arm separately, across visits and across markers for each type of analysis). Analyses were performed using SAS (SAS Institute, Cary, NC; version 9.3 or higher) and R (the R Foundation, Vienna, Austria; version 4.2) software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
2014–2016 Ebola Outbreak in West Africa. https://www.cdc.gov/vhf/ebola/history/2014-2016-outbreak/index.html.
10th Ebola outbreak in the Democratic Republic of the Congo declared over; vigilance against flare-ups and support for survivors must continue. https://www.who.int/news/item/25-06-2020-10th-ebola-outbreak-in-the-democratic-republic-of-the-congo-declared-over-vigilance-against-flare-ups-and-support-for-survivors-must-continue.
Ebola virus disease: Vaccines. https://www.who.int/news-room/questions-and-answers/item/ebola-vaccines.
Pollard, A. J. et al. Safety and immunogenicity of a two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Europe (EBOVAC2): a randomised, observer-blind, participant-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 21, 493–506 (2021).
Kuhn, J. H. et al. A prophylactic multivalent vaccine against different filovirus species is immunogenic and provides protection from lethal infections with Ebolavirus and Marburgvirus species in non-human primates. Plos One 13, e0192312 (2018).
Marzi, A. et al. Antibodies are necessary for rVSV/ZEBOV-GP–mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl Acad. Sci. 110, 1893–1898 (2013).
Menicucci, A. R., Sureshchandra, S., Marzi, A., Feldmann, H. & Messaoudi, I. Transcriptomic analysis reveals a previously unknown role for CD8+ T-cells in rVSV-EBOV mediated protection. Sci. Rep. 7, 919 (2017).
Afolabi MO, I. D. et al. EBL3001 study group. Safety and immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in children in Sierra Leone: a randomised, double-blind, controlled trial. Lancet Infect. Dis. 1, 110–122 (2022).
Anywaine, Z. et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in children and adolescents in Africa: A randomised, placebo-controlled, multicentre Phase II clinical trial. PLOS Med. 19, e1003865 (2022).
Barry, H. et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in healthy and HIV-infected adults: A randomised, placebo-controlled Phase II clinical trial in Africa. PLOS Med. 18, e1003813 (2021).
Gsell, P.-S. et al. Ring vaccination with rVSV-ZEBOV under expanded access in response to an outbreak of Ebola virus disease in Guinea, 2016: an operational and vaccine safety report. Lancet Infect. Dis. 17, 1276–1284 (2017).
Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 389, 505–518 (2017).
Ishola D, M. D. EBL3001 study group. et al. Safety and long-term immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Sierra Leone: a combined open-label, non-randomised stage 1, and a randomised, double-blind, controlled stage 2 trial. Lancet Infect. Dis. 23, e337 (2023).
Kennedy, S. B. et al. Phase 2 Placebo-Controlled Trial of Two Vaccines to Prevent Ebola in Liberia. N. Eng. J. Med. 377, 1438–1447 (2017).
Mutua, G. et al. Safety and Immunogenicity of a 2-Dose Heterologous Vaccine Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Nairobi, Kenya. J. Infect. Dis. 220, 57–67 (2019).
Lee, A. W. et al. PREVAC Study Team. Immunogenicity and vaccine shedding after 1 or 2 doses of rVZVΔG-ZEBOV-GP Ebola vaccine (ERVEBO®): Results from a phase 2, randomized, placebo-controlled trial in children and adults. Clin. Infect. Dis. 78, 870–879 (2024).
Medaglini, D., Santoro, F. & Siegrist, C.-A. Correlates of vaccine-induced protective immunity against Ebola virus disease. Semin. Immunol. 39, 65–72 (2018).
Milligan, I. D. et al. Safety and Immunogenicity of Novel Adenovirus Type 26– and Modified Vaccinia Ankara–Vectored Ebola Vaccines. Jama 315, 1610 (2016).
Winslow, R. L. et al. Immune Responses to Novel Adenovirus Type 26 and Modified Vaccinia Virus Ankara–Vectored Ebola Vaccines at 1 Year. Jama 317, 1075 (2017).
Alexandre, M. et al. Prediction of long-term humoral response induced by the two-dose heterologous Ad26.ZEBOV, MVA-BN-Filo vaccine against Ebola. npj Vaccines 8, 174 (2023).
Bockstal, V. et al. Non-human primate to human immunobridging demonstrates a protective effect of Ad26.ZEBOV, MVA-BN-Filo vaccine against Ebola. npj Vaccines 7, 156 (2022).
Badio, M. et al. Partnership for Research on Ebola VACcination (PREVAC): protocol of a randomized, double-blind, placebo- controlled phase 2 clinical trial evaluating three vaccine strategies against Ebola in healthy volunteers in four West African countries. Trials 22, 86 (2021).
Randomized Trial of Vaccines for Zaire Ebola Virus Disease. New Eng. J. Med. 387, 2411–2424 (2022).
Zimmermann, P. & Curtis, N. Factors That Influence the Immune Response to Vaccination. Clin. Microbiol. Rev. 32, e00084–18 (2019).
Martin, C. A. et al. Ethnic differences in cellular and humoral immune responses to SARS-CoV-2 vaccination in UK healthcare workers: a cross-sectional analysis. eClinicalMedicine 58, 101926 (2023).
Raabe, V. et al. Cellular and humoral immunity to Ebola Zaire glycoprotein and viral vector proteins following immunization with recombinant vesicular stomatitis virus-based Ebola vaccine (rVSVΔG-ZEBOV-GP). Vaccine 41, 1513–1523 (2023).
Rechtien, A. et al. Systems Vaccinology Identifies an Early Innate Immune Signature as a Correlate of Antibody Responses to the Ebola Vaccine rVSV-ZEBOV. Cell Rep. 20, 2251–2261 (2017).
Wiedemann, A. et al. Long-lasting severe immune dysfunction in Ebola virus disease survivors. Nat. Commun. 11, 3730 (2020).
Coulborn, R. M. et al. Case fatality risk among individuals vaccinated with rVSVΔG-ZEBOV-GP: a retrospective cohort analysis of patients with confirmed Ebola virus disease in the Democratic Republic of the Congo. Lancet Infect. Dis. 24, 602–610 (2024).
Kupke, A. et al. Protective CD8+ T Cell Response Induced by Modified Vaccinia Virus Ankara Delivering Ebola Virus Nucleoprotein. Vaccines 10, 533 (2022).
Sullivan, N. J. et al. CD8+ cellular immunity mediates rAd5 vaccine protection against Ebola virus infection of nonhuman primates. Nat. Med. 17, 1128–1131 (2011).
Huttner, A. et al. Antibody responses to recombinant vesicular stomatitis virus-Zaire Ebolavirus vaccination for Ebola virus disease across doses and continents: 5-year durability. Clin. Microbiol. Infect. 29, 1587–1594 (2023).
McLean, C. et al. Persistence of immunological memory as a potential correlate of long-term, vaccine-induced protection against Ebola virus disease in humans. Front. Immunol. 14, 1215302 (2023).
Manno, D. et al. Safety and immunogenicity of an Ad26.ZEBOV booster dose in children previously vaccinated with the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen: an open-label, non-randomised, phase 2 trial. Lancet Infect. Dis. 23, 352–360 (2023).
Jaspard, M. et al. Development of the PREDS score to predict in-hospital mortality of patients with Ebola virus disease under advanced supportive care: Results from the EVISTA cohort in the Democratic Republic of the Congo. eClin. Med. 54, 101699 (2022).
Mulangu, S. et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 381, 2293–2303 (2019).
Sullivan, N. J., Martin, J. E., Graham, B. S. & Nabel, G. J. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat. Rev. Microbiol. 7, 393–400 (2009).
Wong, G. et al. Immune Parameters Correlate with Protection Against Ebola Virus Infection in Rodents and Nonhuman Primates. Sci. Transl. Med. 4, 158ra146 (2012).
Koch, T. et al. Ebola Virus Disease Survivors Show More Efficient Antibody Immunity than Vaccinees Despite Similar Levels of Circulating Immunoglobulins. Viruses 12, 915 (2020).
Diallo, M. S. K. et al. Temporal evolution of the humoral antibody response after Ebola virus disease in Guinea: a 60-month observational prospective cohort study. Lancet Microbe 2, e676–e684 (2021).
Keita, A. K. et al. A 40 months follow-up of Ebola virus disease survivors in Guinea (Postebogui) reveals longterm detection of Ebola viral RNA in semen and breast milk. Open Forum Infec. Dis. 6, ofz482 (2019).
Subtil, F. et al. Dynamics of Ebola RNA Persistence in Semen: A Report From the Postebogui Cohort in Guinea. Clin. Infect. Dis. 64, 1788–1790 (2017).
Etard, J.-F. et al. Multidisciplinary assessment of post-Ebola sequelae in Guinea (Postebogui): an observational cohort study. Lancet Infect. Dis. 17, 545–552 (2017).
Diallo, M. S. K. et al. Understanding Long-term Evolution and Predictors of Sequelae of Ebola Virus Disease Survivors in Guinea: A 48-Month Prospective, Longitudinal Cohort Study (PostEboGui). Clin. Infect. Dis. 73, 2166–2174 (2021).
von Herrath, M. G. et al. ZnT8-Specific CD4+ T Cells Display Distinct Cytokine Expression Profiles between Type 1 Diabetes Patients and Healthy Adults. PLoS ONE 8, e55595 (2013).
Lhomme, E. et al. Analyzing cellular immunogenicity in vaccine clinical trials: a new statistical method including non-specific responses for accurate estimation of vaccine effect. J. Immunol. Methods 477, 112711 (2020).
Benjamini, Y. & Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. The. Ann. Stat. 29, 1165–1188 (2001).
Acknowledgements
The project has been funded by a dedicated Institut national de la santé et de la recherche médicale (Inserm) allocation on behalf the French Research Ministry. This research was supported in part by the National Institutes of Health (NIH), by Institut national de la santé et de la recherche médicale (Inserm) and by the London School of Hygiene and Tropical Medicine (LSHTM). Funding provided in part by NCI contract HHSN261201500003I through the Frederick National Laboratory for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115854, EBOVAC1. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.The dissemination represents only the authors’ views and IMI2JU is not responsible for any use of the information contained in the dissemination. This project is part of the EDCTP2 programme supported by the European Union (grant number RIA2017S-2014 – PREVAC-UP) and by the US National Institute of Allergy and Infectious Diseases of the National Institutes of Health. The clinical trial was conducted with the support of Janssen, Bavarian Nordic and Merck Sharp & Dohme Corp. who provided the vaccines according to EBOVAC 1 grant agreement. The sponsor has no role in study design, data collection and analysis or manuscript writing. We are grateful to the Ministries of Health of Guinea, Liberia, Sierra Leone and Mali who permitted the conduct of the trial. We furthermore thank Alima and all site collaborators for their contribution in the implementation of the trial. We specifically acknowledge Jose-Carlos Fernandez, Adam Nouveau, and Marie Déchenaud for their involvement in establishing the laboratory in Guinea. The authors and study team thank the participants who consented to the trial.
Author information
Authors and Affiliations
Consortia
Contributions
Y.L., A.W., C.L., E.L., R.T., and L.R. conceived and designed the study. Y.L., A.W., E.L., M.H., R.T., and L.R. analyzed and interpreted the data. E.F., L.G., C.R., A.R., M.Y., and J.T. performed the experiments. M.C. and A.B. managed the laboratory in Conakry and participated in sample and clinical data collection. Y.L., A.W., and E.L. drafted the first version and wrote the final version of the manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wiedemann, A., Lhomme, E., Huchon, M. et al. Long-term cellular immunity of vaccines for Zaire Ebola Virus Diseases. Nat Commun 15, 7666 (2024). https://doi.org/10.1038/s41467-024-51453-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51453-z
- Springer Nature Limited