

Abstract
High-throughput sequencing technologies have increasingly led to discovery of disease-causing genetic variants, primarily in postnatal multi-cell DNA samples. However, applying these technologies to preimplantation genetic testing (PGT) in nuclear or mitochondrial DNA from single or few-cells biopsied from in vitro fertilised (IVF) embryos is challenging. PGT aims to select IVF embryos without genetic abnormalities. Although genotyping-by-sequencing (GBS)-based haplotyping methods enabled PGT for monogenic disorders (PGT-M), structural rearrangements (PGT-SR), and aneuploidies (PGT-A), they are labour intensive, only partially cover the genome and are troublesome for difficult loci and consanguineous couples. Here, we devise a simple, scalable and universal whole genome sequencing haplarithmisis-based approach enabling all forms of PGT in a single assay. In a comparison to state-of-the-art GBS-based PGT for nuclear DNA, shallow sequencing-based PGT, and PCR-based PGT for mitochondrial DNA, our approach alleviates technical limitations by decreasing whole genome amplification artifacts by 68.4%, increasing breadth of coverage by at least 4-fold, and reducing wet-lab turn-around-time by ~2.5-fold. Importantly, this method enables trio-based PGT-A for aneuploidy origin, an approach we coin PGT-AO, detects translocation breakpoints, and nuclear and mitochondrial single nucleotide variants and indels in base-resolution.
Similar content being viewed by others


Introduction
The Online Mendelian Inheritance in Man (OMIM) database lists over 4700 genes with known phenotype-causing mutations1 and is expanding by continuous advancements in sequencing technologies as they substantially broaden our understanding of genetic disorders2,3. This expanded understanding has led to increased awareness of the potential benefits of preimplantation genetic testing (PGT) for monogenic disorders (PGT-M). Furthermore, several countries have initiated a population-wide offer of pre-conception carrier testing (PCT) for recessive disease, increasing the number of known carrier couples4,5,6,7. In addition, continuous increases in maternal8 and paternal9 age, contribute to a higher risk for aneuploidies10, and de novo mutations11 and (segmental) chromosomal aberrations in offspring12, respectively. These demographic factors, together with the growing list of identified genetic diseases-causing mutations13, contribute to an increased demand for reproductive care, urging the need for scalable and generic genome-wide PGT approaches.
PGT is an assisted reproductive technology (ART), which is performed on DNA samples from single-cell biopsies from day-3 embryos or 5–10 cell biopsies from day-5/6 in vitro fertilised (IVF) embryos, respectively. PGT aims to minimise the risk of transmitting a known hereditary disease or genetic abnormalities to offspring, thereby reducing the likelihood of miscarriage or the need to contemplate pregnancy termination14,15. Since its inception in 199016, PGT has evolved, encompassing non-hereditary genetic abnormalities i.e. aneuploidies, affecting embryo implantation and viability via PGT for aneuploidies (PGT-A). Yearly, 70,000 PGT cycles are performed globally17,18,19,20, with PGT-A representing over half of these in Europe18. PGT-A typically categorises embryos based on their chromosomal status, ranging from euploid to mosaic to aneuploid. While conventionally PGT-A has deemed embryos with any detectable aneuploidies unsuitable for transfer, randomised controlled trials (RCTs) cast doubt on the clinical utility of PGT-A21,22,23,24,25. Furthermore, there is a growing body of literature demonstrating the birth of healthy, euploid children after the transfer of mosaic aneuploid embryos, as such the practise of discarding these embryos is increasingly criticised26,27. Existing whole genome sequencing PGT-A methods28, which rely on low-coverage embryo sequencing, lack parental information and do not allow determination of the segregational origin of aneuploidies, i.e. whether it originates from meiosis (before fertilisation) or mitosis (after fertilisation). PGT-M is the second most common form of PGT18 and previously required designing family- and locus-specific assays, tailored for the genetic disorder within the family, causing a long waiting list. While genome-wide haplotyping methods, such as Karyomapping29, haplarithmisis30,31,32, (S)Haploseek33,34,35, GENType36 provided a generic approach and alleviated this problem, these methods utilise SNP-genotypes of only a fraction of the genome and require complex laboratory and computational protocols. Currently, a substantial proportion of PGT-M procedures still rely on family- and locus-specific approaches, such as PCR- or fluorescence in situ hybridisation (FISH)-based methods18. Comprehensive chromosome screening methods enable the assessment of chromosomal abnormalities in PGT for structural rearrangements (PGT-SR) with higher throughput than conventional methods including FISH37. Nonetheless, they cannot distinguish embryos with balanced translocations from those that are chromosomally normal38, nor can they detect haploid or triploid embryos. Importantly, transferring an embryo carrying a balanced translocation perpetuates the translocation, including the increased reproductive risk, to future generations. Another form of PGT is PGT for mitochondrial disorders (PGT-MT) that focuses on mutations in the mitochondrial DNA (mtDNA) that contribute to the risk of inheriting a genetic mitochondrial disorder39. These mutations are exclusively maternally inherited and are characterised by heteroplasmy, defined as the coexistence of normal and mutated mtDNA. Clinical symptoms manifest only when the mutation load, the threshold of mutated mtDNA, surpasses a certain level. The mutation load inherited by offspring can be highly variable due to bottleneck principles. PGT-MT allows for the selection of embryos carrying mutation loads below the pathogenic threshold39, minimising the likelihood of clinical manifestation of the associated mtDNA disorder. The current prevailing approach for PGT-MT involves PCR-based method utilising blastomere biopsy39. The integration of PGT-MT within the same laboratory workflow faces challenges due to the limited availability of human data regarding the representativeness of the mutation load in trophectoderm (TE) biopsies for the entire embryo. All presented challenges underscore the need to develop a universal PGT method that streamlines laboratory protocols and provides even genome-wide coverage to identify genetic disorders.
Here, we devised a simplified, scalable, and universal whole genome sequencing-based method for PGT (WGS-PGT) that enables all forms of PGT within a single assay. This approach relies on haplarithmisis30,40, a conceptual workflow that reconstructs and counts the number of haplotypes, inherently traces the origin of aneuploidies to meiosis or mitosis, and determines the level of mosaicism, i.e. proportion of aneuploid cells. Specifically, we demonstrate that WGS-PGT enables (i) PGT in proximity of complex genomic regions, (ii) direct detection of single- and few-base pair genetic variations, (iii) a novel form of PGT-A that uncovers segregational origin (meiotic vs. mitotic) of aneuploidies and their level of mosaicism, called PGT for aneuploidy origin (PGT-AO), (iv) (in)direct detection of the translocation breakpoints and inheritance of normal and derivative chromosomes, allowing the distinction between normal embryos and balanced translocation carriers, and (v) PGT for mtDNA disorders.
Results
Proof-of-concept for WGS-PGT
To establish WGS for haplarithmisis-based PGT, we performed a pilot study in which we carried out deep sequencing (30–40X) to compare current clinical gold standard GBS-PGT (Fig. 1a) in two PGT families (Fig. 1b). The WGS method allows for a 2.5-fold reduction in library preparation times compared to GBS (Fig. 1c). We then performed in silico subsampling at target coverages of 5X, 10X, 20X and 30X, to determine the optimal depth of coverage for accurate diagnosis. Haplarithmisis30,40 is a linkage-based approach that reconstructs haplotype blocks using single nucleotide polymorphism (SNP)-calls. Thus, having more SNP-calls to support a haplotype block reduces the impact of WGA-artefacts and sequencing errors. To measure erroneous SNP-calls, we used Mendelian inconsistencies to probe unexpected SNP-calls that are violating patterns of Mendelian inheritance. Given that 40 informative SNP-calls per Mb are used to perform diagnosis – resulting in 80 informative SNP-calls within 2 Mb upstream and 2 Mb downstream of the region of interest (ROI), the chance of misdiagnosis for a monogenic disorder is close to zero (P = 4.77 × 10−74, Supplementary Note). To this end, we evaluated several key parameters, including breadth (Fig. 1d) and depth of coverage (Supplementary Fig. 1), Mendelian inconsistency rates (Supplementary Fig. 2, Supplementary Data 1) and haplotype concordance (Supplementary Fig. 3). WGS provided a 4-fold higher breadth of coverage, i.e. the proportion of the genome that is sequenced, than GBS, with WGS exhibiting a median breadth of coverage of 90.4% (2.15 s.d.) in all target coverages while GBS exhibits a median breadth of coverage of 22.3% (1.68 s.d.). Comparing different depth of coverage levels revealed a significant difference in breadth of coverage between 5X and 10X (P = 3.52 × 10−3, two-sided Wilcoxon’s rank-sum), while increasing the depth of coverage from 10X to 20X did not lead to a significant increase in breadth of coverage (P = 8.31 × 10−2, two-sided Wilcoxon’s rank-sum, Fig. 1d). The number of genome-wide informative SNPs increased 10-fold for WGS-PGT at 10X coverage compared to GBS-PGT, reaching 2.5 million genome-wide informative SNPs (± 46,000 s.d.) (Supplementary Fig. 4a). Specifically, at 5X coverage there were 1.5 million genome-wide informative SNPs (± 53,422 s.d.), whereas at 30X coverage, this number increased to 2.9 million (± 25,960 s.d.). Typically, a higher number of genome-wide informative SNPs increases the accuracy and reliability of haplotype inference. For all target coverages, the mean genome-wide haplotype concordance between GBS and WGS was higher than 97%. Increasing the depth of coverage from 10X to 20X, revealed no significant improvement (paternal haplotype P = 0.59 and maternal haplotype P = 0.70, two-sided Wilcoxon’s rank-sum, Supplementary Fig. 3). Furthermore, the mean Mendelian inconsistency rates representing the relative proportion of WGA artefacts reduced substantially from 11.3% (± 1.26 s.d.) in GBS-PGT to 4.6% (± 1.35 s.d.) for WGS-PGT at 10X coverage (Supplementary Fig. 2a). Based on these findings, we settled that 10X sequencing provided sufficient data to reliably conduct haplarithmisis-based PGT-M.

a The study’s workflow including embryo biopsy, DNA amplification, library preparation (GBS and WGS), sequencing, analysis, and clinical diagnosis. b study sample inclusion diagram. c library preparation times in hours for GBS and WGS methods. Black outline represents hands-on time per step. d boxplot showing the breadth of coverage for subsampled WGS samples (n = 12 per target coverage) compared to corresponding GBS samples and for 10X WGS samples (n = 84, two-sided Wilcoxon’s rank-sum). e half violin plot, overlayed with boxplot showing depth of coverage for WGAed embryo (MDA) and bulk gDNA samples (BULK) for GBS and WGS. Each data point represents an individual sample (n = 31 for MDA, n = 58 for BULK per method, two-sided Wilcoxon’s rank sum). f Mendelian inconsistency rate for 10X embryo samples (n = 31 per method, two-sided Wilcoxon’s rank sum). g genome-wide haplotype concordance of WGS with GBS indicated per parent (maternal, n = 23; paternal, n = 25). In all boxplots the horizontal lines of the boxplot represent the 25th percentile, median and 75th percentile. The whiskers extend to 1.5 * IQR. Source Data are provided as a Source Data file. GBS: genotyping-by-sequencing; WGS: whole genome sequencing; IQR: interquartile range; WGAed: whole genome amplified; MDA: multiple discplacement amplification. Source data are provided as a Source Data file.
Clinical validation of WGS-PGT
To clinically validate WGS-PGT, we sequenced 31 embryo samples from 16 families at 10X coverage (Fig. 1b, Supplementary Table 1). Specifically, we selected PGT families that posed analytical challenges when using GBS-PGT e.g. when the ROI, representing the genomic location of the pathogenic variant of interest, was located in complex genomic regions, such as telomere regions or when the embryo exhibited haplotype recombination in the proximity of the ROI, when the family was consanguineous, or when the family had multiple genetic disorders, such as monogenic diseases and translocations. We observed a disparity in depth of coverage between Whole Genome Amplified (WGAed) embryo and bulk samples for GBS (P = 1.43 × 10−3, two-sided Wilcoxon’s rank-sum), which is likely attributed to the potential loss of restriction enzyme sites during amplification, leading to a lower library amount (Fig. 1e). However, in WGS, we found a similar depth of coverage between bulk DNA and WGAed samples, indicating a more stable read out from WGS-PGT (P = 0.46, two-sided Wilcoxon’s rank-sum). Results from the key parameters could be replicated in these families, with a mean autosomal Mendelian inconsistency rate of 2.42% (±2.23 s.d.) for WGS and a mean Mendelian inconsistency of 7.66% (±2.33 s.d.) for GBS (n = 31 embryos) (Fig. 1f). The mean autosomal Mendelian inconsistency rate in the validation subset demonstrated a lower rate than in the pilot study. This difference can be attributed to the specific characteristics of family 1, where all embryos had a translocation and two embryos harboured an aneuploid chromosome (Supplementary Fig. 2a, Supplementary Table 3). Moreover, the mean concordance of maternal haplotypes was 97.9% (±3.7 s.d.), and the mean concordance of paternal haplotypes was 98.9% (±1.6 s.d.) (Fig. 1g). One outlier for maternal haplotype concordance was observed at 81.0%, and was from an embryo with a triploid genome (family 18, embryo 37), which originated in meiosis II and was of maternal origin (Supplementary Table 3). Consequently, the maternal haplotypes, derived from combinations of alleles on two chromosomes contributed to the lower haplotype concordance. One of the diagnostic criteria in the analysis of an embryo for a specific monogenic indication include the number of informative SNPs in a 4 Mb interval, i.e. 2 Mb up- and downstream of the mutation, on the maternally and/or paternally inherited haplotypes (Methods and Supplementary Table 2). Since we have deliberately selected challenging PGT families with ROIs in complex genomic regions or high rates of consanguinity, in 13 out of 35 ROI, the haplotype concordance did not meet the assessment criteria in GBS-PGT (Supplementary Data 2). However, using WGS-PGT, in 5 out of the 13 ROIs, the assessment criteria could be met, owing to its inherent higher resolution (Fig. 2a).

a Haplarithms for PGT-M indications with different modes of inheritance, i.e. AD, AR, and X-R. The top panel shows family pedigrees. The lower panel shows haplarithms generated with GBS (gold) and WGS (blue) – from top to bottom – BAF profiles, interpreted paternal haplotypes (H1: darkblue; H2: lightblue), paternal haplarithms, interpreted maternal haplotypes (H1: red; H2: pink), maternal haplarithms, logR values representing the log2 ratio of the observed copy numbers to the expected copy number. The ROI is indicated by an orange line. b Genotypes ascertained by direct mutation detection (obs) compared to the haplarithmisis result (exp) subdivided for genetic indications that included SNVs or single nucleotide deletions. Embryos are numbered, and the indications are indicated in superscript (1: indication 1, 2: indication 2, a or b indicate different genetic loci in case of a compound heterozygous mutation). The mutant allele track shows the type of inheritance indicated by AR or AD, and the triangle is coloured with the mutant allele (A = green, C = orange, G = blue, T = red, del = grey). The concordance of diagnosis and genotype with the haplarithmisis result is colour-coded (concordant = green, not concordant = red, inconclusive = orange). Recessive indications were considered inconclusive if the proportion of reads supporting the reference allele were <30% or >60%. PGT: preimplantation genetic testing; M: monogenic; AR: autosomal recessive; AD: autosomal dominant; X-R: X-linked recessive; H1; haplotype 1; H2: haplotype 2; BAF: B-allele frequency; ROI: region of interest; GBS: genotyping-by-sequencing; WGS: whole genome sequencing; exp: expected, obs: observed; del: deletion. Source data are provided as a Source Data file.
PGT-M with the potential to directly detect pathogenic single nucleotide variants
PGT-M is challenging when a close relative is unavailable for phasing, or in cases when prospective parents present with a de novo pathogenic single nucleotide variant (SNV). Detection of the pathogenic variant shows a promising alternative as it may facilitate a diagnosis in these families. While GBS approaches only cover 20% of the genome and enabled indirect detection of monogenic aberrations, WGS-PGT, at 10X depth of coverage, covers > 80% of the genome (Fig. 1d), thereby facilitating direct SNV detection. For 22 monogenic indications that included single base pair substitutions or deletions, we compared the genotypes and diagnoses ascertained from direct SNV detection with those anticipated based on the haplarithmisis result. Direct SNV detection provided the correct diagnosis in 90% (n = 20) and correct genotype in 82% (n = 18) of the ROI (Fig. 2b, Supplementary Fig. 6). When the number of reads at the ROI was higher than 5, the expected genotype could be correctly identified in all cases. In embryos with a lower read count than 5 reads – embryo 4, embryo 30 and embryo 9 indication 1b – the direct variant detection showed discordant genotype results with the expected results based on haplarithmisis. Remarkably, direct variant detection showed promise in rare instances, resolving pathogenic SNVs in embryos with inconclusive haplarithmisis results. Specifically, in embryo 23, which was assessed for an autosomal dominant pathogenic SNV, the assessment criteria thresholds for haplarithmisis were not met (Supplementary Table 2), resulting in an inconclusive diagnosis of the embryo. Direct variant detection showed presence of the mutant allele at the ROI in 4 out of 5 reads, allowing embryo 23 to be classified as affected (Fig. 2b, Supplementary Fig. 6). To examine whether direct pathogenic variant detection could also be applied to larger deletions, we visualised indications representing deletions of two or more base pairs (n = 11). Deletions spanning two or three base pairs could be identified within the integrated genomics viewer software (IGV) (n = 6, Supplementary Fig. 7a). Bigger deletions presenting as autosomal recessive pathogenic variants showed a loss of coverage (n = 2, Supplementary Fig. 7b), while bigger deletions presenting as autosomal dominant or x-linked variants could be identified using the “view as pairs” option in IGV in two affected embryos from family 7 and two unaffected embryos could be confirmed (Supplementary Fig. 7c). Furthermore, one aberrant embryo did not show the mutation as it has a mitotic trisomy of the unaffected haplotype. Notably, the putative deletion could not be detected directly in one embryo (embryo 5 from family 2) which was a carrier of the deletion (Supplementary Fig. 7c). Following clinical validation, WGS-PGT has been clinically implemented for 489 embryos (n = 115 families, 80 OMIM indications). 87 families were directly assessed for the mutation (pathogenic variant). In 56 families, the variant could be correctly detected, while in 9 families the coverage was lower than 5 reads at the ROI, and 22 families had no affected embryo in the cycle (Supplementary Fig. 7 and Supplementary Data 3).
PGT for aneuploidy origin (PGT-AO): a transformative PGT-A
Haplarithmisis-based WGS-PGT can accurately determine the segregational origin, i.e. meiosis I, meiosis II, or mitosis, of aneuploidies and their degree of mosaicism ( > 10%) and these were all concordant as observed in GBS-PGT (Fig. 3a). Unlike meiotic trisomies, which involve both homologous chromosomes of the contributing parent, mitotic trisomies result from the exact duplication of a single homologue. Distinguishing between mitotic and meiotic II trisomy is possible when a crossover takes place on the chromosome of interest. The crossover rate for human chromosomes varies between 1.07 cM/Mb to 1.76 cM/Mb, ensuring that each chromosome generally experiences at least one crossover depending on the crossover rate and chromosome size41. Distinguishing between mitotic and meiotic II trisomies is not possible in rare cases where there is no crossover, or when the crossover is located in a challenging genomic region (e.g. telomeric or centromeric).

a Schematic representation of trisomies with different segregational origins (meiosis I, meiosis II or mitosis) is shown, using close-relative phasing or using parents-only phasing to identify segregational origin. The segregational origin using parents-only phasing was determined by inspecting the distance between the segmented P1/M1 and P2/M2 at the centromeric and telomeric region. Further explanation is provided in Supplementary Note. b Representative cases of aneuploidies with different degrees of mosaicism. The degree of mosaicism was determined by calculating the distance between paternal and maternal BAF segments as indicated with curly brackets. c Representative haplarithms of embryos with trisomies with a different segregational origin. Top panel: schematic of the chromosomal constitution. Middle panel: haplarithms when using a close relative as referent. Bottom panel: haplarithms when using the same embryo as referent to phase the parents and define the segregational origin. P1/M1 and P2/M2 haplarithms are shown in blue and red dots, respectively. LogR values and segmented logRs are shown in black dots and orange line, respectively. Curly brackets indicate the distance between the paternal and maternal BAF segments. BAF: B-allele frequency. Source data are provided as a Source Data file.
We identified 14 aneuploidies in 11 of the 29 affected embryos analysed, encompassing 10 trisomies, 3 monosomies and 1 triploidy, with some embryos carrying more than one aberration. The segregational origin could be determined for 10 out of 11 detected chromosomal gains (10 trisomies and 1 triploidy, Supplementary Table 3). Only a single embryo (Fig. 3b - embryo 18, family 7) did not have a crossover in the trisomic chromosome, precluding the differentiation between meiotic II and mitotic origins. Furthermore, we observed mosaic meiotic trisomies suggesting that a fraction of the biopsied cells had undergone chromosomal rescue (Supplementary Table 3)28. Three mosaic aberrations with a meiotic origin were identified (Supplementary Table 3). These included two embryos with a trisomy with a mosaicism level of 80% and 90%. The third embryo had a genome-wide triploidy with meiosis II origin and mosaicism level of 100% while chr15 was diploid with a mosaicism level of 50%, suggesting that a fraction of the biopsied cells underwent chromosomal rescue for chr1527. Additionally, the mosaicism level for 3 embryos with monosomy was determined (100%, 100%, 45%). Mosaicism levels of 0% and 100% indicate a non-mosaic zygosity. We further assessed nine aberrant embryos with copy number gains of known segregational origin to validate parents-only haplotyping and subsequent determination of segregational origin (Fig. 3c). In all cases the segregational origin detected by parents-only haplarithmisis was concordant with standard haplarithmisis. These results expand the possibilities of detecting segregational origin in human preimplantation embryos when no close relative is available.
PGT-SR with direct and indirect detection of translocation breakpoints
Nine embryos from three families that underwent PGT-SR with shallow sequencing (Methods) were re-analysed using the WGS-PGT protocol. The copy number state of all embryos could be correctly determined with WGS-PGT and haplarithmisis purely by assessing the segmentation of the logR values, which represent the log2 ratio of the observed to expected copy number (Fig. 4, Supplementary Fig. 9, Supplementary Data 4). Although small duplications, such as the 1.08 Mb segmental duplication of chr16 in embryo 4 of family 1, were not segmented, such embryos could still be correctly diagnosed based on the presence of the reciprocal deletion (Fig. 4), such that each conclusion was concordant with the original copy number variation analysis using VeriSeq. The identification of these segmental deletions and duplications remained consistent across all subsampled sequencing depths, even at 5X coverage (Supplementary Fig. 9). In couples where embryos exhibited no copy number imbalances, the possibility of inheriting either both normal homologues or both derivative chromosomes (balanced translocation) from the carrier parent should be considered. Shallow sequencing proves insufficient to distinguish between these cases. However, our haplarithmisis-based PGT successfully identified embryos with unbalanced translocations. For these cases, the diploid flanking haplotypes of the translocation allowed us to distinguish between normal and derivative chromosomes in all other embryos. Specifically, embryos 1 and 4 from family 1 were phased with an unaffected sibling as a seed for phasing. Haplotypes on the diploid side of the chromosomal breakpoint were distinguished as either dark blue, indicating consistency with the reference haplotype or light blue, indicating the alternative haplotype. Consequently, embryo 1 inherited the normal chr8 and derivative chr16 while embryo 4 inherited the derivative chr8 and normal chr16 (Fig. 4). Importantly, the paired-end sequencing data of the carrier parent and the embryos can be leveraged to identify translocation breakpoints using Manta. We applied Manta because it showed the highest true positive rate for the detection of translocation breakpoints42 for short-read sequencing data as compared to other methods, including DELLY43, LUMPY44, GRIDDS45, and BreakDancer46. In all carrier parents (n = 3) included in this study, we could identify a breakpoint pair that closely corresponded to the expected translocation breakpoints ascertained using karyotyping (Supplementary Data 4). Unlike the karyotyping results, the breakpoints derived from paired-end sequencing data could be determined at approximately base pair resolution, allowing more precise regions of interest to be defined for haplarithmisis. It is important to note that the breakpoint locations obtained may exhibit variation among embryos, and in some cases, multiple hits may be identified. Subsampled data from family 1 showed that a minimum genome-wide depth of coverage of 10X is required to accurately call these breakpoints (Supplementary Data 4). We could identify corresponding breakpoint pairs in six of the eight unbalanced embryos from families 1 and 19. The two embryos in which no breakpoints were identified both inherited a derivative chr8 carrying the small segment of chr16 (1.08 Mb). Although paired-end sequencing analysis did not identify a breakpoint pair in embryo 23 of family 9, which could be attributable to a chaotic copy number profile (Supplementary Data 4), we could correctly identify the relevant breakpoint pair in the unbalanced embryos 24 and 25. For the paternal ins(10;7) of family 19 (Supplementary Data 4), one would expect to find a single breakpoint on chr10 and two corresponding breakpoints on chr7, specifically a position on the q arm of chr7 and the end of chr7. However, this could not accurately be detected using Manta. While Manta did identify breakpoint pairs from between chr10 and chr7 in the carrier parent, the chr10 coordinates do not match the findings from the diagnostic karyotyping and only one corresponding chr7 position was identified which lies in the centre of the expected inserted segment. Whether the same breakpoints would be found in unbalanced embryos could not be assessed as neither of the unbalanced embryos, embryos 38 and 39, inherited the derivative chr10. Based on the segmented logR information from Haplarithmisis conducted on embryos 38 and 39, it was however possible to determine that the chr7 breakpoint would be around 129,135,000 bp, which is in line with the findings from the diagnostic karyotyping of the father.

Representative haplarithms for embryos with structural rearrangements. Two embryos from family 1 are shown where the structural rearrangement is a translocation between chr8 and chr16 as indicated by the schematic chromosome representation. Haplarithms include BAFs, paternal haplotypes, paternal haplarithm and copy number indicated by logR with breakpoints as identified by Manta (green) and flanking haplotypes H1 (darkblue) and H2 (lightblue) indicated by the dashed box. P1/M1 and P2/M2 haplarithms are shown in blue and red dots, respectively. LogR values and segmented logRs are shown in black and orange, respectively. PGT: preimplantation genetic testing; BAFs: B-allele frequencies; Pat: paternal; H1: haplotype 1; H2: haplotype 2. Source data are provided as a Source Data file.
PGT for mitochondrial DNA disorders
The current gold standard for PGT for mtDNA disorders (PGT-MT) requires a specialised PCR-based restriction fragment length polymorphism (PCR-RFLP) workflow that is carried out on a day-3 blastomere biopsy39. We compared the heteroplasmy levels from the blastomere biopsy to heteroplasmy levels obtained by applying the same protocol to day-5 TE biopsies (n = 4, Fig. 1b) and found that the heteroplasmy levels differed by 1% to 4% (Fig. 5a). Furthermore, the PCR-RFLP protocol applied to the corresponding DNA derived from the surplus embryos, i.e. the remaining embryo, yielded heteroplasmy levels that differed by 0% to 4% and 1% to 3% to the day-3 and day-5 biopsy results, respectively (Fig. 5a). Subsequently, we applied our WGS-PGT protocol to day-5 TE biopsies (n = 4), and DNA derived from the corresponding surplus embryo material, i.e. 100–200 cells, with a target genome-wide sequencing depth of 30X (Fig. 1b). The mitochondrial genome was highly covered at all levels of subsampling with only one site being covered less than 100 times in the TE-biopsy data after subsampling to 10X coverage (Fig. 5b). Importantly, 105 mtDNA sites with known pathogenic variants had a minimum coverage of 1,944X in the 10X subsampled TE-biopsy data (n = 4) (Supplementary Fig. 10). To assess reproducibility of our PGT-MT approach, we calculated the mitochondrial genome coverage of the samples sequenced at 10X for PGT-MT (n = 2) or PGT-M and PGT-SR (n = 23) indications and found that the coverage across all WGAed embryo samples was comparable (two sites with coverage under 100X in any sample, minimum coverage of any pathogenic variant site 826X in any sample) (Fig. 5b, Supplementary Fig. 10). Compared to the heteroplasmy levels elucidated from the day-3 biopsies by PCR-RFLP, the day-5 10X WGS-PGT results varied by 1.5% lower to 3.3% higher (Fig. 5a). Subsampling the sequencing data had little effect on the calculated heteroplasmy levels with estimates varying up to a maximum of 1.6% in any sample at different levels of genome-wide coverage (Supplementary Tables 4–5). The subsampled 10X WGS-PGT heteroplasmy levels from the surplus embryo were 0% to 3.6% higher compared to the day-3 biopsy and between 1.6% lower and 1.5% higher compared to the day-5 biopsy (Fig. 5a, Supplementary Tables 4–5). Similarly, when TE biopsy and surplus embryo were sequenced directly at 10X (n = 2, Fig. 1b), the heteroplasmy levels obtained for PCR-RFLP on day 3 differed by 4% from the WGS TE biopsy result and there was a 1 to 1.8% difference between the WGS results from the TE biopsy and the surplus embryo material (Fig. 5a, Supplementary Table 6). In two cases, where the day 3 biopsy with PCR-RFLP did not yield a result, a heteroplasmy level could be obtained using WGS-PGT.

a Heteroplasmy levels in m.3243 A > G embryos as determined by PCR-RFLP for day-3 blastomere biopsy, day-5 TE biopsy and surplus embryo material (embryos 41–44) or using ~10X WGS-PGT (embryos 45–50). b Mean depth of coverage per mtDNA position is shown for ~10X WGS-PGT data from embryos sequenced for PGT-M or PGT-SR (outer ring, n = 23), deep sequencing with subsequent sub-sampling for PGT-MT (middle ring, n = 4) and direct ~10X sequencing for PGT-MT (inner ring, n = 2). Position 0 is at the top with the positions ordered clockwise and colour representing the number of reads per position. The MELAS mutation position (m.3243) is highlighted as a separate segment and magnified 30x compared to all other positions. PGT: preimplantation genetic testing; MT: mitochondrial DNA disorders; RFLP: restriction fragment length polymorphism; TE: trophectoderm biopsy; WGS: whole genome sequencing; M: monogenic; SR: structural rearrangement; mtDNA: mitochondrial DNA. Source data are provided as a Source Data file.
Discussion
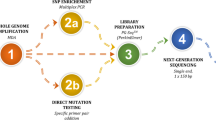
We present WGS-PGT, a clinical whole genome sequencing method for all forms of PGT that outperforms traditional and state-of-the-art PGT technologies (Fig. 6). While a plethora of sequencing-based PGT methods has emerged recently31,32,35,36,47,48,49,50,51,52,53,54, none enabled all forms of nuclear and mitochondrial PGT in a single assay (Supplementary Table 7). Moreover, the advent of SNP-array and GBS-based PGT methodologies enabled generic assays, however these approaches still faced challenges in complex genomic regions, e.g. telomeres and centromeres, in consanguineous couples, and when there is haplotype recombination in proximity of the ROI. The increased resolution through WGS-PGT allowed more embryo samples to meet the diagnostic assessment criteria thresholds in these complex genomic regions for PGT-M. In parallel, PGT-M via haplotyping can be complemented with direct pathogenic variant detection. WGS-PGT enabled parents-only PGT-AO by identifying aneuploidies, their segregational and parental origin and their level of mosaicism without requiring a close relative for phasing. We demonstrate the ability to detect structural rearrangements and distinguish normal from balanced embryos through both a direct approach, leveraging paired-end sequencing information from the embryo samples, and an indirect approach, which involves analysing flanking haplotypes. Even though various strategies exist to differentiate normal from balanced embryos, including MaReCs52, a method based on shallow sequencing55, and a method based on Nanopore sequencing technology56, they cannot be integrated with other workflows for other PGT purposes as each workflow requires different technologies. Finally, we show the comparability and reproducibility of heteroplasmy levels between traditional day-3 blastomere biopsy with PCR-RFLP and our WGS-PGT method. We demonstrate that all mtDNA positions in the samples sequenced at 10X coverage have a mtDNA coverage of more than 100X except for two sites. While prior studies have demonstrated the ability to identify mtDNA variants and heteroplasmy levels from WGS data57,58, integrating these into an all-in-one PGT method is an important step forward that makes PGT scalable and accessible.

Illustrations and characteristics of PGT methods. WGS-PGT serves as an all-in-one method that captures all the functions within a single approach, while more traditional PGT methods are each designed for a specific purpose. WGS: whole genome sequencing; PGT: preimplantation genetic testing. PCR: polymerase chain reaction.
A key feature of WGS-PGT is the ability to directly detect the pathogenic variant of interest. Direct variant detection can complement haplarithmisis by resolving uncertain or inconclusive findings and by offering a solution in families with a de novo variant in one of the parents or when a suitable close relative for phasing is unavailable. In all cases where the depth of coverage exceeded 5X, we could correctly detect the SNV. A prior study that conducted WGS on embryos to identify de novo pathogenic variants for known diseases and polygenic risk score analysis58 did not account for allelic dropout or drop-in issues related to amplification as they did not include parental sequencing information in their analysis. An advantage of our method is that it can be performed both in a direct and indirect fashion using haplarithmisis principles, to be certain of our pathogenic variant of interest.
Apart from the primary objective of PGT-A–i.e. prioritising embryos with the highest implantation potential–we introduced PGT-AO, which determines the segregational origin of aneuploidies and their degree of mosaicism in parallel. The distinction between PGT-AO and PGT-A underscores the divergence between the objectives of selecting a chromosomally euploid embryo or an embryo with the highest implantation potential for transfer. Importantly, our approach permits the analysis of haploid or triploid embryos and embryos with meiotic aneuploidies, particularly those involving chromosomes assessed in non-invasive prenatal testing (NIPT)59. Mitotic aneuploid embryos are often mosaic and the abnormality may not be uniformly distributed throughout the blastocyst60,61,62,63. Furthermore, previous studies have shown that mosaic embryos can result in healthy offspring26,27, and chromosomal mosaicism may disappear via a self-correction mechanism where aneuploid cells are depleted in the inner cell mass and remain present in the TE lineage64. Current copy number quantitation methods fail to differentiate between true mosaic embryos and uniformly euploid or aneuploid embryos with technical noise65. These findings underscore the importance of assessing the parental and segregational origin of aneuploidies besides the degree of mosaicism. The ability of PGT-AO to select an embryo without meiotic aneuploidies, most likely helps reducing the risk of failed implantation and miscarriage of a conceptus with a chromosomal aberration66,67. Longitudinal non-selection studies are needed to explore the connection between the segregational origin of aneuploidies, implantation and viable pregnancy outcomes. This innovation raises ethical, legal, and social issues that need further scrutiny with respect to proportionality, justice and reproductive and informational autonomy68.
WGS-PGT has some inherent limitations. The coverage levels may not suffice (< 5X) for accurate de novo pathogenic variants detection, which is recommended to be 30–40X69. To address this, we propose increasing the depth of coverage to 30X when there is a de novo pathogenic variant in the parents. The genome-wide Mendelian inconsistency rate of 2.42%, representing both amplification errors, sequencing errors and putative de novo pathogenic variants. Notably, when specifically examining bulk samples from trios, others have observed Mendelian inconsistency rates of 1.92%70. Additionally, short-read sequencing may limit the ability of WGS-PGT to directly assess pathogenic SNVs that are located in highly repetitive regions of the genome, which can be tackled by long-read sequencing. While incorporating long-read sequencing into our method could enhance the ability to directly detect de novo SNVs, it will increase the costs and complexity of the workflow.
The Mendelian incosistency rates highly exceed the average rate of de novo pathogenic variants of 1.20 × 10−8 per nucleotide per generation (~0.3% per generation)71, probably attributable to sequencing errors. Longitudinal validation of the direct pathogenic variant detection is essential. Although incorporating PGT-MT within the same workflow is possible, we need further validation of WGS-PGT for mtDNA disorders on day-5 biopsies due to the limited sample size of PGT-MT families. Another limitation of our method and PGT in general is the need for embryo biopsy, as biopsy procedures demand specialised technical expertise, costly equipment and might impede embryo viability. In response, developments in the field of PGT have focused on utilising non-invasive DNA sources like cfDNA in the spent culture medium72,73 that originates from inner cell mass, trophectoderm cells74, cumulus cells, and polar bodies75. Potential contamination from cells or maternal origin should be assessed in future methods. Haplarithmisis can distinguish between contributions from maternal and foetal genomes in placental DNA samples40, therefore our WGS-PGT method could be used to tackle the maternal contamination and thus WGS-PGT is future-proof for development of non-invasive PGT.
In summary, we developed and applied a new PGT method, named WGS-PGT, that can capture all forms of PGT in a single assay. WGS-PGT enables a simplified, scalable, and universal PGT that outperforms current state-of-the-art PGT methods and has the capacity to enhance reproductive genetic care.
Methods
Study participants and ethical approval
Couples were counselled by clinical geneticists at Maastricht University Medical Centre + (MUMC+) and enrolled in the diagnostic preimplantation genetic testing (PGT) procedure (licensed by the Dutch Ministry of Health, Welfare and Sport CZ-TSZ-291208) after signing an informed consent form. Couples who underwent PGT, provided written informed consent for the use of affected embryos for the development of PGT methods. Full ethics approval was waived by the ethics committee from the Maastricht UMC+ (file number 2023-0091) due to the retrospective design of the study, which involved the use of spare (amplified) DNA and the anonymized handling of the data according to the Dutch Medical Treatment Contracts Act (WGBO). Genetic and clinical data shared in the context of this study cannot be used to identify individuals. We included 21 families who had undergone PGT for (double) monogenic, structural, or mitochondrial indications where spare (amplified) DNA from all samples was available. No additional embryo biopsies were performed for this study specifically, except for PGT-MT (see in section PGT for mitochondrial DNA disorders).
GBS-PGT sample collection and processing
For PGT-M the standard clinical procedure in our facility involved genotyping-by-sequencing (GBS), as described previously31,76. These procedures are part of standard clinical practice and were not performed for this study specifically. Briefly, peripheral blood samples were collected from prospective parents and close relatives from which DNA was isolated using the QIAsymphony DSP DNA Midi kit (Qiagen, Germany). A TE biopsy, i.e. 5–10 cells, was taken from sufficiently developed embryos on day-5 and the collected material was subjected to multiple displacement amplification (MDA) using the REPLI-g Single Cell kit (Qiagen, Germany), following the manufacturer’s instructions. Library preparation, using the OnePGT solution (Agilent Technologies) was then carried out on genomic DNA samples from parents and close relative(s), along with the WGAed samples from embryos, following the manufacturer’s instructions and as previously described31. The libraries were sequenced on a NextSeq 500 sequencing system. All excess DNA not used for library preparation was stored at −20 °C, in accordance with clinical standards.
Whole genome sequencing sample processing
DNA from parents and close relative(s) and WGAed DNA from embryos, (see above) that was stored according to clinical standards, was subjected to whole genome sequencing (WGS) library preparation. Paired-end short-reads of 150 bp (300 cycles) were used with an average insert size of 450 bp. Briefly, a minimum input of 20 μl, with a concentration of 30 ng/μl, was supplemented with 0.12 ng of embryo tracking system (ETS) fragments (concentration: 0.03 ng/μl)76. Subsequently, bead-linked transposome (BLT) PCR-free library preparation (Illumina, San Diego) was carried out according to the manufacturer’s instructions for input quantities ranging from 300 to 2000 ng. The resulting libraries were purified using a double-sided bead purification process. Sequencing was performed using a NovaSeq 6000 in the Radboudumc to a target depth of coverage of ~30X-40X or 10X.
Sequencing data processing and quality control
The raw sequencing data were demultiplexed and aligned to the human reference genome (complemented with the sequences of all ETS amplicons) using bwa-mem2 (v. 2.2.1)77. The WGS data were aligned to hg38 while the GBS data were aligned to hg37, and positions were then converted to hg38 using liftOver from the Rtracklayer package78. The quality of the resulting alignment was assessed using Qualimap (v.2.2.1.)79 to determine the breadth and depth of coverage as well as the purity of the expected ETS fragment.
Samples that underwent deep (~40X) sequencing, were subsampled using the “view” function from SAMtools (v. 1.15.1)80. The fraction of the original bam file required to generate different subsets was calculated by dividing the target coverage (5X, 10X, 20X and 30X) by the original coverage. Wilcoxon Rank-Sum Tests were used to compare the GBS and WGS groups and Kruskal-Wallis test with Dunn’s multiple comparisons test to compare the different target coverage groups.
Haplarithmisis-based PGT
GBS and WGS data were analysed using a modified version of the siCHILD analytical pipeline that is equipped with haplarithmisis30 and has been further adapted for sequencing data76. Initially, a preparatory test was conducted for the parents and close relatives (s) to assess whether the couple was eligible for PGT. Subsequently, an “embryo test” was run in which embryo haplotypes were reconstructed to ascertain a diagnosis. Using this pipeline, aligned sequencing data was processed using Joint HaplotypeCaller from GATK (v. 3.4-46)81 to extract the genomic locations in the dbSNP database (v. 150). All subsequent processing was carried out in R (version 3.3.1)82 as previously described30,76. Briefly, the GATK output was used to determine the genotype per position of each sample using vcfR (v. 1.8.0.9000)83 R package. This was used to calculate B allele frequencies (BAF), which were subsequently phased by leveraging information from the parents and a close relative. Segmentation of the parent-specific phased BAFs was used to determine the haplotypes. Copy number states for 100kb-sized genomic bins were assessed using the QDNAseq (v. 1.10.0)84 R package and segmented using piecewise constant fitting (PCF)85 with a gamma value of 50. GBS-PGT requires eight informative SNPs at either 2 Mb side of the ROI (Supplementary Table 2). The proportion of these informative SNPs should be at least > 80% concordant with either the affected or unaffected haplotypes as determined by parental phasing.
Haplarithmisis comparison between GBS and WGS
Haplarithmisis output was evaluated from subsampled data generated at ~10X depth of coverage by assessing mendelian inconsistency level, number of informative SNPs, and haplotype concordance. The assessment criteria for the preparatory test and embryo test are listed (Supplementary Table 2). Mendelian inconsistency rate was defined as the proportion of inconsistent genotypes out of the total number of genotypes that were analysed. These rates were calculated for individual chromosomes and then the mean for all autosomes was calculated. Haplotype concordance between GBS and WGS was determined by comparing the interpreted haplotypes per parent. Subsequently, the haplotype concordance of WGS-PGT with GBS-PGT was assessed for each target coverage using Kruskal-Wallis test followed by Dunn’s multiple comparisons test.
Direct mutation detection
SNVs and deletions in affected embryos were evaluated by examining the nucleotides at the base level. We analysed genetic loci that entailed single nucleotide alterations such as SNVs and deletions. In total 22 genetic loci were included, of which 19 SNVs and three deletions. Moreover, 11 genetic loci were analysed that entailed deletions ranging 2 bp from 398 kb. Reads mapping the relevant genetic location were extracted from bam files by indicating the chromosome, start and end position of a genomic interval using the SAMtools “view” function80. The resulting bam files were visualised in the Integrated Genomics Viewer (IGV) (version 2.11.9) to ascertain the nucleotides at the indicated position. PGT results determined by haplarithmisis (affected, not affected, carrier, inconclusive) were compared with the putative diagnosis based on direct variant detection. Moreover, the expected genotype based on the haplotyping result was compared to the genotype as ascertained by direct variant detection.
PGT-AO: classification of (segmental) chromosomal abnormalities, their segregational origin and their level of mosaicism
Copy number variation (CNV) calls were visualised employing haplarithms. Copy number state of the embryos was determined by analysing log2 ratio of the observed copy number to the expected copy number, as indicated by logR ratios, alongside shifts in genotype frequencies of the reads, measured by BAFs. Subsequently, the aberrations were classified based on several criteria. (i) the copy number aberration detected (i.e. gain or loss), (ii) the size of the aberration (i.e. genome-wide, chromosomal, or sub-chromosomal), (iii) the parental origin of the aberration (i.e. paternal or maternal), (iv) the segregational origin of the aberration (i.e. meiosis I, meiosis II, or mitosis), and (v) the degree (> 10%) of mosaicism. To determine the degree of mosaicism, the genomic coordinates at the logR shift were used to extract the segmented parental phased BAF of the location of interest. BAF values were then compared to the reference dataset by Conlin et al.86. Besides conventional haplarithmisis that includes a close relative to phase the parents, “parents-only phasing” was performed by phasing the parental genome with the embryo itself.
PGT for structural rearrangements
Three families with PGT-SR indications were included. Copy number variation was originally analysed using the VeriSeq-PGS kit (Illumina Inc., Santa Clara, CA) according to the manufacturer’s instructions. For the family that only had a PGT-SR indication, an additional TE biopsy was taken from the affected surplus embryos to generate a WGAed DNA sample using the REPLI-g Single Cell kit (Qiagen, Germany). In the case of families with both PGT-SR and PGT-M indications, the excess WGAed DNA was used and re-processed with our WGS-PGT approach as described above. The data for families with dual PGT-M and PGT-SR indications (family 1 and 9), for which close relative(s) were also sequenced, were processed and visualised as described for PGT-M cases. Where deep sequencing (30–40X) was undertaken, the subsampling strategy was also applied, and structural rearrangements were assessed at all target coverages. For one family (family 19) no referent individual was sequenced, in this case each embryo was used to phase the remaining embryos. Derivative chromosome breakpoints were ascertained using Manta (v1.1.0) with default settings87. The resulting variants were then filtered to include only break points (“BND”) where pairs of mates were identified on the expected chromosomes. In cases where Manta did not identify identical breakpoints in the embryo, the breakpoints were estimated from the haplarithmisis output, specifically from the segmentation of the phased parental BAFs and the segmentation of the logRs.
PGT for mtDNA disorders
10 embryos from 2 families were included (Fig. 1b) that were deemed affected (>transfer threshold 15%)39,88 for mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS, m.3243 A > G) based on results from the current gold standard blastomere-biopsy (day-3) testing. The embryos were re-biopsied on day-5 to obtain a TE biopsy sample and the remaining embryo defined as surplus embryo was also analysed to gain an accurate representation of the true heteroplasmy level. Four TE biopsies and their corresponding surplus embryos from family 20 were re-analysed using the PCR-based restriction fragment length polymorphism (PCR-RFLP) protocol that was also used to analyse the day-3 biopsy samples. The protocol was implemented as previously described by Sallevelt et al.39. Briefly, the biopsy material was subjected to cell lysis followed by two rounds of PCR. The first amplification PCR was carried out with unlabelled primers for the m.3243 A > G mutation, after which a fluorescently labelled primer was added for the second PCR round. The resulting product was enzymatically digested, purified, and analysed by capillary electrophoresis. The mutation load was determined by dividing the area of the mutation peak by the sum of both the wild type and mutation peak. The remaining four embryos from family 20 (TE biopsy and surplus embryo) were processed with the WGS-PGT protocol described above. These samples underwent deep sequencing with a target sequencing depth of 30–40X. The sequencing data were processed and subsampled as described above. The two embryos from family 21 (TE biopsy and surplus embryo) were also processed with WGS-PGT and sequenced to a depth of ~10X. Sequencing depth per position was determined using the “depth” function from SAMtools80. Known pathogenic variants in the mitochondrial genome were extracted from the MITOMAP’s confirmed pathogenic mutations database89. The “HaplotypeCaller” function form GATK was used to determine the number of reads supporting the reference and alternative alleles at the indicated position90, from which the heteroplasmy percentage was calculated.
Data Visualisation
Data were visualised using visualisation modules of siCHILD/haplarithmisis pipeline30,40, R packages ggplot91, circlize92, ggpubr93, and cowplot94.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The WGS and GBS data cannot be shared publicly to protect the privacy of the families that participated in the study. Specifically, the consent of the samples from which data was generated does not allow for broad sharing of the raw GBS and WGS data. We are not allowed to share the raw anonymized sequencing WGS and GBS data as patients with rare variants and/or pathogenic mutations may be traceable. The anonymized raw data may be requested through the corresponding author (masoud.zamaniesteki@mumc.nl) and via application to the data access committee of MUMC+ (helpdesk.ctcm@mumc.nl). The turn-around-time for processing initial applications is 2 weeks. After the final assessment by the data access committee of MUMC+ and following a signed data transfer agreement with the academic institution of the requester, the data can be shared. The data will be available in MUMC+ during the legal data retention timeframe (15 years) from the start of the study (2021). All other data supporting the findings described in this manuscript are available in the article and its Supplementary Information files. Source data are provided in this paper.
Code availability
The code of haplarithmisis and the scripts used in the analysis of this study are available via Github (https://github.com/CellularGenomicMedicine/WGSPGT) and are available as a Code Ocean capsule (https://doi.org/10.24433/CO.6619222.v1).
References
McKusick, V. A. Mendelian inheritance in man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604 (2007).
Satam, H. et al. Next-generation sequencing technology: current trends and advancements. Biology 12, 997 (2023).
Mastrorosa, F. K., Miller, D. E. & Eichler, E. E. Applications of long-read sequencing to Mendelian genetics. Genome Med 15, 42 (2023).
Sallevelt, S. et al. Diagnostic exome-based preconception carrier testing in consanguineous couples: results from the first 100 couples in clinical practice. Genet Med 23, 1125–1136 (2021).
Sallevelt, S. C. E. H. et al. A comprehensive strategy for exome-based preconception carrier screening. Genet Med 19, 583–592 (2017).
Van Steijvoort, E. et al. Interest in expanded carrier screening among individuals and couples in the general population: systematic review of the literature. Hum. Reprod. Update 26, 335–355 (2020).
Zhang, T. et al. Expanded preconception carrier screening in clinical practice: review of technology, guidelines, implementation challenges, and ethical quandaries. Clin. Obstet. Gynecol. 62, 217–227 (2019).
Eurostat. Women are having their first child at an older age. Eurostat (2020). Available at: https://ec.europa.eu/eurostat/web/products-eurostat-news/-/ddn-20200515-2.
Khandwala, Y. S., Zhang, C. A., Lu, Y. & Eisenberg, M. L. The age of fathers in the USA is rising: an analysis of 168 867 480 births from 1972–2015. Hum. Reprod. 32, 2110–2116 (2017).
Gruhn, J. R. et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science 365, 1466–1469 (2019).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet 48, 126–133 (2016).
Sloter, E. D. et al. Frequency of human sperm carrying structural aberrations of chromosome 1 increases with advancing age. Fertil. Steril. 87, 1077–1086 (2007).
Amberger, J. S., Bocchini, C. A., Scott, A. F. & Hamosh, A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 47, D1038–d1043 (2019).
Hughes, T. et al. A review on the motivations, decision-making factors, attitudes and experiences of couples using pre-implantation genetic testing for inherited conditions. Hum. Reprod. Update 27, 944–966 (2021).
Handyside, A. H. et al. Biopsy of human preimplantation embryos and sexing by DNA amplification. Lancet 1, 347–349 (1989).
Handyside, A. H., Kontogianni, E. H., Hardy, K. & Winston, R. M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 344, 768–770 (1990).
Roche, K., Racowsky, C. & Harper, J. Utilization of preimplantation genetic testing in the USA. J. Assist Reprod. Genet 38, 1045–1053 (2021).
Spinella, F. et al. ESHRE PGT consortium data collection XXI: PGT analyses in 2018. Hum. Reprod. Open 2023, hoad010 (2023).
van Montfoort, A. et al. ESHRE PGT consortium data collection XIX-XX: PGT analyses from 2016 to 2017. Hum. Reprod. Open 2021, hoab024 (2021).
Bai, F. et al. Assisted reproductive technology service availability, efficacy and safety in mainland China: 2016. Hum. Reprod. 35, 446–452 (2020).
Gleicher, N., Barad, D. H., Patrizio, P. & Orvieto, R. We have reached a dead end for preimplantation genetic testing for aneuploidy. Hum. Reprod. 37, 2730–2734 (2022).
Simopoulou, M. et al. PGT-A: who and when? a systematic review and network meta-analysis of RCTs. J. Assist Reprod. Genet 38, 1939–1957 (2021).
Munné, S. et al. Preimplantation genetic testing for aneuploidy versus morphology as selection criteria for single frozen-thawed embryo transfer in good-prognosis patients: a multicenter randomized clinical trial. Fertil. Steril. 112, 1071–1079.e1077 (2019).
Yan, J. et al. Live Birth with or without preimplantation genetic testing for aneuploidy. N. Engl. J. Med 385, 2047–2058 (2021).
Mastenbroek, S., de Wert, G. & Adashi, E. Y. The Imperative of responsible innovation in reproductive medicine. N. Engl. J. Med 385, 2096–2100 (2021).
Greco, E., Minasi, M. G. & Fiorentino, F. Healthy babies after intrauterine transfer of mosaic aneuploid blastocysts. N. Engl. J. Med 373, 2089–2090 (2015).
Viotti M. et al. Chromosomal, gestational, and neonatal outcomes of embryos classified as a mosaic by preimplantation genetic testing for aneuploidy. Fertil Steril, (2023).
Viotti, M. Preimplantation genetic testing for chromosomal abnormalities: aneuploidy, mosaicism, and structural rearrangements. Genes (Basel) 11, 602 (2020).
Handyside, A. H. et al. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med Genet 47, 651–658 (2010).
Zamani Esteki, M. et al. Concurrent whole-genome haplotyping and copy-number profiling of single cells. Am. J. Hum. Genet 96, 894–912 (2015).
Masset, H. et al. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum. Reprod. 34, 1608–1619 (2019).
Masset, H. et al. Single-cell genome-wide concurrent haplotyping and copy-number profiling through genotyping-by-sequencing. Nucleic Acids Res 50, e63 (2022).
Backenroth, D. et al. Haploseek: a 24-h all-in-one method for preimplantation genetic diagnosis (PGD) of monogenic disease and aneuploidy. Genet Med 21, 1390–1399 (2019).
Zeevi, D. A. et al. Expanded clinical validation of Haploseek for comprehensive preimplantation genetic testing. Genet Med 23, 1334–1340 (2021).
Backenroth D. et al. SHaploseek: a sequencing-only high-resolution implementation of comprehensive preimplantation genetic testing. Sci. Rep. 13, 18036 (2023).
De Witte, L. et al. GENType: all-in-one preimplantation genetic testing by pedigree haplotyping and copy number profiling suitable for third-party reproduction. Hum. Reprod. 37, 1678–1691 (2022).
Griffin, D. K. & Ogur, C. PGT-SR: a comprehensive overview and a requiem for the interchromosomal effect. DNA 3, 41–64 (2023).
Gui, B. et al. Chromosomal analysis of blastocysts from balanced chromosomal rearrangement carriers. Reproduction 151, 455–464 (2016).
Sallevelt, S. C. et al. Preimplantation genetic diagnosis in mitochondrial DNA disorders: challenge and success. J. Med Genet 50, 125–132 (2013).
Zamani Esteki, M. et al. In vitro fertilization does not increase the incidence of de novo copy number alterations in fetal and placental lineages. Nat. Med 25, 1699–1705 (2019).
Haenel, Q., Laurentino, T. G., Roesti, M. & Berner, D. Meta-analysis of chromosome-scale crossover rate variation in eukaryotes and its significance to evolutionary genomics. Mol. Ecol. 27, 2477–2497 (2018).
Gong T., Hayes V. M., Chan E. K. F. Detection of somatic structural variants from short-read next-generation sequencing data. Brief Bioinform. 22, bbaa056 (2021).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014).
Cameron, D. L. et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res 27, 2050–2060 (2017).
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009).
Kumar, A. et al. Whole-genome risk prediction of common diseases in human preimplantation embryos. Nat. Med 28, 513–516 (2022).
Xie, P. et al. A novel multifunctional haplotyping-based preimplantation genetic testing for different genetic conditions. Hum. Reprod. 37, 2546–2559 (2022).
Yuan, P. et al. A whole-genome sequencing-based novel preimplantation genetic testing method for de novo mutations combined with chromosomal balanced translocations. J. Assist Reprod. Genet 37, 2525–2533 (2020).
Murphy, N. M., Samarasekera, T. S., Macaskill, L., Mullen, J. & Rombauts, L. J. F. Genome sequencing of human in vitro fertilisation embryos for pathogenic variation screening. Sci. Rep. 10, 3795 (2020).
Chen, S. et al. Comprehensive preimplantation genetic testing by massively parallel sequencing. Hum. Reprod. 36, 236–247 (2021).
Xu, J. et al. Mapping allele with resolved carrier status of Robertsonian and reciprocal translocation in human preimplantation embryos. Proc. Natl Acad. Sci. USA 114, E8695–e8702 (2017).
Yan, L. et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc. Natl Acad. Sci. USA 112, 15964–15969 (2015).
Peters, B. A. et al. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res 25, 426–434 (2015).
Zhai, F. et al. Preimplantation genetic testing for structural rearrangement based on low-coverage next-generation sequencing accurately discriminates between normal and carrier embryos for patients with translocations. Reprod. Biomed. Online 45, 473–480 (2022).
Xia, Q. et al. Nanopore sequencing for detecting reciprocal translocation carrier status in preimplantation genetic testing. BMC Genomics 24, 1 (2023).
Xia, Y. et al. The first clinical validation of whole-genome screening on standard trophectoderm biopsies of preimplantation embryos. F&S Reports 5, 63–71 (2024).
Xia Y. et al. Genome-wide disease screening in early human embryos with primary template-directed amplification. Preprint at bioRxiv, 2021.2007.2006.451077 (2021).
van Prooyen Schuurman, L. et al. Clinical impact of additional findings detected by genome-wide non-invasive prenatal testing: follow-up results of the TRIDENT-2 study. Am. J. Hum. Genet 109, 1140–1152 (2022).
Sachdev, N. M., McCulloh, D. H., Kramer, Y., Keefe, D. & Grifo, J. A. The reproducibility of trophectoderm biopsies in euploid, aneuploid, and mosaic embryos using independently verified next-generation sequencing (NGS): a pilot study. J. Assist Reprod. Genet 37, 559–571 (2020).
Orvieto, R. The reproducibility of trophectoderm biopsies - the chaos behind preimplantation genetic testing for aneuploidy. Eur. J. Obstet. Gynecol. Reprod. Biol. 254, 57–58 (2020).
Victor, A. R. et al. Assessment of aneuploidy concordance between clinical trophectoderm biopsy and blastocyst. Hum. Reprod. 34, 181–192 (2019).
Gleicher, N., Patrizio, P., Mochizuki, L. & Barad, D. H. Previously reported and here added cases demonstrate euploid pregnancies followed by PGT-A as “mosaic” as well as “aneuploid” designated embryos. Reprod. Biol. Endocrinol. 21, 25 (2023).
Yang, M. et al. Depletion of aneuploid cells in human embryos and gastruloids. Nat. Cell Biol. 23, 314–321 (2021).
Rana, B. et al. Identifying parental and cell-division origins of aneuploidy in the human blastocyst. Am. J. Hum. Genet 110, 565–574 (2023).
van der Meij, K. R. M. et al. Experiences of pregnant women with genome-wide non-invasive prenatal testing in a national screening program. Eur. J. Hum. Genet 31, 555–561 (2023).
Essers, R. et al. Prevalence of chromosomal alterations in first-trimester spontaneous pregnancy loss. Nat. Med 29, 3233–3242 (2023).
de Wert G., & Dondorp, W. Human Reproductive Genetics in Medically Assisted Reproduction: Ethical Considerations. In: Textbook Human Reproductive Genetics. (eds) Viville S., Sermon, K. D. (Cambridge University Press, 2023).
Souche, E. et al. Recommendations for whole genome sequencing in diagnostics for rare diseases. Eur. J. Hum. Genet 30, 1017–1021 (2022).
Kothiyal, P., Wong, W. S. W., Bodian, D. L. & Niederhuber, J. E. Mendelian inconsistent signatures from 1314 ancestrally diverse family trios distinguish biological variation from sequencing error. J. Comput Biol. 26, 405–419 (2019).
Kong, A. et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012).
Shamonki, M. I., Jin, H., Haimowitz, Z. & Liu, L. Proof of concept: preimplantation genetic screening without embryo biopsy through analysis of cell-free DNA in spent embryo culture media. Fertil. Steril. 106, 1312–1318 (2016).
Schobers, G. et al. Liquid biopsy: state of reproductive medicine and beyond. Hum. Reprod. 36, 2824–2839 (2021).
Rubio, C. et al. Multicenter prospective study of concordance between embryonic cell-free DNA and trophectoderm biopsies from 1301 human blastocysts. Am. J. Obstet. Gynecol. 223, 751.e751–751.e713 (2020).
Chen Y. et al. DNA methylome reveals cellular origin of cell-free DNA in spent medium of human preimplantation embryos. J. Clin. Invest. 131, e146051 (2021).
van Dijk, W. et al. Embryo tracking system for high-throughput sequencing-based preimplantation genetic testing. Hum. Reprod. 37, 2700–2708 (2022).
Vasimuddin M., Misra S., Li H., Aluru S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 314–324 (2019).
Lawrence, M., Gentleman, R. & Carey, V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842 (2009).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Danecek P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Van der, Auwera, Geraldine. A., Brian, D. OC. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra 1st edn. (O’Reilly Media, 2020).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, https://www.R-project.org/ (Vienna, Austria, 2021).
Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Scheinin, I. et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res 24, 2022–2032 (2014).
Nilsen, G. et al. Copynumber: efficient algorithms for single- and multi-track copy number segmentation. BMC Genomics 13, 591 (2012).
Conlin, L. K. et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet 19, 1263–1275 (2010).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Chinnery, P. F., Howell, N., Lightowlers, R. N. & Turnbull, D. M. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain 120, 1713–1721 (1997).
Lott, M. T. et al. mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinforma. 44, 1.23.21–26 (2013).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Wickham H. ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag New York, 2016).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
A. K. ggpubr: ‘ggplot2’ Based Publication Ready Plots (2023).
Wilke C. O. cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’ (2020).
Acknowledgements
We are grateful to all families that participated in this study. This study was funded by The EVA (Erfelijkheid Voortplanting & Aanleg) specialty programme (grant no. KP111513) of Maastricht University Medical Centre (MUMC+), the Horizon Europe (NESTOR, grant no. 101120075) and the Horizon 2020 innovation (ERIN, grant no. EU952516) grants of the European Commission. We would like to specially thank F.J.M. Snepvangers, L.E.C. Meers, W. Loneus, K. Daenen, S. Spierts, B.M. Reuters and M. Kurvers-Alofs for their support with all clinical wet-lab related procedures. Also, we would like to thank C. Gilissen and R.C. Derks from the Radboudumc Genome Technology Center for WGS expertise in computational and laboratory analyses. We thank L. Brandts for statistical consultations. We would like to thank D. Christopikou and F. Spinella from the PGT steering committee of the European Society of Reproduction and Embryology (ESHRE) for the critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
A.J., A.P., and M.Z.E. conceived and designed the study. M.D., J.D., D.H., S.S., S.M.S., J.L., L.H., L.V., A.W., A.P., and M.Z.E. devised and implemented the wet-lab experiments. A.J., R.K., R.E., P.C., J.M., B.Y., M.V., B.K., and M.Z.E. contributed to data processing and bioinformatics. M.H., S.M., R.G., and C.D. consulted and recruited the patients. A.W., A.P., W.D., and M.Z.E. contributed to WGS-PGT project management. W.D., M.D., J.M., B.Y., M.V., B.K., D.H., S.S., S.M.S., J.A., L.H., C.U., J.D., E.C., H.B., A.W., A.P. and M.Z.E. contributed to clinical implementation. M.N., G.W. devised ethical and psychological implications. M.Z.E. oversaw and supervised the study. A.J. wrote the initial draft of the manuscript. A.J., R.K., R.E., S.S., J.D., H.B., A.P., and M.Z.E. contributed to data interpretation and manuscript writing. All the authors read and approved the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
M.Z.E. is co-inventor on patent applications: ZL910050-PCT/EP2011/ 060211-WO/2011/157846 ‘Methods for haplotyping single cells’ and ZL913096-PCT/EP2014/068315-WO/2015/028576 ‘Haplotyping and copy-number typing using polymorphic variant allelic frequencies’. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daniel Ariad who co-reviewed with Ajinkya Kokate; and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Janssen, A.E.J., Koeck, R.M., Essers, R. et al. Clinical-grade whole genome sequencing-based haplarithmisis enables all forms of preimplantation genetic testing. Nat Commun 15, 7164 (2024). https://doi.org/10.1038/s41467-024-51508-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51508-1
- Springer Nature Limited