

Abstract
DNA methylation (DNAm) has been implicated in acute coronary syndrome (ACS), but the causality remains unclear in cross-sectional studies. Here, we conduct a prospective epigenome-wide association study of incident ACS in two Chinese cohorts (discovery: 751 nested case-control pairs; replication: 476 nested case-control pairs). We identified and validated 26 differentially methylated positions (DMPs, false discovery rate [FDR] <0.05), including three mapped to known cardiovascular disease genes (PRKCZ, PRDM16, EHBP1L1) and four with causal evidence from Mendelian randomization (PRKCZ, TRIM27, EMC2, EHBP1L1). Two hypomethylated DMPs were negatively correlated with the expression in blood of their mapped genes (PIGG and EHBP1L1), which were further found to overexpress in leukocytes and/or atheroma plaques. Finally, our DMPs could substantially improve the prediction of ACS over traditional risk factors and polygenic scores. These findings demonstrate the importance of DNAm in the pathogenesis of ACS and highlight DNAm as potential predictive biomarkers and treatment targets.
Similar content being viewed by others


Introduction
Acute coronary syndrome (ACS) is one of the most severe subtype of coronary heart disease (CHD), including a group of conditions characterized by a sudden reduction or blockage of blood flow to the heart muscle, such as acute myocardial infarction (AMI) and unstable angina pectoris (UAP)1. Despite substantial progress in the prevention, diagnosis, and treatment of ACS, it remains a major cause of morbidity and mortality in China and worldwide2. While genome-wide association studies (GWAS) have identified over 150 genetic variants associated with CHD3, few studies specifically focus on ACS subtype and the biological mechanisms of the GWAS loci remain largely unknown4. The DNA methylation (DNAm), which integrates environmental factors and genetic susceptibility to regulate gene expression without altering the DNA sequence, has gained increasing research interests5. In addition to providing mechanistic insights into the development of ACS, DNAm alterations have the potential to serve as preventive and therapeutic targets because the epigenome can be modified by environmental factors6,7.
A growing body of evidence, including our previous work8, supports an important role of DNAm in the pathogenesis of ACS9. However, most previous studies could not distinguish the direction of causality between changes in DNAm and ACS due to their cross-sectional design. In contrast, while prospective cohort design is more reliable to establish causality due to the time-ordering of the associations, there were five epigenome-wide association studies (EWAS) of incident CHD or its subtypes based on prospective cohorts10,11,12,13,14. These studies, however, showed little overlap in their reported differentially methylated positions (DMPs), possibly due to heterogeneity in population, CHD subtypes, and the adjusted covariates. In particular, the lack of validation in independent populations and different mechanisms underlying subtypes of CHD might limit the robustness of these findings. Targeting homogeneous subtypes of CHD, such as ACS, in a large EWAS is likely to generate robust associations and to elucidate the regulatory role of DNAm in disease pathogenesis.
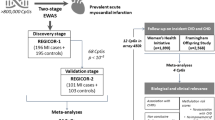
In this study, we conduct a two-stage EWAS of incident ACS based on two Chinese cohorts. We first perform genome-wide DNA methylation analysis of incident ACS at 777,307 cytosine-phosphate-guanine (CpG) positions in the Dongfeng-Tongji (DFTJ) cohort15, then externally validate significant DMPs in the China Kadoorie Biobank (CKB)16. We further explore the clinical applications of these DMPs as prevention and treatment targets and risk prediction biomarkers for ACS.
Results
Characteristics of the study populations
After quality control (QC), the discovery dataset (DFTJ) consisted of 751 incident ACS cases (376 men) with a mean age of 64.93 years and 751 matched controls (376 men) with a mean age of 64.84 years; the replication dataset (CKB) included 476 incident ACS cases (267 men) with a mean age of 50.97 years and 476 matched controls (267 men) with a mean age of 49.79 years. In both datasets, ACS cases had higher body mass index (BMI) and higher prevalence of hypertension, dyslipidemia, and diabetes (P < 0.05) (Table 1).
The Shiyan multi-omics dataset consisted of 156 healthy participants (78 men) with a mean age of 41.93 years (Supplementary Table 1). The ACS case-control expression dataset consisted of 12 ACS patients and 12 matched controls, who had a mean age of 53.75 and 53.82 years, respectively (Supplementary Table 2). The overall study design flowchart is presented in Supplementary Fig. 1.
Differentially methylated CpGs associated with incident ACS
In the discovery stage, after adjusting for major risk factors of ACS and the proportion of six leukocytes, we identified 72 DMPs (false discovery rate [FDR] <0.05) and 27 DMPs (Bonferroni-corrected P value < 0.05) that were associated with incident ACS (Fig. 1 and Supplementary Data 1). Ten out of these 72 DMPs were annotated to previously reported cardiovascular disease (CVD) susceptibility genes (Supplementary Data 2). The most significant DMP was cg00937402 mapped to TUSC3, which had a P value of 2.08 × 10−24. This DMP, however, together with four other DMPs, failed to pass QC in the replication dataset. Replication was conducted on the remaining 67 significant DMPs, among which 26 showed directionally consistent associations at FDR < 0.05 (all Pmeta < 2.1 × 10−7, Table 2). The top hit was cg04869583 mapped to DNM1L (odds ratio [OR] = 0.61, 95% confidence interval [CI]: 0.55–0.68, Pmeta = 1.42 × 10−18). Three of the 26 DMPs were mapped to known CVD susceptibility genes, including cg00660626 to PRKCZ, cg24395386 to PRDM16, and cg16749093 to EHBP1L1 (Supplementary Data 2). In addition, 20 of the 26 DMPs were mapped to genes relevant to CVD related traits, such as obesity, blood lipid levels, and inflammatory factors (Supplementary Data 3). Sensitivity analysis stratified by ACS subtypes showed no evidence of heterogeneity between AMI and UAP (P for heterogeneity >0.2), except for cg01550915 (P for heterogeneity = 0.04, Supplementary Fig. 2). Further adjustment of physical activity and dietary intakes had little impact on the EWAS results (Supplementary Fig. 3), and all of the 26 validated DMPs remained significant (FDR < 0.05) in both discovery and replication cohorts (Supplementary Data 4).

a Manhattan plot. The 26 validated CpGs associated with incident ACS are shown in red dots. We labelled genes mapped to the 26 validated CpGs, with 3 mapped to known cardiovascular disease genes labelled in red text. b QQ plot. The QQ plot illustrates the distribution of observed P values compared to expected values under the null hypothesis of no association. λ is the genomic inflation factor. c Volcano plot. The x-axis shows the effect size of each CpG (raw M value) associated with incident ACS, whereas the y-axis indicates –log10 (P) of the associations. The 7 hypermethylated CpGs with FDR < 0.05 are shown in red dots, and the 65 hypomethylated CpGs with FDR < 0.05 are shown in blue dots. For Manhattan plot and volcano plot, the horizontal solid line corresponds to the genome-wide significance threshold after multiple testing correction (FDR < 0.05), and the horizontal dashed line corresponds to the significance threshold of Bonferroni-corrected P < 0.05. The P value and FDR are calculated using two-sided tests. ACS acute coronary syndrome, CpG cytosine-phosphate-guanine, FDR false discovery rate.
DMPs identified in the discovery stage showed significant enrichment in eight Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (P < 0.05), with the top being Hedgehog signaling pathway, inositol phosphate metabolism, circadian rhythm, and phosphatidylinositol signaling system (Supplementary Data 5). Furthermore, many of the top enriched Gene Ontology (GO) pathways were related to phosphoinositol signaling pathways (Supplementary Fig. 4).
For each validated DMP, we selected the most significant cis-methylation-quantitative trait locus (cis-meQTL) as the instrumental variable (IV) for Mendelian randomization (MR) analysis (Supplementary Data 6). Causal evidences were reinforced between four CpGs and CHD subtypes, including cg00660626 mapped to PRKCZ (UAP, β ± SE = −0.463 ± 0.234, P = 0.048), cg27100266 to TRIM27 (UAP, −1.071 ± 0.412, P = 0.009), cg22293416 to EMC2 (UAP, −0.234 ± 0.116, P = 0.044), and cg16749093 to EHBP1L1 (MI, −0.385 ± 0.175, P = 0.028) (Supplementary Data 7). In addition to UAP, cg27100266 mapped to TRIM27 was also significantly associated with the overall CHD risk in the MR analysis (β ± SE = −0.560 ± 0.208, P = 0.007, Supplementary Data 8).
Expression of genes nearby 26 validated DMPs in ACS-related tissues
We found three significant negative associations between validated DMPs with the expression of their mapped genes in leukocytes, including cg03609847 and PIGG, cg12853539 and HDDC2, and cg16749093 and EHBP1L1 (P < 0.05) (Supplementary Data 9). In addition, we observed that the methylation levels at ten DMPs were significantly correlated with the expression levels of at least one proximal gene (P < 0.05) (Supplementary Data 10). The low expression of several genes, such as PRDM16 and NRXN3, in the leukocytes might limit the statistical power to detect their correlation with DNAm (Supplementary Fig. 5).
Combining evidence from different datasets (Supplementary Figs. 6–8), we found significant three-way associations at cg03609847 to PIGG (Fig. 2) and cg16749093 to EHBP1L1 (Fig. 3). Consistent with hypomethylation in incident ACS cases, the expression levels of both PIGG and EHBP1L1 were elevated in the leukocytes of ACS patients. Furthermore, overexpression of PIGG and EHBP1L1 in atherosclerotic plaques suggested that they may increase the risk of ACS by promoting atherosclerosis.

a The schematic diagram depicting association directions between DNAm, mRNA expression, and ACS risk. b The correlation between DNAm of cg03609847 and PIGG gene expression levels in leukocytes of 156 healthy participants was examined by Pearson correlation test. c The comparison of PIGG gene expression levels in leukocytes between 12 pairs of ACS cases and matched controls was analyzed using the “limma” package. d The comparison of PIGG gene expression levels in 32 pairs of normal carotid tissue and carotid atheroma plaques was performed by GEO2R online tool. e The comparison of PIGG gene expression in 13 early atherosclerotic carotid artery segments and 16 advanced atherosclerotic carotid artery segments was performed by GEO2R online tool. All P values were two-sided. All box plots show median value, IQR, up to 1.5 IQR (whiskers). RNA expression was quantified by TPM across all genes, followed by log2(TPM + 1) transformation for subsequent analysis. ACS acute coronary syndrome, DNAm DNA methylation, IQR interquartile range, TPM transcripts per million.

a The schematic diagram depicting association directions between DNAm, mRNA expression and ACS risk. b The correlation between DNAm of cg16749093 and EHBP1L1 gene expression levels in leukocytes of 156 healthy participants was examined by Pearson correlation test. c The comparison of EHBP1L1 gene expression levels in leukocytes between 12 pairs of ACS cases and matched controls was analyzed using the “limma” package. d The comparison of EHBP1L1 gene expression levels in 32 pairs of normal carotid tissue and carotid atheroma plaques was performed by GEO2R online tool. e The comparison of EHBP1L1 gene expression in 13 early atherosclerotic carotid artery segments and 16 advanced atherosclerotic carotid artery segments was performed by GEO2R online tool. All P values were two-sided. All box plots show median value, IQR, up to 1.5 IQR (whiskers). RNA expression was quantified by TPM across all genes, followed by log2(TPM + 1) transformation for subsequent analysis. ACS acute coronary syndrome, DNAm DNA methylation, IQR interquartile range, TPM transcripts per million.
Identification of transcription factors and their potential regulatory roles
Based on transcription factor binding site prediction, the top 30 transcription factors that may bind to cg03609847 and cg16749093 were listed in Supplementary Data 11. In particular, ChIP-seq experiments provided strong evidence that POLR2A, the largest subunit of RNA polymerase II, could bind to promoter regions of both PIGG and EHBP1L1 (Supplementary Data 12). Furthermore, hTFtarget database suggested that POLR2A could regulate the expression of both PIGG and EHBP1L1. In our Shiyan multi-omics dataset, the mRNA expression level of POLR2A had moderate negative correlations with the methylation level of cg03609847 (R = −0.115; P = 0.152) and cg16749093 (R = −0.193; P = 0.016), and strong positive correlations with the mRNA expression levels of PIGG (R = 0.876; P < 0.001) and EHBP1L1 (R = 0.816; P < 0.001) (Supplementary Fig. 9).
Prediction of ACS risk by MRS and PRS
Based on 67 DMPs, we constructed a methylation risk score (MRS) and evaluated its predictive performance for ACS in both DFTJ and CKB cohorts. As shown in Fig. 4 and Table 3, by adding MRS to the reference model of traditional risk factors, the area under the receiver operating characteristic curves (AUC) increased significantly from 0.628 to 0.686 (P < 0.001, Delong’s test) in the DFTJ cohort, and from 0.625 to 0.729 (P < 0.001) in the CKB cohort. In addition, MRS significantly improved risk discrimination and reclassification of ACS in both cohorts (Table 3).

a Evaluation in the DFTJ cohort. b Evaluation in the CKB cohort. Traditional risk factors include age, sex, BMI, smoking status, drinking status, hypertension, dyslipidemia, and diabetes. The MRS was calculated by adding the M-values of 67 DMPs identified in the discovery cohort, weighted by their β coefficients from the discovery cohort. ACS acute coronary syndrome, AUC area under ROC curve, DMP differentially methylated position, MRS methylation risk score, PRS polygenic risk score, PRSBBJ published PRS based on the BioBank Japan cohort, PRSCHN published PRS based on Chinese populations, ROC receiver operating characteristic curve.
For comparison, we evaluated the predictive value of two polygenic risk scores (PRSs) of CHD and found that there was little improvement in the prediction of ACS by adding PRSs to the reference model (Fig. 4 and Table 3). Although PRS significantly improved risk discrimination and reclassification of ACS in CKB cohort, the improvement was much lower than those of MRS (Table 3).
Discussion
We have conducted a two-stage EWAS of incident ACS based on two large prospective Chinese cohorts and the Illumina EPIC array covering more than 850,000 CpGs. We identified 26 reproducible DMPs associated with incident ACS, three of which were mapped to known CVD susceptibility genes (PRKCZ, PRDM16, and EHBP1L1). Further comparison of gene expression data from leukocytes, atherosclerotic plaques, and adjacent normal tissues pinpointed cg03609847 to PIGG and cg16749093 to EHBP1L1 with potential regulatory roles in the pathogenesis of ACS. Finally, we constructed an MRS, leading to substantial improvement in the risk prediction of ACS over traditional risk factors and PRS. These findings provide insights into the pathogenesis of ACS and have important implications for the prevention and treatment of ACS.
To our knowledge, there were five prospective EWASs on CHD10,11,12,13,14, of which none focused on ACS and only two had performed independent validation10,12. However, no reproducible DMPs have been reported. Several reasons might contribute to the low replication rate. First, DNAm is sensitive to environmental exposures, which can result in substantial heterogeneity in the DNAm patterns in different populations12. Second, CHD has many subtypes with distinct pathogenesis and clinical presentations. Third, unreliable results were common in studies with small sample sizes or no replication data. We overcome these limitations by conducting a large-scale two-stage EWAS of ACS in the Chinese population, followed by in-depth investigations of the potential gene regulatory role and the disease risk prediction value of the significant DMPs.
We observed significant three-way associations among DNA hypomethylation, up-regulated gene expression, and increased ACS risk at cg03609847 (PIGG) and cg16749093 (EHBP1L1). Increased expression levels of their mapped genes were observed not only in the leukocytes of ACS patients but also in atherosclerotic plaque tissues, supporting their potential role in the pathogenesis of ACS. PIGG is a key gene involved in the biosynthesis and transport of glycosylphosphatidylinositol-anchored proteins (GPI-Aps)17, which are involved in heart development18. In addition, overexpression of PIGG may affect the phosphoinositide signaling pathway, which has been implicated in cardiovascular disease19. EHBP1L1 is involved in actin cytoskeleton organization and has been identified to associate with CHD20 and AMI21. Our findings further support the involvement of EHBP1L1 in the pathogenesis of atherosclerosis and ACS. Moreover, we showed that DNAm at these two CpGs might regulate the expression of PIGG and EHBP1L1 by affecting the binding affinity of POLR2A, the largest subunit of RNA polymerase II22.
Among the 26 validated loci, EHBP1L1 (cg16749093), PRKCZ (cg00660626), and PRDM16 (cg24395386) have been reported to associate with cardiovascular disease by previous GWAS20,21,23,24. Our MR analysis further supported causal associations between hypomethylation of these DMPs and increased risk of incident ACS. But we did not observe significant association between methylation and gene expression levels at PRKCZ and PRDM16, possibly due to their low expression levels in leukocytes and the limited sample size of the Shiyan multi-omics dataset. PRKCZ has been implicated in cardiomyopathy25. Functionally, PRKCZ is involved in the proliferation, differentiation, and secretion of cardiac myocytes, as well as the phosphoinositide 3-kinase (PI3K), inflammatory response, and chemokine signaling pathways26,27. In addition, Infante et al.28 have identified PRKCZ methylation as potential epigenetic-sensitive target for ACS via network analysis, while Steenaard et al.29 have found that smoking-related demethylation of cg05603985 might be involved in development of CHD by regulating PRKCZ expression in whole blood. The other gene, PRDM16, plays a crucial role in adipocyte transformation and thermogenesis30, and has been reported to associate with obesity31, lipid levels32, insulin resistance33, blood pressure34, and CHD20.
Our findings provide additional insights into the mechanisms underlying ACS pathogenesis. For example, PLCD3 encodes a member of the phospholipase C family that catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to the diacylglycerol and inositol 1,4,5-triphosphate (IP3), both of which mediate diverse cellular responses to extracellular stimuli by inducing protein kinase C and increasing cytosolic Ca2+ concentrations35. The IP3/Ca2+ signaling pathway could generate Ca2+ signals to control diverse biological processes and has been implicated in atrial arrhythmias and congestive heart failure36. Our finding of ACS-related DMP at PLCD3 suggests that the InsP3/Ca2+ signaling pathway may be involved in the pathogenesis of ACS, which needs to be confirmed by future studies.
AMI and UAP are two subtypes of ACS. Our subgroup analysis found no heterogeneity in the effect sizes for 25 out of the 26 validated DMPs between AMI and UAP, except for cg01550915 mapped to ABCA3 (P for heterogeneity = 0.04). Thus, AMI and UAP likely share most of their pathophysiological basis. For cg01550915, we observed a much stronger effect size in UAP (β ± SE = −0.41 ± 0.07) than in AMI (−0.09 ± 0.14). ABCA3 plays a crucial role in maintaining the normal function of pulmonary surfactant, which has potential implications on cardiac function37. Our results suggested that the potential effect of ABCA3 in UAP might be more pronounced than in AMI. However, the underlying mechanism remains unclear and needs to be investigated in the future.
We proposed and validated an MRS as a weighted sum of the ACS-related DMPs, in a way like the PRS based on GWAS variants38,39,40,41,42,43. In contrast to PRS, which often showed statistically significant but small improvement in the risk prediction of incident CHD over traditional risk factors, our MRS substantially improved the prediction accuracy of ACS risk in both DFTJ and CKB cohorts, demonstrating the potential clinical applications of MRS. Because DNAm is responsive to environmental stimuli, an MRS may capture information of one’s health conditions over time, and is therefore more predictive of future disease risk than a PRS. Consistently, Zheng et al.44 showed that an MRS of cardiovascular health from young adulthood to midlife was predictive of an individual’s future risk of subclinical and clinical CVD independent of traditional risk factors. Several previous studies also demonstrated the potential clinical value of MRS for CVD risk stratification in European populations10,45,46. Our study strengthens and extends these previous findings by demonstrating the predictive value of MRS for ACS in the Chinese populations.
Several strengths of this study, including the large sample size, independent replication, and targeting the most severe subtype of CHD, have substantially enhanced the robustness of our findings. Our prospective study design together with MR analysis provides strong evidence to support causality of several DMPs in the development of ACS. By combining information from multiple gene expression datasets, we highlight the gene regulatory function of several DMPs and their potential role in atherosclerosis and ACS. With these merits, our study represents a remarkable progress towards uncovering the important role of DNAm in the pathogenesis of ACS. Furthermore, our proposed MRS may serve as a powerful predictive biomarker to facilitate early prevention and intervention of ACS.
We also acknowledge several limitations in the present study. First, compared to the DFTJ and CKB cohorts, the participants in the Shiyan multi-omics dataset are younger and have excluded various diseases. The potential differences in health status and age distribution may affect the generalizability of our findings. Additionally, the relatively small sample size and cross-sectional study design of the Shiyan dataset limit our ability to infer causality and detect moderate effect associations. In addition, we used the most significant cis-meQTL as the IV in MR analysis to minimize potential horizontal pleiotropy and LD between cis-meQTLs, however, the small contribution of a single SNP to DNAm levels may limit the statistical power of MR. Furthermore, we compared our proposed MRS with the PRS for CHD, under the assumption that ACS, as a subtype of CHD, would share similar genetic basis with CHD. Ideally, a direct comparison with the PRS for ACS should be conducted when large-scale GWAS targeting ACS become available in the future. Finally, our association analyses have suggested potential gene regulatory functions of the validated DMPs in the pathogenesis of ACS, but the underlying mechanisms should be further explored by experimental studies.
We have identified significant and reproducible associations between incident ACS and blood DNAm at 26 CpGs. Particularly, evidence from multi-omics data of different sources suggested cg03609847 at PIGG and cg16749093 at EHBP1L1 were likely involved in the pathogenesis of ACS by transcriptional regulation and thus could serve as promising prevention and treatment targets for ACS. Moreover, we have demonstrated remarkable predictive value of DNAm markers for incident ACS. Further validation in other populations and animal models are warranted to ensure the generalizability of our findings and uncover the in-depth biological mechanism.
Methods
Study population
Our study adopted a two-stage 1:1 matched nested case-control study design. The discovery stage was based on a nested case-control study of incident ACS within the DFTJ cohort15. Details of the DFTJ cohort have been described in the Supplementary methods. Briefly, the DFTJ cohort is an ongoing prospective cohort of retired employees of the Dongfeng Motor Corporation in Shiyan, Hubei, China. The DFTJ cohort initially enrolled 27,009 subjects during 2008–2010 and was expanded to enroll a total of 38,295 subjects during April to October 2013. The present study was based on subjects enrolled in 2013, of whom the baseline information was collected by questionnaire and physical examination. Information on disease incidence and mortality until December 31, 2018 was collected through electronic medical records of the health insurance network of Dongfeng Motor Company. ACS was defined as AMI coded as ICD-10 I21 and UAP coded as ICD-10 I20.0. Participants were diagnosed by an expert panel of physicians based on symptoms, clinical examinations according to the ACCF/AHA guidelines47. Incident cases were defined as those with first ACS diagnosis between enrollment and December 31, 2018.
In the present study, we started with 38,295 participants enrolled in 2013 and excluded those with prevalent CHD (n = 6457), stroke (n = 2406), cancer (n = 2686), and severely abnormal electrocardiogram (n = 838), as well as those with unavailable blood samples (n = 3626), leaving 24,415 participants. DNA methylation was measured from whole blood samples at baseline for 785 incident ACS. For each incident ACS case, we randomly selected a control who was free of CVD and cancer at the time of the case event. Each pair was matched on age (within one year), sex, and blood draw time (within 6 months). Thirty-four case-control pairs were excluded during the QC of DNAm data. Finally, 751 pairs of incident ACS case-control samples were included in the discovery stage.
Replication was based on a nested case-control study of incident CHD within the CKB cohort16. Details of the study design, inclusion and exclusion criteria, and disease diagnosis were available in Si et al.13. Briefly, baseline DNAm was measured for 494 incident CHD case-control pairs, of which CHD was diagnosed during the follow-up period until December 31, 2015. Incident CHD cases were defined as nonfatal acute myocardial infarction coded as I21 and fatal ischemic heart disease (IHD) coded as ICD-10 I20-I25. Each case was matched to one control free of CHD, cancer, or cerebrovascular diseases throughout follow-up. Cases and controls were one-to-one matched by birth year (within 3 years), age at baseline (within 3 years), sex, study area, and fasting time prior to blood draw (0–6, 6–8, 8–10, and ≥10 h). To be consistent with our discovery cohort, we excluded 10 non-ACS cases and their matched controls. In addition, eight case-control pairs were excluded by QC of DNAm data. Thus, we retained 476 pairs of incident ACS case-control samples for the replication analysis.
We evaluated correlation between DNAm and RNA expression of the annotated genes in an independent set of 156 healthy participants (Shiyan multi-omics dataset) recruited from the same city as the DFTJ cohort between June and July 202148. We performed gene differential expression analysis of leukocytes between 12 ACS cases and 12 age- (±5 years) and sex-matched healthy controls collected in Wuhan, Hubei, China (ACS case-control expression dataset)8. A detailed description of the study populations and covariates was provided in the Supplementary Methods.
The study protocols have been approved by the Ethics Committee of School of Public Health, Tongji Medical College (Wuhan, China), the Ethics Review Committee of the Chinese Center for Disease Control and Prevention (Beijing, China), the Oxford Tropical Research Ethics Committee, University of Oxford (UK), and Peking University Institutional Review Board (Beijing, China). All participants provided written informed consent.
Laboratory methods
DNAm measurement and QC
DNAm in leukocytes was profiled by the Infinium Methylation EPIC BeadChip (Illumina, USA) for samples from the DFTJ cohort, the CKB cohort, and the Shiyan multi-omics dataset. To minimize batch effects, blood samples were randomized prior to testing. Cohort-specific details of laboratory procedures, data processing, QC, and normalization are provided in the Supplementary Methods.
RNA sequencing data
For the Shiyan multi-omics dataset, RNA samples extracted from leukocytes were sequenced using NovaSeq6000 (Illumina, USA). Details of RNA sequencing are provided in the supplementary Methods. RNA expression was quantified by transcripts per million (TPM) across all genes, followed by log2(TPM + 1) transformation for subsequent analysis.
Gene expression microarray data
Gene expression profiles of 12 ACS case-control pairs were assayed by HumanHT-12 v4 BeadChip (Illumina, USA). Detailed laboratory procedures, data processing, QC, and normalization can be found in the Supplementary Methods.
Statistical analyses
Genome-wide methylation analysis of incident ACS
In the discovery stage (DFTJ cohort), we tested association of DNA methylation (M-value) at each CpG site with incident ACS using conditional logistic regression models, adjusted for BMI, smoking, drinking, hypertension, dyslipidemia, diabetes, and six leukocyte proportions (CD4+ T cell, CD8+ T cell, natural killer cell, monocyte, neutrophil, and B cell). Leukocyte proportions were estimated with the FlowSorted.Blood.EPIC function in the “bigmelon” R package. The association between each CpG and incident ACS in the replication stage was tested using conditional logistic regression with adjustment of the same covariates as in the discovery stage as well as age (because the 3-year matching range might introduce residual confounding) and region (10 regional centers). Statistical significance was defined as FDR < 0.05 based on the tested loci at each stage separately11,49. Association results were combined using the inverse variance weighted fixed-effect meta-analysis by METAL software. For each CpG, we reported the OR per standard deviation (SD) increase in the M-value, and the corresponding 95% CI. Sensitivity analysis was performed for ACS subtypes (AMI and UAP). In the sensitivity analyses, we further adjusted for physical activity, defined as total energy expenditure measured in metabolic equivalent (MET)-hours/week for the DFTJ cohort and MET-hours/day for the CKB cohort, and major dietary intake of meats, fish, vegetables, and fruits. We queried the public database to check if DMPs (EWAS catalog and EWAS Atlas) and their annotated genes (GWAS catalog and PhenoScanner) had been reported to associate with CVD or cardiovascular traits.
Gene set enrichment analysis
Gene set enrichment analysis was performed for genes mapped to ACS-related DMPs identified in the discovery stage (FDR < 0.05), in comparison to the background set of genes mapped by CpGs on the Illumina EPIC array. We used the gometh function in the “missMethyl” package50 to identify enriched KEGG and GO pathways.
Cis-meQTL and MR analyses
Two-sample MR analysis was applied to estimate the causal effects of DMPs on incident ACS risk (see details in the Supplementary Methods). Firstly, we performed cis-meQTL analysis for SNPs within 1 Mb of each CpG based on 4411 unrelated samples that have both genotyping data and DNAm data from the DFTJ cohort (see details in the Supplementary Methods). Briefly, the methylation M-value of each CpG was modeled as a linear function of the SNP genotype, with adjustment of age, sex, smoking status, six leukocyte proportions (CD4+ T cell, CD8+ T cell, natural killer cell, monocyte, neutrophil, and B cell), the top four methylation principal components, and the top four genotype principal components. The most significant SNP in the cis-meQTL analysis was selected as the IV for the MR analysis of the corresponding CpG. GWAS summary statistics were obtained from the BioBank Japan (BBJ) cohort for MI (14,992 cases and 146,214 controls)23, UAP (5891 cases and 146,214 controls)23, and CHD (25,892 cases and 142,336 controls)38. We used the Wald ratio method implemented in the “TwoSampleMR” package to estimate the causal effect of each DMP on MI, UAP, and CHD.
Correlations between DNA methylation and gene expression
Expression quantitative trait methylation (eQTM) analysis was performed to identify which DMPs were associated with expression levels of targeted or nearby genes. For the validated DMPs, we extracted mRNA expression levels of their annotated genes as well as genes within 1 Mb in leukocyte samples of 156 participants in the Shiyan multi-omics dataset. We examined the correlation between DNAm and mRNA levels using the Pearson correlation test. Because LCE5A and IGFN1 were expressed in less than 30% of the samples, they were replaced by the second nearest genes of the CpGs (FLG2 and TMEM9, respectively). A two-sided P < 0.05 was considered statistically significant. We used the GENE2FUNC function in the FUMA GWAS tool to examine the expression of genes annotated to DMPs in 54 tissues51,52.
Differential gene expression analysis
For the validated DMPs, we performed differential expression analyses of their annotated genes in leukocytes and atherosclerotic tissues. First, we compared gene expression in leukocytes in 12 pairs of ACS cases and matched-healthy controls using linear regression models implemented in the “limma” package, with no adjustment of covariates8. Next, we analyzed gene expression data in relevant tissues, downloaded from the Gene Expression Omnibus (GEO) database under the accession numbers GSE43292 and GSE28829. The GSE43292 dataset included gene expression profiles of atherosclerotic plaques and the adjacent normal tissues from 32 hypertensive patients, assayed by the Affymetrix Human Gene 1.0 ST Array (Illumina, USA)53. The GSE28829 dataset included gene expression profiles of 13 early atherosclerotic carotid artery segments and 16 advanced atherosclerotic carotid artery segments, assayed by the and the Affymetrix Human Genome U133 Plus 2.0 Array (Illumina, USA)54. The analyses of GSE43292 and GSE28829 datasets were performed using the GEO2R online tool with default settings (https://www.ncbi.nlm.nih.gov/geo/geo2r/).
Evaluation of the transcriptional regulatory role of DNAm
We explored whether the association between DNAm and gene expression was related to transcriptional regulation. We first predicted the transcription factor binding site within 50 bp upstream and downstream of the CpG sites through the AnimalTFDB 3.0 database55. We further selected transcription factors that were likely to bind to the target genes by searching the ENCODE database, which displays a list of transcription factors supported by evidence from ChIP-seq experiments56. Then we checked whether the selected transcription factors have potential regulatory effects on the target genes by searching the hTFtarget database57. Finally, we examined Pearson’s correlations between mRNA expression levels of candidate transcription factors, methylation levels of CpGs, and mRNA expression levels of target genes in Shiyan multi-omics dataset.
Construction and evaluation of a methylation risk score for incident ACS
Based on the EWAS results of discovery stage, we constructed a MRS for incident ACS in a way similar to the PRS. The MRS was calculated as a weighted sum of M-values of the 67 significant CpGs identified in the discovery stage (FDR < 0.05): MRS = β1M1 + β2M2 +… + β67M67, where βi is the effect size of the ith CpG on incident ACS obtained from the discovery EWAS and Mi is the M-value of the ith CpG. This MRS construction method was applied to both DFTJ and CKB cohorts.
For comparison, we also computed PRS based on imputed genome-wide array genotyping data for participants from both the DFTJ and the CKB cohorts. Since there are limited large-scale GWAS studies specifically targeting ACS in Asian populations or other populations, we chose to construct the PRS for CHD instead. We used two published PRS models calibrated for East Asians: PRSBBJ reported by Koyama et al.38 and PRSCHN reported by Lu et al.39 PRSBBJ was constructed from 75,028 genetic variants based on meta-analysis of three GWAS of CHD (BBJ, CARDIoGRAMplusC4D, and the UK Biobank). PRSCHN consisted of 540 genetic variants and was calibrated for the Chinese population.
We evaluated the performance MRS and PRS in predicting the risk of incident ACS based on logistic regression models. Evaluation was based on participants with both DNA methylation and genotype data (n = 1886 from DFTJ and n = 947 from CKB). The reference model consisted of traditional risk factors, including age, sex, BMI, smoking status, drinking status, hypertension, dyslipidemia, and diabetes. We evaluated the improvement of performance when adding MRS or PRS to the reference model. We trained the models using data from the DFTJ cohort and evaluated model performance using the CKB cohort. The predictive performance was measured by the AUC58, the integrated discrimination improvement (IDI), and net reclassification index (NRI)59. In order to reduce over-fitting in the discovery stage, the AUC, IDI, and NRI of the DFTJ cohort were calculated using the 5-fold cross-validation method.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The full EWAS summary statistics of generated in this study have been deposited in the OMIX, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, under accession code OMIX006068 (https://ngdc.cncb.ac.cn/omix). The phenotypes associated with validated CpGs were extracted from MRC-IEU EWAS catalog database (http://www.ewascatalog.org/) and EWAS Atlas (https://ngdc.cncb.ac.cn/ewas/atlas/index). The phenotypes associated with genes mapped to validated CpGs were extracted from GWAS catalog database (https://www.ebi.ac.uk/gwas/home) and PhenoScanner database (http://www.phenoscanner.medschl.cam.ac.uk). In the Mendelian randomization analysis, the GWAS summary statistic data for MI (Sakaue et al., PMID: 34594039), UAP (Sakaue et al., PMID: 34594039), and CHD (Koyama et al., PMID: 33020668) are derived from the BBJ cohort. Lookup of the transcription factors with CpGs and target genes was conducted using the public database of AnimalTFDB 3.0 (https://guolab.wchscu.cn/AnimalTFDB#!/), ENCODE, and hTFtarget (https://guolab.wchscu.cn/hTFtarget/#!/). The GSE43292 and GSE28829 datasets used in this study are publicly available in the GEO database. Construction of PRS was based on two published PRS models calibrated for East Asians: PRSBBJ reported by Koyama et al. (PMID: 33020668) and PRSCHN reported by Lu et al. (PMID: 35195259). The raw individual participant data included in this project are protected and are not available due to data privacy laws. All other processed data supporting the key findings of this study are available within the article and its Supplementary Information/Source Data files. Source data are provided with this paper.
References
Bhatt, D. L., Lopes, R. D. & Harrington, R. A. Diagnosis and treatment of acute coronary syndrome: a review. JAMA 327, 662–675 (2022).
Bergmark, B. A., Mathenge, N., Merlini, P. A., Lawrence-Wright, M. B. & Giugliano, R. P. Acute coronary syndromes. Lancet 399, 1347–1358 (2022).
Erdmann, J., Kessler, T., Munoz Venegas, L. & Schunkert, H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res. 114, 1241–1257 (2018).
Bjorkegren, J. L. M., Kovacic, J. C., Dudley, J. T. & Schadt, E. E. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll. Cardiol. 65, 830–845 (2015).
Rakyan, V. K., Down, T. A., Balding, D. J. & Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541 (2011).
Martin, E. M. & Fry, R. C. Environmental influences on the epigenome: exposure- associated DNA methylation in human populations. Annu. Rev. Public Health 39, 309–333 (2018).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Li, J. et al. Genome-wide analysis of DNA methylation and acute coronary syndrome. Circ. Res. 120, 1754–1767 (2017).
Fernández-Sanlés, A., Sayols-Baixeras, S., Subirana, I., Dégano, I. & Elosua, R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis 263, 325–333 (2017).
Guarrera, S. et al. Gene-specific DNA methylation profiles and LINE-1 hypomethylation are associated with myocardial infarction risk. Clin. Epigenetics 7, 133 (2015).
Agha, G. et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation 140, 645–657 (2019).
Navas-Acien, A. et al. Blood DNA methylation and incident coronary heart disease: evidence from the Strong Heart Study. JAMA Cardiol. 6, 1237–1246 (2021).
Si, J. et al. Epigenome-wide analysis of DNA methylation and coronary heart disease: a nested case-control study. Elife 10, e68671 (2021).
Hillary, R. F. et al. Blood-based epigenome-wide analyses of 19 common disease states: A longitudinal, population-based linked cohort study of 18,413 Scottish individuals. PLoS Med. 20, e1004247 (2023).
Wang, F. et al. Cohort Profile: the Dongfeng-Tongji cohort study of retired workers. Int. J. Epidemiol. 42, 731–740 (2013).
Chen, Z. et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int. J. Epidemiol. 40, 1652–1666 (2011).
Lane, W. J. et al. PIGG defines the Emm blood group system. Sci. Rep. 11, 18545 (2021).
Lukacs, M., Roberts, T., Chatuverdi, P. & Stottmann, R. W. Glycosylphosphatidylinositol biosynthesis and remodeling are required for neural tube closure, heart development, and cranial neural crest cell survival. Elife 8, e45248 (2019).
Krajnik, A. et al. Phosphoinositide signaling and mechanotransduction in cardiovascular biology and disease. Front. Cell. Dev. Biol. 8, 595849 (2020).
van der Harst, P. & Verweij, N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 122, 433–443 (2018).
Hartiala, J. A. et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 42, 919–933 (2021).
Sainsbury, S., Bernecky, C. & Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 129–143 (2015).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Song, Y. et al. Identification of susceptibility loci for cardiovascular disease in adults with hypertension, diabetes, and dyslipidemia. J. Transl. Med. 19, 85 (2021).
Zaveri, H. P. et al. Identification of critical regions and candidate genes for cardiovascular malformations and cardiomyopathy associated with deletions of chromosome 1p36. PLoS One 9, e85600 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Sjostedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, 6482 (2020).
Infante, T. et al. ABCA1, TCF7, NFATC1, PRKCZ and PDGFA DNA methylation as potential epigenetic-sensitive targets in acute coronary syndrome via network analysis. Eur. Heart J. 42, 1367–1367 (2021).
Steenaard, R. V. et al. Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin. Epigenetics 7, 54 (2015).
Jiang, N., Yang, M., Han, Y., Zhao, H. & Sun, L. PRDM16 regulating adipocyte transformation and thermogenesis: a promising therapeutic target for obesity and diabetes. Front. Pharmacol. 13, 870250 (2022).
Liu, L. et al. The relationship between PRDM16 promoter methylation in abdominal subcutaneous and omental adipose tissue and obesity. Clin. Nutr. 40, 2278–2284 (2021).
AlAmrani, A., AbdelKarim, M. & AlZoghaibi, M. PRDM16 gene polymorphism is associated with obesity and blood lipids profiles in Saudi population. J. Clin. Med. 7, 6 (2018).
Chen, P. et al. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia 60, 645–655 (2017).
Liu, C. et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat. Genet. 48, 1162–1170 (2016).
Runkel, F. et al. Alopecia in a viable phospholipase C delta 1 and phospholipase C delta 3 double mutant. PLoS One 7, e39203 (2012).
Berridge, M. J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 (2016).
Shulenin, S. et al. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N. Engl. J. Med. 350, 1296–1303 (2004).
Koyama, S. et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 52, 1169–1177 (2020).
Lu, X. et al. A polygenic risk score improves risk stratification of coronary artery disease: a large-scale prospective Chinese cohort study. Eur. Heart J. 43, 1702–1711 (2022).
Abraham, G. et al. Genomic prediction of coronary heart disease. Eur. Heart J. 37, 3267–3278 (2016).
Hindy, G. et al. Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscl. Throm. Vas 40, 2738–2746 (2020).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Lambert, S. A., Abraham, G. & Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 28, R133–R142 (2019).
Zheng, Y. et al. Association of cardiovascular health through young adulthood with genome-wide DNA methylation patterns in midlife: the CARDIA study. Circulation 146, 94–109 (2022).
Westerman, K. et al. Epigenomic assessment of cardiovascular disease risk and interactions with traditional risk metrics. J. Am. Heart Assoc. 9, e015299 (2020).
Fernandez-Sanles, A. et al. DNA methylation and age-independent cardiovascular risk, an epigenome-wide approach: the REGICOR study (REgistre GIroní del COR). Arterioscl. Throm. Vas. 38, 645–652 (2018).
O’Gara, P. T. et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127, e362–e425 (2013).
Li, W. et al. Circulating metals, leukocyte microRNAs and microRNA networks: a profiling and functional analysis in Chinese adults. Environ. Int. 169, 107511 (2022).
Zhang, Y. et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun. 8, 14617 (2017).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ayari, H. & Bricca, G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J. Biosci. 38, 311–315 (2013).
Doring, Y. et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 125, 1673–1683 (2012).
Hu, H. et al. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 47, D33–D38 (2019).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Zhang, Q. et al. hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genom. Proteom. Bioinf. 18, 120–128 (2020).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 12, 77 (2011).
Pencina, M., D’Agostino, R., D’Agostino, R. & Vasan, R. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat. Med. 27, 157–172 (2008).
Acknowledgements
We thank all study participants, researchers, and students who contributed to this study. This study was supported by the National Natural Scientific Foundation of China (82021005 [T.W.], 82325044 [C.W.], 82192903 [T.W.], 82192904 [J.L.], 82192900 [L.L.], and 82203997 [P.L.]), the Natural Science Fund for Distinguished Young Scholars of Hubei Province (2022CFA046 [C.W.]), the Chief Scientist Research Project of Hubei Shizhen Laboratory (HSL2024SX0003 [T.W.]), and the Fundamental Research Funds for the Central Universities (2019kfyXMBZ015 [T.W.] and 2023BR030 [C.W.]).
Author information
Authors and Affiliations
Contributions
T.W., C.W., and J.L. conceptualized and supervised the project. P.L., and J.S. conducted the analysis and contributed equally to this work. P.L., J.S., Z.Z., Y.J., Y.W., Q.J., W.L., X.X., Y.Y., M.Q., H.W., T.M., K.L., J.J., Q.W., C.Y., Y.G., Y.Y., H.W., and X.H. contributed to data generation and curation. X.Z., M.H., H.G., I.M., R.W., Z.C., L.L., J.L., C.W., and T.W. contributed samples. P.L., and C.W. drafted the manuscript, T.W., C.W., J.L., P.L., J.S., and all the primary authors reviewed, edited and approved manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shaowei Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Long, P., Si, J., Zhu, Z. et al. Genome-wide DNA methylation profiling in blood reveals epigenetic signature of incident acute coronary syndrome. Nat Commun 15, 7431 (2024). https://doi.org/10.1038/s41467-024-51751-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51751-6
- Springer Nature Limited