

Abstract
P2X receptors, a subfamily of ligand-gated ion channels activated by extracellular ATP, are implicated in various physiopathological processes, including inflammation, pain perception, and immune and respiratory regulations. Structural determinations using crystallography and cryo-EM have revealed that the extracellular three-dimensional architectures of different P2X subtypes across various species are remarkably identical, greatly advancing our understanding of P2X activation mechanisms. However, structural studies yield paradoxical architectures of the intracellular domain (ICD) of different subtypes (e.g., P2X3 and P2X7) at the apo state, and the role of the ICD in P2X functional regulation remains unclear. Here, we propose that the P2X3 receptor’s ICD has an apo state conformation similar to the open state but with a less tense architecture, containing allosteric sites that influence P2X3’s physiological and pathological roles. Using covalent occupancy, engineered disulfide bonds and voltage-clamp fluorometry, we suggested that the ICD can undergo coordinated motions with the transmembrane domain of P2X3, thereby facilitating channel activation. Additionally, we identified a novel P2X3 enhancer, PSFL77, and uncovered its potential allosteric site located in the 1α3β domain of the ICD. PSFL77 modulated pain perception in P2rx3+/+, but not in P2rx3−/−, mice, indicating that the 1α3β, a “tunable” region implicated in the regulation of P2X3 functions. Thus, when P2X3 is in its apo state, its ICD architecture is fairly ordered rather than an unstructured outward folding, enabling allosteric modulation of the signaling of P2X3 receptors.
Similar content being viewed by others

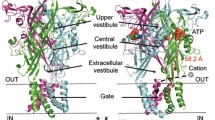

Introduction
The intracellular domain (ICD) of ion channels is crucial for the regulation of channel activity and signal transduction1,2. Several ion channels are directly activated by endogenous and exogenous substances inside cells. These include cyclic nucleotide-gated (CNG) channels, which are activated by cyclic nucleotides3, transient receptor potential melastatin 2 (TRPM2) receptors, which are activated by intracellular adenosine diphosphate ribose (ADPR) and calcium ions4,5, and TRPA1’s abundance of cysteine residues at the N-terminal, which allows it to sense mustard oil, cinnamic aldehyde, and other chemicals6. The ICD is frequently capable of interacting directly with other proteins that influence channel function. For example, one mechanism of N-methyl-D-aspartate (NMDA) receptor-mediated neurotoxicity is the intracellular interaction of TRPM4 with NMDA receptors7,8. ICD also provides ion permeation-independent signaling pathways of ion channels, e.g., the TRPM7 channel’s ICD includes a kinase region that gives the TRPM7 ion channel and kinase dual functionality9,10. Acidic neuronal necrosis is facilitated by acid-sensing ion channels (ASIC1a) via its C-terminus, which interacts with receptor-interacting protein kinase 1 (RIPK1)11,12.
P2X receptors are a class of non-selective cation channels that detect extracellular ATP levels ranging from nanomolar to millimolar13,14. A total of seven distinct subtypes of P2X (P2X1–7) have been identified, each of which consists of three subunits in a homo- or heterotrimer13,14. P2X receptors are found in many tissues of both higher and lower organisms15. They play a role in a variety of physiological and pathological processes, such as synaptic transmission16, inflammation17,18, neuropathic pain19,20, chronic cough21, blood-pressure control22,23,24, and immune regulation25,26,27. As a result, P2X receptors have drawn a lot of interest as a cutting-edge class of therapeutic targets. Among them, the P2X3 receptor has the potential to be a new therapeutic target for endometriosis28, neuropathic pain29,30, hypertension22, idiopathic interstitial pulmonary fibrosis31 and chronic cough21,32; the P2X4 receptor, for alcohol use disorders and neuropathic pain33,34,35; and the P2X7 receptor, for rheumatoid arthritis36,37, neuralgia38, and a variety of psychiatric disorders and neurodegeneration diseases39,40.
In recent years, the structures of about thirty P2X receptors from higher or lower organisms have been determined by X-ray diffraction or single-particle cryo-electron microscopy (cryo-EM) methods41. From these structures, it is found that P2X has a very conserved three-dimensional extracellular architecture and almost the same gating mechanism. Notably, the seven subtypes of P2X receptors differ in their intracellular termini (N- and C-termini) sequence, particularly the C-terminal, which can vary in length from 41 amino acids for the shortest P2X4 to 240 amino acids for the longest P2X742,43. This variation in length may cause the P2X receptor’s ICD to different assembly and may contribute to inter-subtype functional variability. Among available P2X structures, only ICD structure of rat P2X7 (rP2X7) receptor in the apo and open states, and of the hP2X3 in the open state, have been determined44,45 (see also Fig. 1A, B and Supplementary Fig. 1). The N- and C-termini of the P2X7 receptor fold together to form a “cytoplasmic cap” architecture and a unique GDP-binding domain45 (Fig. S1). Notably, comparisons of open and closed structures as well as fluorescence changes in voltage-clamp fluorometry (VCF) indicate that the ICD of the P2X7 receptor undergoes little conformational change during channel gating45,46. Additionally, the open crystal structure of the human P2X3 (hP2X3) receptor takes a similar ICD assembly to that of the open cryo-EM structure of P2X7 (F7-W21 and N345-E363, numbered by hP2X3). However, the architecture of the ICD is missing in the apo structure of the hP2X3 receptor, and it has been assumed that the P2X3’s ICD folded in a different manner44 with that of P2X745 in the apo state (Fig. 1A, B and below). This assumption is somewhat reasonable because P2X7’s sequence length differs from that of other P2X subtypes, including P2X3. However, it is still unclear whether P2X3’s ICD indeed adopts distinctly in the apo and open states. Doubts about this point were reinforced when we refined the hP2X3 crystal structure in the resting state with bound inhibitor AF-219 (Supplementary Fig. 2A) and found an inward-folded ICD (similar to the conformation of ICD in the open state, Fig. 1C and Supplementary Fig. 2B). Meanwhile, it is still unknown how P2X3’s ICD functions in channel gating and how is the ICD involved in the regulation of physiopathological function of P2X3.

A, B Cartoon representation of potential architectures for the human P2X3 (hP2X3) receptor in its apo/antagonist-bound state (A) and open state (B), as suggested by structural studies. In the proposed apo or antagonist-bound state (A), the hP2X3 receptor exhibited loosely folded ICD extending outward. Conversely, in the open state (B), these intracellular regions were finely folded inward. C Zoom-in view of the refined crystal structure of hP2X3 (PDB ID: 5YVE) in its antagonist-bound closed state, featuring the inclusion of 8 amino acids (13-PKVIVVKS-20) appended to the N-terminus of the receptors.
Here, we suggest that the P2X3 receptor’s ICD could move in coordination with the transmembrane region and facilitate channel activation. We also identified the novel P2X3 receptor enhancer PSFL77, which acts at the ICD of P2X3. Using PSFL77 as a chemical tool, in combination with engineered disulfide bonds, and changes in emission spectra of fluorescent unnatural amino acids (flUAA), we found that the ICD of the P2X3 receptor adopts a well-assembled, open-state-like, but slightly different conformation in the apo state. Furthermore, PSFL77 was able to affect nociception in wild-type (WT) mice but not in P2X3 knockout animals, suggesting a potential role for ICD allosteric regulation in P2X3 receptor signaling.
Results
Interrelated movements of intracellular and transmembrane domains possibly contribute to the activation of P2X3 receptors
Within the ICD of rat P2X3 (rP2X3), the amino acids from three subunits, T9-S13 (subunit A), K14-S20 (subunit B), and F358-E363 (subunit C), create three longitudinally organized β-sheets, flanked by an α-helix from subunit B (L347-K357) (Fig. 2A). We have designated this particular region as the 1α3β domain, and the formation of the “cytoplasmic cap” of the trimeric P2X3 receptor is achieved by the combination of the three 1α3β domains (Fig. 2A, B).

A The homology model of the rP2X3 receptor at the open state, observed in parallel to the membrane, with the membrane position indicated by pink dots. Side and bottom perspectives of the ICD of rP2X3 in the right panel, including the delineation of amino acid residue ranges 9–13, 14–20, 347–357, and 358–363. Each of the three subunits is distinctly colored, with the “cytoplasmic cap” composed of three 1α3β domains. B Zoom-in view of critical residues within the 1α3β domain of P2X3 receptors (numbered according to rat P2X3, rP2X3), displayed in sticks for emphasis. C Side and bottom views of the interrelated motions of ICD and transmembrane domains in the mode derived from employing membrANM analysis on the hP2X3 receptor. Motion direction is visually emphasized through the use of blue arrows. D Pooled data comparing rP2X3 current density in response to ATP (10 μM) for WT and mutant receptors. Each open circle in the scatter plot represents an individual measurement. Data are expressed as mean ± S.E.M. n = 5 (rP2X3Y10A, rP2X3T12A, rP2X3V16A, rP2X3I23A, rP2X3L343A, rP2X3Y353A, rP2X3R356A, rP2X3K357A and rP2X3E363A), 6 (rP2X3V18A, rP2X3S20A, rP2X3V361A and rP2X3E359A), or 10 (rP2X3WT). *P < 0.05 and **P < 0.01 vs. WT, one-way ANOVA with Dunnett’s multiple comparisons test, P = 0.003 (rP2X3Y10A, rP2X3T12A, rP2X3V16A, rP2X3I23A, rP2X3Y353A, rP2X3K357A), 0.002 (rP2X3V18A), 0.9743 (rP2X3S20A), 0.005 (rP2X3L343A), 0.9996 (rP2X3R356A), 0.0058(rP2X3V361A), 0.0001 (rP2X3E359A), and 0.0459 (rP2X3E363A).
Since the function of the 1α3β domain in channel activity remains unexplored, we initially investigated its potential role in P2X3 gating. Normal mode analysis was performed on the open hP2X3 structure (PDB ID: 5SVK) in both channels that were embedded in cell membranes (membrANM, 20 modes) and in the absence of cell membranes (ANM, 100 modes). In all membrANM and ANM motion models, there was a high association observed between the motions of the transmembrane domain (TMD) and the 1α3β domain (Fig. 2C and Supplementary Fig. 3). Particularly, in membrANM’s low-frequency mode 3, 1α3β remained intact as a single unit (Fig. 2C). As the TMD underwent a twist, the 1α3β domain likewise underwent rotation, accompanied by an increase in pore size (Supplementary Movie 1), implying a crucial role of 1α3β in channel gating of P2X3 receptors.
Indeed, mutations in key amino acids (Fig. 2B) in the 1α3β domain revealed that rP2X3Y10A, rP2X3T12A, rP2X3V16A, rP2X3V18A, rP2X3I23A, rP2X3L343A, rP2X3Y353A, rP2X3K357A and rP2X3E359A nearly abolished the ATP response of P2X3 receptors (P < 0.001, vs. WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (13, 65) = 7.668, n = 5–10). While ATP-induced currents were significantly reduced at rP2X3V361A and rP2X3E363A, but not rP2X3R356A and rP2X3S20A (current density = 13.9 ± 6.6, 20.2 ± 5.0, 52.9 ± 16.9, 69.4 ± 17.1 and 57.4 ± 14.0 pA/pF for rP2X3V361A, rP2X3E363A, rP2X3R356A, rP2X3S20A and rP2X3WT, respectively, P > 0.05 or <0.05 vs. rP2X3WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (13, 65) = 7.668, n = 5–10, Fig. 2D). Remarkably, the total and surface expression levels of mutants with significantly reduced ATP currents (Fig. 2D) were only slightly different or not different from those of WT P2X3 receptors (Supplementary Fig. 4A, B). We introduced supplementary mutations (T12A, V16A, L343A, and Y353A) into the S15C/F358C mutant. Trimeric bands of mutant rP2X3T12A/S15C/F358C, rP2X3V16A/S15C/F358C, rP2X3L343A/S15C/F358C and rP2X3Y353A/S15C/F358C were still discernible in non-reducing protein immunoblots (Supplementary Fig. 4C). Additionally, we evaluated the role of the trimeric assembly of the mutated P2X3 intracellular region using bioluminescence resonance energy transfer (BRET)47,48,49 (Supplementary Fig. 4D, E). RLUC and EYFP were fused to the C-terminus of P2X3, and its ATP responsiveness was validated through electrophysiological measurements (Supplementary Fig. 4D). Point mutations (rP2X3T12A, rP2X3Y353A, and rP2X3K357A) were individually introduced into P2X3-RLUC and P2X3-EYFP, and their intracellular interactions were examined via BRET in HEK293T cells post-transfection. Significant BRET interactions for rP2X3T12A, rP2X3Y353A and rP2X3K357A were observed (BRET ratios for rP2X3WT, rP2X3T12A, rP2X3Y353A and rP2X3K357A were 0.11 ± 0.02, 0.10 ± 0.02, 0.08 ± 0.01, 0.08 ± 0.02, respectively, *P < 0.05 and **P < 0.01 vs. pCDNA3, one-way ANOVA with Dunnett’s multiple comparisons test; Supplementary Fig. 4E). As a negative control, WT rP2X4 (rP2X4-EYFP) demonstrated no significant effect with WT P2X3-RLUC (BRET ratio = 0.04 ± 0.01, P = 0.5909 vs. pCDNA3, one-way ANOVA with Dunnett’s multiple comparisons test; Supplementary Fig. 4E). These results suggested that altered 1α3β internal interactions as well as altered channel activations, may be responsible for the loss of channel gating in mutations, rather than affecting channel surface expressions and/or channel assembly.
flUAA incorporation combined with VCF analysis confirm the allosteric change of ICD during P2X3 activation
Thus, in contrast to P2X745,46, the ICD of the P2X3 receptor possibly undergo significant conformational changes upon activation. The gating allosteric changes of several ion channels have been effectively studied using ANAP (Fig. 3A), a flUAA capable of detecting conformational changes in adjacent residues throughout VCF analysis32,50,51,52,53. Therefore, we introduced ANAP at several sites within the ICD including rP2X3T9, rP2X3S15, rP2X3F358, rP2X3V361, rP2X3E363 and rP2X3T364 (Fig. 3B, left). YFP, fused at the end of the rP2X3 plasmid, exhibited a normal fluorescent signal, confirming the P2X3 receptor’s incorporation of flUAA at the specific site for normal expression (Fig. 3C). Since V143 is exposed to the solution and is located in the extracellular head domain of P2X3 (Fig. 3B, right), it was used as a negative control32. The maximal absorption spectrum wavelength of ANAP for rP2X3V143ANAP remained relatively unchanged subsequent to the administration of 10 μM ATP (1.08 ± 0.33 nm, Fig. 3D, E), consistent with previous findings32. Compared to rP2X3V143ANAP, rP2X3T9ANAP, rP2X3F358ANAP, rP2X3E363ANAP and rP2X3T364ANAP (Fig. 3D–H), but not rP2X3S15ANAP and rP2X3V361ANAP, showed significant emission peak shifts (2.58 ± 0.40, 0.50 ± 0.31, 2.33 ± 0.21, 2.25 ± 0.55, 2.89 ± 0.48 and 3.00 ± 0.32 nm for rP2X3T9ANAP, rP2X3S15ANAP, rP2X3F358ANAP, rP2X3V361ANAP, rP2X3E363ANAP and rP2X3T364ANAP, respectively; *P < 0.05 and **P < 0.01 vs. rP2X3V143ANAP, unpaired t test, n = 5–13, Fig. 3D). These results indicate that the ICD (Fig. 2A) of the P2X3 receptor underwent allosteric changes during the transition of the channel from the resting state to the open state, which differs from the unchanged conformation of ICD revealed by structural and VCF studies of the P2X7 receptor46.

A Schematic representation of the insertion of fluorescent unnatural amino acid (flUAA) ANAP into the P2X3 receptor. B ANAP-incorporated sites located in the 1α3β domain of P2X3-ICD and the negative control site in the head domain, which are shown in sticks form for emphasis. V143 is situated in the extracellular head domain of rP2X3 and is fully exposed to the solution. C Representative images showing negative control cells (without the addition of pANAP vector or L-ANAP) and positive cells (expressing ANAP- rP2X3-YFP. Pseudo-color was employed for ANAP, and ANAP fluorescence was observed in positive cells but not in negative control cells. D–H Pooled data (D) and representative ANAP emission peak shifts (E–H) of ANAP from the cells expressing ANAP-mutant rP2X3 receptors. The emission spectra in the absence (control, Ctrl) and presence (ATP) of ATP are represented in black and blue, respectively. Each open circle in the scatter plot represents an individual measurement. Box plots indicate data distribution of the second and third quartiles (box), median (line) and Min to Max (whiskers). n = 5 (rP2X3T364ANAP), 6 (rP2X3F358ANAP), 9 (rP2X3E363ANAP), 10 (rP2X3S15ANAP), 12 (rP2X3T9ANAP and rP2X3V361ANAP) or 13 (rP2X3V143ANAP), *P < 0.05 and **P < 0.01, vs. rP2X3V143ANAP, two-side unpaired t test, P = 0.0075 (rP2X3T9ANAP), 0.2263 (rP2X3S15ANAP), 0.0251 (rP2X3F358ANAP), 0.0759 (rP2X3V361ANAP), 0.0044 (rP2X3E363ANAP), and 0.004 (rP2X3T364ANAP). I Sites of covalent modification within the 1α3β domain (shown with stick for emphasis). J, K Representative traces (J) and pooled data (K, mean ± S.E.M., n = 4 (rP2X3V361C/C3A), 5 (rP2X3V361C and rP2X3C3A), 6 (rP2X3WT) or 7 (rP2X3V16C)) to illustrate the impact of MTSEA (1 mM) on ATP (10 µM)-induced currents in both WT and mutated rP2X3. This ratio signifies the ATP-induced current observed following MTSEA treatment normalized by the ATP current measured before MTSEA administration. rP2X3V16C/C3A did not react to ATP (n.d.). Each open circle in the scatter plot represents an individual measurement. **P < 0.0001 vs. WT; ##P < 0.0001 vs. rP2X3C3A, one-way ANOVA with Dunnett’s multiple comparisons test.
Covalent occupancy-induced reduced channel activity underscores the critical role of allosteric changes of ICD during P2X3 activation, rather than other P2X subtypes
To further study the role of the ICD’s allostery in P2X3 activation, a click chemical covalent occupancy was used to prevent the ICD from conformational changes when the channel was activated. We introduced free cystine into the ICD of P2X3, and used MTSEA, which can cross the cell membrane54, to covalently attach to the free sulfhydryl group in the mutated channel (Fig. 3I). ATP currents of rP2X3V16C and rP2X3V361C were significantly reduced after MTSEA (1 mM)’s covalent modification (the ratio of the ATP current after/before = 0.160 ± 0.045, 0.058 ± 0.019 and 1.07 ± 0.11, for rP2X3V16C, rP2X3V361C and rP2X3WT, respectively; **P < 0.01 vs. rP2X3WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (4, 22) = 60.39, n = 4–7; Fig. 3J, K). The decreased ATP current was restored by the removal of covalently attached MTSEA using the reduced agent DTT (10 mM, Fig. 3J). On the other hand, the ATP current of WT rP2X3 was not significantly affected by either MTSEA or DTT treatment (Fig. 3J). Therefore, rather than being the result of other nonspecific occupancy, the considerable attenuation of rP2X3V16C and rP2X3V361C’s ATP current by MTSEA is caused by its disturbance of the ICD conformation.
Given the presence of free cysteine C3 in the P2X3 receptor, we introduced additional C3A mutation in rP2X3V16C and rP2X3V361C. This was taken to more effectively avoid the potential interference of nonspecific covalent occupancy of the endogenous free sulfhydryl group at C3. rP2X3V16C/C3A did not react to ATP, we therefore did not continue the validation using MTSEA (Fig. 3K). Notably, MTSEA’s application still reduced the ATP current of rP2X3V361C/C3A (ratio after/before = 0.124 ± 0.029, n = 4, P < 0.01, vs. WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (4, 22) = 60; Fig. 3K), suggesting that the decrease in ATP current resulting from the covalent occupancy of MTSEA on rP2X3V361C is not attributed to concurrent covalent modification of the free sulfhydryl group of the residue C3.
The superposition of the P2X7 open and resting conformations suggests that the ICD is not engaged in P2X gating (Supplementary Fig. 1), implying that the intracellular domains of P2X3 and P2X7 may exhibit distinct roles in channel activation. To test this idea, we performed covalent occupancy at the corresponding sites of the P2X7 ICD (rP2X7T19 and rP2X7I391, Supplementary Fig. 5A). Indeed, ATP-induced currents were unaffected by the modification of MTSEA in rP2X7T19C and rP2X7I391C (ratio = 0.812 ± 0.041, 0.987 ± 0.077 and 0.925 ± 0.057 for rP2X7T19C, rP2X7I391C and rP2X7WT, respectively; P > 0.05, vs. WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (2, 12) = 1.665, n = 4–6; Supplementary Fig. 5B, C).
Additionally, we performed covalent occupancy by introducing free cysteine at identical sites of other P2X subtypes (Supplementary Fig. 5A). Interestingly, MTSEA’s covalent occupancy at the corresponding site in hP2X1 (ratio = 1.15 ± 0.08 and 1.01 ± 0.04 for hP2X1A371C and hP2X1WT, respectively; P > 0.05, unpaired t test, n = 3–7; Supplementary Fig. 5D, E), rP2X2 (ratio = 0.885 ± 0.124, 0.979 ± 0.125 and 1.02 ± 0.05 for rP2X2C9A/C430A, rP2X2V370C/C9A/C430A and rP2X2WT, respectively; P > 0.05, one-way ANOVA with Dunnett’s multiple comparisons test, F (2, 11) = 0.5560, n = 3–7, Fig. S5F, G), and hP2X4 (ratio = 1.17 ± 0.10 and 1.04 ± 0.04, for hP2X4V375C and hP2X4WT, respectively; P > 0.05, unpaired t test, n = 4–6, Fig. S5H, I), did not affect channels’ activation. Here, the purpose of introducing mutation in residues (e.g., rP2X2C9A/C430A) other than the corresponding one (e.g., rP2X2V370C) is to abolish the nonspecific covalent occupancy of MTSEA by neutralizing the endogenous cystine of P2X-ICD. Based on these data, it appears that the P2X3 receptor’s ICD has an atypical structure or function within the P2X family, setting it apart from P2X1, P2X2, P2X4, and P2X7 subunits.
Engineered disulfide bonds incorporated into the ICD suggest that the 1α3β domain in its apo state has undergone precisely inward folding rather than unstructured outward folding
The observed differences in the P2X3-ICD compared to other P2X subtypes (Supplementary Fig. 5) prompted us to investigate whether there are significant differences in the three-dimensional architectures of P2X3-ICD between its open and apo states. By comparing the crystal structures of P2X3 in its open and apo states, an assumption has been made regarding the apo or inhibitor-bound resting state of the ICD in hP2X344,45, which may exhibit unstructured outward folding (Fig. 1A). On the other hand, the finely folded ICD of rP2X7 remains unchanged in both its apo and open states45 (Supplementary Fig. 1). Furthermore, we employed the COOT and PHENIX software tools (see Methods) to refine the hP2X3-ICD structure in the resting state with the bound inhibitor AF-219 (Supplementary Fig. 2A). Based on the electronic density, eight amino acids (13-PKVIVVKS-20) were added to the N-terminal of the improved structure. Remarkably, all newly added N-termini are folded inward (Fig. 1C and Supplementary Fig. 2B), which is compatible with the P2X3 open structure’s N-terminal orientation (Fig. 1B). When view collectively, these results further cloud our understanding of the true three-dimensional structure of apo P2X3-ICD.
To better elucidate this point, we built engineered disulfide bonds in the ICD (Fig. 4A) based on the open conformation of rP2X3 (the ICD is a disorder region in the apo crystal structure of P2X344). If the designed disulfide bonds can spontaneously form in the absence of ATP, it implies that the conformation of the ICD of P2X3 in the apo state is not loosely folded but rather resembles the open state of P2X3 or the apo/open conformation of P2X7. We chose S15 and F358 of the ICD for engineered disulfide bond introducing due to their 6.3 Å Cβ-Cβ distance in the open state (Fig. 4A), which is close to the Cβ-Cβ distance of 5.5 Å in natural disulfide bond55. Non-reducing gel analysis of rP2X3S15C/F358C revealed more distinct trimerization bands than β-Me treatment (Fig. 4B). As controls, rP2X3WT, rP2X3S15C and rP2X3F358C did not display any trimerization bands regardless of whether they were treated with β-Me or not (Fig. 4B). These results demonstrate that the Cβ-Cβ distance between rP2X3S15 and rP2X3F358 is close enough when they are at apo state. Because rP2X3S15 and rP2X3F358 are located on two rigid β-sheet structures (Fig. 4A), their proximity suggests that the ICD may be in a well-folded state, as opposed to the disordered N- and C-termini found in structural studies44.

A Location of engineered disulfide bonds within the 1α3β domain of P2X3-ICD. B The S15C-F358C intersubunit disulfide bond formation identified using non-reducing gel analysis. The experiment was repeated thrice with similar results. C, D Representative current traces (C) and pooled data (D) recorded from transfected cells containing either the mutant or WT P2X3 receptors. H2O2 (0.3%) and dithiothreitol (DTT, 10 mM) were employed to facilitate and disrupt the disulfide bond, respectively. The ratio compares the current generated by ATP (10 µM) following DTT treatment to H2O2 treatment. Every measurement is represented by the scatter of the open circles. Data are expressed as mean ± S.E.M. n = 4 (rP2X3F358C), 5 (rP2X3S15C), 6 (rP2X3WT) or 10 (rP2X3 S15C/F358C), **P < 0.0001 vs. rP2X3WT, one-way ANOVA with Dunnett’s multiple comparisons test. E ATP concentration-response curves for rP2X3WT and rP2X3S15C/F358C after the treatment of H2O2 or DTT. Data are expressed as mean ± S.E.M. n = 5 (0.03, 0.1, 0.3, 1, 3 and 10 μM) independent cells for rP2X3WT-H2O2, n = 4 (0.03, 0.1, 0.3 and 10 μM), or 3 (1 μM) independent cells for rP2X3WT-DTT, n = 3 (0.03, 0.1, and 0.3 μM), or 5 (1, 3 and 10 μM) independent cells for rP2X3S15C/F358C-H2O2, n = 4 (0.03, 0.1, 0.3, 1 and 3 μM), or 3 (10 μM) independent cells for rP2X3S15C/F358C-DTT. F A schematic showing how residue 356’s ANAP emission peak shifts as a result of chemically modifying rP2X3V16C in the absence of ATP. G–I Effects of MTSEA (1 mM)-treatment on ANAP fluorescence. Pooled data (G) and representative emission peak shifts of ANAP from the cells expressing rP2X3R356ANAP (H) and rP2X3V16C/R356ANAP (I). Each pair of connected open circles in the scatterplot signifies a single measurement in the before-and-after comparison. n = 16 (rP2X3R356ANAP) or 26 (rP2X3V16C/R356ANAP), *P = 0.0214 and **P = 0.0041, two-way ANOVA with Sidak’s multiple comparisons test.
Furthermore, the addition of 0.3% H2O2, which resulted in an increased number of disulfide bonds being formed in rP2X3S15C/F358C, led to an decrease in the 10 μM ATP-induced P2X3 activation, and this effect was reversed by 10 mM DTT (ATP current ratio (DTT/H2O2) = 2.36 ± 0.23, 0.958 ± 0.086, 1.01 ± 0.11 and 0.857 ± 0.063 for rP2X3S15C/F358C, rP2X3S15C, rP2X3F358C and rP2X3WT, respectively, P < 0.01, rP2X3S15C/F358C vs. WT, P > 0.05, rP2X3S15C and rP2X3F358C vs. rP2X3WT, one-way ANOVA with Dunnett’s multiple comparisons test, F (3, 21) = 17.28, n = 4–10; Fig. 4C, D). Furthermore, DTT and 0.3% H2O2 treatments did not affect the apparent affinity of ATP in both WT and rP2X3S15C/F358C (the concentration of ATP that produces half of the maximum response (EC50) in rP2X3S15C/F358C = 0.182 ± 0.036 and 0.117 ± 0.038 μM, with DTT and H2O2 treatments, nh = 1.15 ± 0.39 and 1.03 ± 0.21, respectively, P = 0.4554, comparison of fits with extra sum of squares F test, F (1, 43) = 0.5675, n = 3–5; the EC50 in rP2X3 WT = 0.364 ± 0.083 and 0.247 ± 0.084 μM, for with DTT and H2O2 treatments, nh = 0.85 ± 0.24 and 1.04 ± 0.40, respectively, P = 0.0915, F (1, 42) = 2.982, n = 3–5, Fig. 4E). This suggests that the impact of the engineered S15C/F358C disulfide bonds is on the conformational change in the 1α3β domain of P2X3’s ICD, rather than altering the apparent ATP affinity. These findings imply that the P2X3 receptor’s ICD in the apo state is similar to that in the open state, but slightly different because disulfide bond formed under oxidizing conditions reduced channel function of P2X3.
In the absence of ATP, covalent occupancy in V16 by MTSEA alters the ANAP emission peak of rP2X3V16C/R356ANAP
Further evidence of the closeness of R356 and V16 at the apo state was established through changing the fluorescence of ANAP in the absence of ATP by introducing ANAP at R356 and replacing V16 with Cys (rP2X3V16C/R356ANAP, Fig. 4F). Treatment with MTSEA (1 mM) did not or only induced mild fluorescence changes in rP2X3R356ANAP (P = 0.9545, after MTSEA’s treatment vs. before, two-way ANOVA with Sidak’s multiple comparisons test, F (1,81) = 6.271, n = 16; Fig. 4G, H), indicating that nonspecific covalent modification in endogenously expressed membrane proteins of HEK293T cells and intracellular aggregation of positively charged MTSEA would not cause alterations in the ANAP spectrum. In contrast, rP2X3V16C/R356ANAP had a considerable shift in ANAP’s maximum absorption wavelength before and after MTSEA administrations (P = 0.0041, rP2X3V16C/R356ANAP vs. rP2X3R356ANAP, two-way ANOVA with Sidak’s multiple comparisons test, F (1, 81) = 6.271, n = 26, Fig. 4G, I). This indicates that the N- and C-terminals of P2X3 are finely organized rather than disordered in the apo state.
PSFL77, a novel enhancer of rP2X3, may function at the 1α3β domain of rP2X3
Next, we discovered small molecules that specifically target the 1α3β domain of P2X3, and we used these molecules to modulate P2X3’s physiological and pathological functions. Notably, amino acid mutations in the interface at the N- and C-termini significantly altered the desensitization and current amplitude of the P2X receptors56,57,58. Consequently, regarding the identification of novel molecules, we are particularly interested in small compounds that are capable of modulating P2X3 desensitization and are of a small molecular size that allows them to cross membranes effortlessly. With this in mind, we discovered that the chemical 3,4-dihydroxybenzaldehyde (Fig. 5A), which we named PSFL77, could greatly increase ATP currents and slow down the desensitization of P2X3 (Fig. 5B, C). PSFL77 increased the ATP current of P2X3 by a factor of two, which we designated one of as R1 (Fig. 4B), the ratio of the ATP current after PSFL77 treatment to the current before treatment. Another parameter, denoted as R2 (Fig. 5C), indicates the extent of channel desensitization. It is defined as the ratio of the current at 1 s after reaching the maximum current to the maximal current. Notably, for PSFL77, the altered EC50 of increasing current R1 was 6.45 ± 1.57 μM at 0.1 μM ATP and 7.49 ± 0.19 μM at 10 μM ATP (nh = 1.61 ± 0.71 and 1.50 ± 0.06), whereas the EC50 of R2 was 2.15 ± 1.69 μM and 2.59 ± 2.21 μM at 0.1 and 10 μM ATP (nh = 2.00 ± 1.64 and 2.00 ± 2.85), respectively (Fig. 5B, C). The EC50 values for PSFL77 at 0.1 μM ATP and 10 μM ATP for its functions as an enhancer and desensitization regulator were nearly identical, indicating that the efficacy of PSFL77 is independent of ATP concentration. Nevertheless, the EC50 of PSFL77 enhancing (R1) and desensitizing alterations (R2) differed by approximately 3–5-fold at the same ATP concentration (Fig. 5B, C). In addition to PSFL77, we examined 12 other chemicals with similar structures (Supplementary Fig. 6A). 2,3-dihydroxybenzaldehyde and methyl caffeate were the other two effective ones (Supplementary Fig. 6B). PSFL77, 2,3-dihydroxybenzaldehyde and methyl caffeate had R1 values of 2.75 ± 0.31, 2.87 ± 0.27 and 1.64 ± 0.23, and R2 values of 0.939 ± 0.0123, 0.940 ± 0.009 and 0.938 ± 0.014 (Supplementary Fig. 6C, D), respectively, indicating a significant impact on the P2X3 current amplitude and channel desensitization. Among them, PSFL77 was chosen as the chemical tool for subsequent action site analysis and functional validation.

A Chemical structure of PSFL77. B Concentration-response curves showing effects of PSFL77 on the current amplitude of P2X3 induced by 0.1 μM or 10 μM ATP. The y axis signifies the normalized fold of current increase, calculated as the ratio of the current observed with PSFL77 to that recorded without PSFL77. Data are expressed as mean ± S.E.M. n = 6 (1, 3 and 10 μM), or 5 (30 and 100 μM) independent cells for 10 μM ATP, n = 4 (1, 3, 10, 30 and 100 μM) independent cells for 0.1 μM ATP. C Concentration-response curves employed to illustrate the influence of PSFL77 on the desensitization of P2X3 receptors at 0.1 μM or 10 μM ATP. The y axis shows the altered normalized ratio, calculated by dividing the current at 1 s after reaching the maximum by the maximum current amplitude. Data are expressed as mean ± S.E.M. n = 6 (1, 3 and 10 μM), or 5 (30 and 100 μM) independent cells for 10 μM ATP, n = 4 (1, 3, 10, 30 and 100 μM) independent cells for 0.1 μM ATP. D Pooled data showing the effects of PSFL77 (100 μM) on P2X3, P2X1, P2X2, P2X4 and P2X7 receptors. The y axis indicates the fold increase in current induced by PSFL77, with each open circle representing an individual measurement (mean ± S.E.M., n = 5 (P2X1, P2X2, P2X4 and P2X7) or 8 (P2X3). E Application of PSFL77 (100 μM) via patch pipettes on rP2X3 receptors. Consistent traces across three repetitions were obtained. F Schematic diagram illustrates the strategy employed for constructing chimeras of rP2X2 and rP2X3. G, H Representative current traces (G) and summarized data (H) to show the effect of PSFL77 (100 μM) on ATP (10 μM)-induced activation of the rP2X2/rP2X3 chimeras. The scatter of each open circle represents an individual measurement (mean ± S.E.M., n = 5 (rP2X2WT and CH2), 7 (CH2) or 8(rP2X3WT)), **P < 0.01 vs. rP2X3WT; #P < 0.05 vs. rP2X2WT, one-way ANOVA with Dunnett’s multiple comparisons test, P = 0.0050 (rP2X2WT vs. rP2X3WT), 0.0044 (CH1 vs. rP2X3WT), and 0.0016 (CH2 vs. rP2X2WT). Data from WT P2X3 in D were replotted in H for comparison. I Concentration-response curve of PSFL77 on the current amplitude of the chimera CH2 activated by 10 μM ATP. The y axis shows the normalized current increase, derived from the ratio of ATP- current with and without PSFL77. Data are expressed as mean ± S.E.M. n = 4 (1, 3, 10, 30 and 100 μM) independent cells for CH2.
Furthermore, we assessed the influence of PSFL77 on each of the seven subtypes of P2X receptors, excluding the two non-functional subtypes, namely P2X5 and P2X6. The lack of effect of PSFL77 even at 100 μM for hP2X1, rP2X2, hP2X4, and rP2X7 (ratio = 1.14 ± 0.05, 0.912 ± 0.059, 1.08 ± 0.07, and 0.950 ± 0.045, respectively, n = 4–8; Fig. 5D) indicates that PSFL77 is a P2X3-selective positive allosteric modulator.
With this knowledge, we can validate whether PSFL77 acts directly at the 1α3β domain of the ICD. PSFL77 (100 μM) exhibited its activity when applied directly on the intracellular side (Fig. 5E), suggesting that PSFL77 may act on P2X3’s ICD. Furthermore, if PSFL77 works solely through stimulating the 1α3β domain, a typical PSFL77 response could be transferable from P2X3 to P2X2 subtypes, where PSFL77 is ineffective (Fig. 5D). Hence, we generated two chimeras using rP2X2 and rP2X3 (Fig. 5F): CH1, consisting of rP2X2’s ICD and rP2X3’s TMD and extracellular domain (ECD), and CH2, composed of rP2X2’s ECD and TMD, and rP2X3’s ICD. Indeed, CH2 had the same effect as rP2X3 WT (R1 = 3.22 ± 0.76, n = 5), but CH1 did not have any effect at all (R1 = 1.06 ± 0.06, n = 7; Fig. 5G, H). Additionally, PSFL77’s EC50 (R1) for chimeric CH2 action was nearly identical to that of rP2X3 (3.86 ± 0.34 μM, nh = 4.29 ± 1.30, n = 3–5, Fig. 5I). These data collectively suggested that the ICD of rP2X3 may be the action site of PSFL77.
To reinforce our findings, we induced point mutations into certain amino acids of the 1α3β domain (Fig. 6A). Among these, rP2X3 showed inactivity against ATP in rP2X3T12A, rP2X3V16A and rP2X3Y353A mutants (Fig. 2D), precluding further validation of the PSFL77 effect. Notably, rP2X3R356A mutation caused PSFL77 functionally inactive (R1 = 0.820 ± 0.078, P = 0.0006, vs. rP2X3WT, unpaired t test, n = 5, Fig. 6B, C). The potency of PSFL77 was significantly reduced on rP2X3V16I (R1 = 1.67 ± 0.17, P = 0.0114, vs. rP2X3WT, unpaired t test, n = 7), while it was notably enhanced on rP2X3V18A (R1 = 16.7 ± 3.7, P = 0.0014, vs. rP2X3WT, two-side unpaired t test, n = 7) and rP2X3Y353W (R1 = 10.2 ± 2.2, P = 0.0013, vs. rP2X3WT, two-side unpaired t test, n = 5), when they are compared to that of rP2X3WT (R1 = 2.75 ± 0.31, n = 8; Fig. 6C).

A Possible acting mode of PSFL77 within the1α3β domain of P2X3-ICD. B, C Representative current traces (B) and pooled data (C) showing the effect of PSFL77 (100 μM) on WT rP2X3 and its mutants. The y axis denotes the fold increase in current induced by PSFL77, with each open circle on the scatter plot representing an individual measurement (mean ± S.E.M., n = 3 (rP2X3E363A), 5 (rP2X3V361A, rP2X3R356A and rP2X3Y353W), 7 (rP2X3V16I, rP2X3V18A and rP2X3H352A) or 8 (rP2X3WT)). *P < 0.05 and **P < 0.01 vs. rP2X3WT, two-side unpaired t test, P = 0.0114 (rP2X3V16I), 0.0014 (rP2X3V18A), 0.0006 (rP2X3R356A), and 0.0013 (rP2X3Y353W). D Concentration-response curves of PSFL77 on the current amplitude of WT rP2X3 and its mutants induced by 10 μM ATP. The y axis signifies the normalized fold of current increase, expressed as the ratio of the current in the presence of PSFL77 to that in the absence of PSFL77. Data are expressed as mean ± S.E.M. n = 5 (0.3 and 100 μM), 6 (3 and 30 μM) or 7 (1 and 10 μM) independent cells for rP2X3V16I. n = 3 (1, 3, 10, 30, 100 and 300 μM) independent cells for rP2X3V18A. n = 5 (1, 3, 10 and 100 μM), or 4 (30 μM) independent cells for rP2X3V19A. n = 4 (1, 10, 30 and 100 μM), or 3 (3 μM) independent cells for rP2X3V361A. n = 4 (1, 3 and 30 μM), or 6 (10 μM and 100 μM) independent cells for rP2X3Y353W. E Concentration-response curves of PSFL77 to show the impact of PSFL77 on the delay of desensitization in P2X3 induced by 10 μM ATP. The y axis signifies the normalized ratio, derived by dividing the current at 1 s after reaching the maximum current by the maximum current. Curve fitting was performed using solid lines in accordance with the Hill 1 equation (mean ± S.E.M., n = 5 (0.3 μM), 6 (3 and 30 μM) or 7 (1, 10 and 100 μM) independent cells for rP2X3V16I. n = 3 (1, 3, 10, 30, 100 and 300 μM) independent cells for rP2X3V18A. n = 5 (1, 3, 10 and 100 μM), or 4 (30 μM) independent cells for rP2X3V19A. n = 4 (1, 10, 30 and 100 μM), or 3 (3 μM) independent cells for rP2X3V361A. n = 3 (1 μM), 4 (3 and 30 μM), or 5 (10 μM and 100 μM) independent cells for rP2X3Y353W). F Effects of PSFL77 on the concentration-response curve of ATP in P2X3 receptors. Solid lines were fitted to the Hill 1 equation (mean ± S.E.M., n = 6 (3 and 10 μM) or 7 (0.03, 0.1, 0.3 and 1 μM) independent cells for ATP only, n = 3 (1 μM), 5 (0.01, 0.03, 0.1 and 0.3 μM) or 6 (0.003 μM) independent cells for ATP + PSFL77).
Further validation of the apparent affinity of PSFL77 was conducted with these mutations, revealing a 2–15-fold reduction in potency in enhancing ATP current of rP2X3V18A, rP2X3Y353W and rP2X3V361A (EC50 (R1) = 108 ± 38, 13.5 ± 2.9 and 18.8 ± 1.4 μM for them, nh = 0.81 ± 0.12, 4.62 ± 3.25 and 2.25 ± 0.35, respectively, n = 3–5). rP2X3V16I (6.99 ± 1.97 μM (R2), nh = 1.52 ± 0.48) and rP2X3V361A (7.58 ± 3.36 μM (R2), nh = 1.71 ± 0.71) altered desensitization by a 3–4-fold rightward shift in the EC50 of PSFL77 (Fig. 6D, E). Notably, in the resting state, the 1α3β domain is relatively relaxed and unstructured, causing rP2X3V16I to have a minor impact on PSFL77 functionality, with only a slight modification in current magnitude (R1) (Fig. 6D). Conversely, in the open state, the 1α3β domain adopts a more tense conformation, allowing rP2X3V16I to significantly influence PSFL77, leading to a marked rightward shift in the affinity of R2 (Fig. 6E), reflecting the degree of desensitization. These findings, combined with the observation that R356A abolished the enhancing effect of PSFL77 on P2X3, suggest that PSFL77 may interact with the 1α3β domain, specifically at R356, V16, V361 and Y353.
PSFL77 possibly acts on both the open and resting states of rP2X3, affirming the similarity in the three-dimensional architectures of the ICD in these two states
As shown above (Fig. 5B, C), the results imply a subtle distinction in how PSFL77 influences both current amplitude and desensitization mechanisms. Examining the role of PSFL77 at the unitary current of P2X3, it was found that it not only significantly prolonged open duration and increased the channel open probability (Supplementary Fig. 7A–D) but was also able to increase the unitary current amplitudes from 0.710 ± 0.032 pA to 1.22 ± 0.07 pA (P = 0.0021, unpaired t test, n = 3–4; Supplementary Fig. 7E). These findings indicate that, in the absence of PSFL77, the channel rapidly closes upon opening, leading to macroscopic current desensitization. Conversely, PSFL77 enhances unitary current conductance and opening probability, thereby amplifying the current response. Furthermore, prolonged open duration stabilizes the channel in its open state, delaying the transition to desensitization and reducing macroscopic current desensitization. Our findings suggest a sequential role for PSFL77: it initially acts in the resting state, subsequently regulates ATP-induced conformational changes in the transmembrane region, and enhances channel conductance. When the channel is fully open, PSFL77 binding in the ICD may stabilize it in the open state, thereby prolonging the duration of openness (see below, Fig. 8).
Moreover, stimulation with 10 nM or 30 nM ATP in solution failed to induce P2X3 activation, whereas PSFL77 successfully elicited P2X3 receptor responses to 10 nM or 30 nM ATP (Fig. 6F). Accordingly, there was a rightward shift in the ATP concentration-response curves caused by PSFL77 (EC50 = 0.048 ± 0.005 μM and 0.271 ± 0.065 μM for 100 μM and no PSFL77 applications, nh = 3.17 ± 0.67 and 1.19 ± 0.24, respectively; Fig. 6F). Our data indicate that PSFL77’s influence on P2X3 function is not limited to the fully open state of P2X3, but that it can also function in P2X3’s resting or certain substates. Notably, the Hill coefficient differs between the ATP and ATP + PSFL77 curves (Fig. 6F), increasing from approximately 1–2 to around 3. The P2X receptor, a trimeric channel with three independent ATP binding sites59, typically activates with ATP binding to just two of these sites60,61. PSFL77 may modulate the intracellular 1α3β domain, affecting cooperative interactions among these sites. However, further studies are needed to elucidate the specific mechanism of this modulation. Thus, for PSFL77 to bind, the 1α3β domain should be folded similarly across various states, including the apo state; without this uniformity, PSFL77 could not effectively bind at different states.
PSFL77 causes nociception in P2rx3 +/+, but not in P2rx3 −/−, mice
Finally, we evaluated PSFL77’s in vivo regulatory effects on P2X3 function. Given the abundance of P2X3 receptors in dorsal root ganglia (DRG) neurons and their association with nociception62,63,64, we study effects of PSFL77 on pain sensations in both P2rx3+/+ and P2rx3−/− mice (Fig. 7A, B). Without separating the males from the females, the mice were split into three groups: the vehicle group (3.33‰ DMSO + saline), the group administered 10 μM AF-353 (AF-353), and the group receiving 300 μM PSFL77 (PSFL77).

A An illustration of the von Frey test procedure in mice. B Effects of PSFL77 on the mechanical withdrawal threshold in P2rx3+/+ and P2rx3−/− mice. Data are presented as mean ± S.E.M. (n = 6 (Vehicle P2rx3+/+ and AF-353 P2rx3+/+), 7 (PSFL77 P2rx3−/−), or 10 (PSFL77 P2rx3+/+). * P < 0.05 and ** P < 0.01 vs. vehicle P2rx3+/+, two-way ANOVA with Dunnett’s multiple comparisons test, P = 0.0039 (10 min), 0.0196 (20 min) for AF-353 P2rx3+/+, P < 0.0001 (10, 20, 30, 45, 60, 75, 90, 105 and 120 min), 0.0004 (150 min), and 0.0446 (180 min) for PSFL77 P2rx3+/+. C Representative images depicting cells expressing rP2X2-pIRES-eGFP and rP2X3-pIRES-mCherry, with pseudo-colors assigned to eGFP (green) and mCherry (red). D, E Representative current traces (B) and pooled data (C) showing the effect of PSFL77 (100 μM) on rP2X2/3 heterotrimers (mean ± S.E.M., n = 4 (rP2X2/3), 5 (rP2X2) or 8 (rP2X3). * P = 0.0121 and ** P = 0.0008 vs. rP2X2WT, two-side unpaired t test).
The nociceptive sensitivity in mice was examined by Von Frey test (Fig. 7A), and the mean paw withdraw threshold (PWT) was roughly similar in all three mouse groups (2.89 ± 0.12, 2.76 ± 0.09, and 2.82 ± 0.10, respectively, Fig. 7B). Following the intrathecal injection of 10 μL of 300 μM PSFL77, mice exhibited a notable increase in nociceptive sensitivity. After 10 min, PWT dropped to 1.53 ± 0.13 g (P < 0.001 vs. vehicles, two-way ANOVA with Dunnett’s multiple comparisons test, F (33, 300) = 6.834, n = 10), and then it slowly rose again. There was still a small effect after 150 min (2.34 ± 0.12 g, P = 0.0004 vs. vehicles, two-way ANOVA with Dunnett’s multiple comparisons test, F (33, 300) = 6.834).
This nociceptive effect was decreased by 10 μM AF-353, a specific P2X3 inhibitor65, and knocking out of the P2rx3 gene resulted in the loss of PSFL77 activity (Fig. 7B). Serving as a control, a higher pain threshold and analgesia were observed at 10–20 min after administering an injection of the same concentration of AF-353 alone (3.32 ± 0.13, and 3.23 ± 0.06 g for 10 min and 20 min, respectively, P < 0.05, vs. vehicles, two-way ANOVA with Dunnett’s multiple comparisons test, F (33, 300) = 6.834, n = 6, Fig. 7B). Additionally, injection of the same volume solvent (3.33‰ DMSO + saline) had no effect on PWT of mice (n = 6, Fig. 7B).
Previous studies have highlighted the pivotal roles of both P2X3 monomeric receptors and P2X2/3 heteromeric receptors in pain sensation66. To evaluate PSFL77’s impact on P2X2/3, we co-transfected rP2X2-pIRES-EGFP and rP2X3-pIRES-mcherry using a validated heterotrimers expression method32, allowing simultaneous visualization of cells expressing both fluorophores (Fig. 7C). Confirmation that recorded currents originated from P2X2/3 was based on their desensitization kinetics and selective inhibition by AF-353 (Fig. 7D). Notably, PSFL77 significantly potentiated P2X2/3 heterotrimers (ratio = 1.78 ± 0.29, P = 0.0121, vs. rP2X2, unpaired t test, n = 4, PSFL77 100 μM; Fig. 7E), implicating its role in pronociceptive effects involving both P2X3 homotrimers and P2X2/3 heterotrimers. This effect was absent in P2rx3−/− animals, confirming the involvement of the 1α3β domain, which encompasses both P2X2 and P2X3. These findings underscore PSFL77’s modulatory potential in pain sensation through both receptor types and suggest allosteric modulation of the 1α3β domain under physiopathologic conditions. However, the presence of endogenous substances targeting P2X3 homotrimers or P2X2/3 heterotrimers akin to PSFL77, and their mode of modulation on P2X receptor function, remain unclear.
Discussion
The ICD of ion channels plays a crucial role in their activity and regulation1,2,10. In the case of the P2X3 receptor, its ICD consists of N- and C-terminals. Structural studies suggest that the ICD of the P2X3 receptor forms a “cytoplasmic cap” when the channel is in an open state44. However, the assembly of the ICD in the apo state of P2X3 remains unclear44,45. We demonstrated that the P2X3 receptor’s ICD is important for channel activation. Furthermore, we suggest that, the assembly of the P2X3 receptor’s ICD in the apo state follows a pattern comparable to, but slightly different from, the open state. Our findings indicate that the P2X3 receptor’s ICD contains intracellular allosteric sites. Moreover, PSFL77 acts at this site to modify nociception in mice, suggesting an allosteric mechanism that could be implicated in P2X3 receptor function regulation.
Our identification of an ICD assembly mode in the apo state of rP2X3 that differs from the structural studies is supported by the evidence listed below (Fig. 8). First, in the apo state, S15 and F358 exhibit close proximity, facilitating the spontaneous formation of a disulfide bond between C15 and C358 in rP2X3S15C/F358C (Fig. 4B). Secondly, due to the near proximity of V16 and R356 at the apo state, the ANAP emission spectrum shift of rP2X3V16C/R356ANAP is going to be impacted by the covalent modification by MTSEA at V16C (Fig. 4G–I). Third, our previously determined crystal structure of the P2X3 receptor in complex with AF-219/gefapixant shows a distribution of electron density in the ICD (Fig. 1C and Supplementary Fig. 2). Furthermore, the N-terminal’s electron density in the hP2X3/gefapixant complex is comparable to that of the P2X3 receptor’s N-segment in the open state (Fig.1C and Supplementary Fig. 2B). Fourth, despite conformational differences, PSFL77 can function in both the open and resting states, suggesting that the N- and C-termini of P2X3 have comparable three-dimensional structures in both states (Fig. 6). If the apo state features a loosely folded outward ICD, PSFL77 binding would be hindered. Although there is a possibility of PSFL77 binding to distinct pockets in both the open and apo states, the likelihood is diminished by the observation that R356A mutation abolishes both functions of PSFL77 on the apo (R1) and open states (R2) (Fig. 6B, C). Moreover, both the mutation affecting the enhancing effect of PSFL77 (R1) and desensitization (R2) are within the same allosteric site (Fig. 6D, E). The disparity observed between our findings and those of crystal structural studies could be clarified in the following way: In the structural determinations, the ordered open structure suggests that the N- and C-terminals are in a relatively thermodynamically stable state. In contrast, the apo state exhibits a thermodynamically unstable conformer, leading to a disordered ICD. Nevertheless, the available evidence supporting the notion of a distinction in the assembly of N- and C-terminals between the apo and open states may not be robust. This perspective is reinforced by the structural studies of P2X7, where the three-dimensional structures of the corresponding regions in the apo and open states are nearly identical45.

Contrary to what structural investigations have found, we suggest that even in the apo state, the N- and C-termini are still folded inward and near to one other, just like in the open state, but with a less rigid three-dimensional architecture. In the presence of PSFL77, the “cytoplasmic cap”/1α3β of the ICD remains in a stable open conformation, sustaining the increased pore radius of the channel via influencing the conformation of the transmembrane domain. The facilitation of the channel opening by PSFL77 may play a role in modulating pain perception.
Different P2X receptor subtypes have unique ICD primary sequences, which contribute to subtype differences in activation or regulation. This variety offers a chance to find drugs that are specially targeted by each P2X subtype67. We identified the novel subtype-selective allosteric modulator, PSFL77, which effectively enhanced ATP currents and delayed channel desensitization (Fig. 5). To the best of our knowledge, this marks the first instance of an intracellular allosteric site and regulatory small molecule for the P2X3 receptor. PSFL77 exhibited subtle distinct affinities for both increasing the maximum current and delaying desensitization (Fig. 5B, C), suggesting potential action through two separate mechanisms. Nevertheless, despite their distinct characteristics, both mechanisms function within the identical site (Fig. 6D, E). This emphasizes the complex interplay between the ICD of P2X3 and the TMD. Such interplay suggests the potential for diverse and more efficient regulatory mechanisms, offering an avenue for modulating P2X function in different physiopathological conditions. Notably, the conserved phosphorylation site T18 in the P2X2 receptor corresponds to T12 in the P2X3 receptor68, both located within our proposed allosteric site. This suggests that this site could potentially serve as a structural basis for endogenous substances to modulate P2X3 receptor function, although further exploration is required. The identification of other endogenous intracellular substances, regulatory proteins, or post-translational modifications of the P2X receptor ICD may unveil its physiopathological activities in the future.
In summary, we have presented an ICD assembly model that clarifies the P2X3 receptor’s apo conformation and illuminates its role in the P2X3 gating mechanism. We also emphasized the role that an intracellular allosteric site plays in regulating the physiological and pathological functions of the P2X receptor.
Methods
Ethics
The study was approved by the ethics committee of the China Pharmaceutical University.
Cell culture and transfection
The Human Embryonic Kidney 293 cells (HEK293) were procured from the Cell Bank at the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. The cells were cultured with DMEM (Corning) containing 10% FBS (PAN), 1% Glutamax (Gibco), and 1% penicillin/streptomycin (HyClone) at 37 °C under 5% CO2 atmosphere. The plasmids of rP2X2, rP2X3, hP2X4, and rP2X7 were generously supplied by Drs. Alan North and Lin-Hua Jiang. The cDNA of hP2X1 was synthesized from JiKai Gene and subcloned into the pcDNA3.1 vector. All mutations were introduced using QuikChange mutagenesis kit and subsequently verified by DNA sequencing. Chimeras were generated using the ClonExpress II One Step Cloning Kit (Vazyme). Both wild-type and mutant plasmids were transfected into HEK293 cells by calcium phosphate transfection69.
Patch-clamp electrophysiology
Both the perforated patch clamp and whole-cell patch clamp were employed in this study70. Experiments were conducted 24 to 48 h post-transfection at room temperature (25 ± 2 °C). Patch pipettes were pulled from glass capillaries using a two-stage puller (PC-100, Narishige, Japan) and had a resistance ranging from 3 to 5 megaohms. The extracellular solution consisted of 150 mM NaCl, 5 mM KCl, 10 mM glucose, 2 mM CaCl2, 10 mM HEPES, and 1 mM MgCl2, with the pH adjusted to 7.35–7.40. The intracellular solution contains 120 mM KCl, 30 mM NaCl, 0.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 5 mM EGTA, with the pH ranging from 7.2 to 7.4 used in the whole-cell patch clamp, while contains 75 mM K2SO4, 55 mM KCl, 5 mM MgSO4, 10 mM HEPES, and pH 7.2 used in the perforated patch clamp. The data were acquired using an Axopatch 200B (Molecular Devices) in conjunction with a Digidata 1550B digitizer (Molecular Devices). Prior to data acquisition, the amplifier was set to a holding potential of −60 mV. Signals were sampled at 10 kHz and filtered at 2 kHz. To prevent channel desensitization, ATP was administered at intervals of 8–10 min. Single-channel recordings were conducted using the outside-out configuration. Recording pipettes, pulled from borosilicate glass (World Precision Instruments, Inc.) and fire-polished, had a resistance of 8–12 megohms. The holding potential was set at −80 mV. The extracellular and intracellular solutions used were the same as those employed in whole-cell recordings. For single-channel recordings, the signals were sampled at 50 kHz with a 2 kHz filter, and low-pass filtered at 200 Hz. Clampfit software (Molecular Devices) was used for analysis of the data.
Refinement of the crystal structure of P2X3 in complex with AF-219
Building upon the determined hP2X3 structure featuring bound inhibitor AF-219 (PDB: 5YVE), a refined hP2X3-ICD structure at the resting state was generated through molecular addition using the COOT software package71. Based on the original density, 8 amino acids (13-PKVIVVKS-20) were introduced at the N-terminus of the model. The rest of the protein model remained the same as the original. Subsequent refinement cycles with PHENIX contributed to the improvement of the new structural model72. The R-work/R-free values for the final ligand-free model stand at 0.2467/0.3247, showing a slight improvement compared to the original model’s 0.235/0.296. It is noteworthy that the electron density of the ICD is relatively low, but given that the original model’s values were already close to 0.3, this minor increase is considered acceptable.
Membrane ANM analysis
Explicit Membrane ANM (membrANM) is a method employed to investigate the overall conformational dynamics of membrane proteins in the presence of a lipid bilayer73,74. In this study, the position of the membrane, which is composed of POPC, is determined using the OPM website (https://opm.phar.umich.edu/)75. A total of 20 models were generated using ProDy76. The cross-correlation coefficient Cij of residue pairs was calculated by the Eqs. (1) and (2):
where kB is the Boltzmann constant, T is temperature, mi and mj are the masses of atoms i and j; vk is the vibration frequency of mode k; aik and ajk denote the ith and jth components of the kth eigenvector and the symbol < > represents an average over the simulation trajectory.
flUAA incorporation and VCF analysis
HEK293T cells were cultured in the confocal dish with DMEM (Corning) containing 10% FBS (PAN), and 1% Glutamax (Gibco). An amber stop codon (TAG) was introduced at a designated position of rP2X3, and the mutant gene was co-transfected into HEK293 cells with the tRNA/ANAPRS pair. L-ANAP (AsisChem Inc.) was directly added to the culture medium to the final concentration of 20 µM. After 12–24 h, The L-ANAP-containing culture medium was completely replaced with L-ANAP-free culture medium. Cells were further cultured in ANAP-free medium for 24 h before experiments. Fluorescence of ANAP was excited with a wLS LED light source (Photometrics) using BP340-390 excitation filters, DM410 dichroic filters and BA420-IF emission filters. Emission spectrum of ANAP was imaged with an Acton SpectraPro SP-2150 spectrometer (Princeton Instruments) and a Prime 95B CCD camera (Photometrics). Cells with both YFP fluorescence and ANAP fluorescence were selected, and 50 frames were recorded with 0.65 s intervals in MetaMorph. The data values were normalized after subtracting the background and then the emission peaks were determined by fitting the spectra using a tilted Gaussian distribution32,50.
Total and surface expressions of P2X receptors
HEK293T cells expressing rP2X3 or its mutants were first washed with chilled PBS three times and then incubated with sulfo-NHS-LC-biotin. The reaction was halted by incubating the cells with 1 mM glycine in PBS. After collecting the cells, they were lysed with RIPA buffers. Following centrifugation, 20% of the supernatant volume was diluted with SDS loading buffer to obtain the total protein fraction, whereas the remaining supernatant was incubated at 4 °C with NeutrAvidin agarose resin for 4–5 h. The resin was then washed five times with chilled PBS, and diluted with SDS loading buffer to obtain the surface proteins fraction. The protein samples were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with 5% milk at room temperature for 1–2 h and then incubated overnight at 4 °C with anti-EE-tag, anti-GAPDH, or anti-Na+/K+-ATPase antibodies dissolved in 5% milk. Subsequently, the PVDF membranes were washed 4–5 times with TBST and incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. Finally, protein expression was visualized by exposing the PVDF membranes to ECL solution (Thermo) for 1–3 min using automated chemiluminescence-fluorescence image analysis systems (Tanon 5200, Multi). The quantification of protein expression was conducted using the ImageJ software. The analysis of protein expression was repeated in at least three independent experiments.
BRET analysis
HEK293T cells were transfected with rP2X3-RLUC and rP2X3-EYFP (wild-type or mutant variants) and seeded in 96-well plates for 24 h. The next day, cells were washed with 1× PBS, followed by addition of 90 μL 1× PBS and 10 μL coelenterazine H (final concentration 5 μM) per well. Luminescence of Rluc8 at 475 nm and eYFP fluorescence at 535 nm were measured for 1 s using a Molecular Devices ID5 microplate reader48,49.
Animals
Adult mice (8–12 weeks old) were used in all experiments. WT C57BL/6 mice were obtained from Huachuang Sino Company (Taizhou, China). All animals were maintained in a controlled environment (23 ± 2 °C, 50 ± 10% humidity, 12 h light/dark cycle) with access to standard food and water. Animal experiments were approved by the Animal Ethics Committee of China Pharmaceutical University and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Mechanical sensitivity (von Frey) test
The mechanical withdrawal threshold is defined as the force exerted by the Von Frey filament that elicits a response in the hind paw. This was determined following established procedures64. Briefly, the mouse was situated on a wire mesh surface and allowed to acclimate for 30–60 min before testing. Electronic von Frey filaments were applied to the plantar surface of the mouse’s hind paw from beneath the mesh. The filaments ascended at a consistent velocity, with progressively increasing forces applied. Upon reaching the threshold, the mouse withdrew its paw from the stimulus, and the force at withdrawal was automatically recorded. Each animal underwent five assessments of mechanical sensitivity, spaced at least 1 min apart. The average value was then computed based on the five measurements and presented as the paw withdrawal threshold (g).
Statistical analysis
All results are expressed as Mean ± SEM, and statistics are analyzed using Student’s t test or ANOVA, for which *P < 0.05 and **P < 0.01 were considered as significant difference. Concentration-response relationships were fitted to the equation as I/Imax = A1 + (A2-A1)/[1 + (EC50/[agonist])n], where I is the normalized current at a given concentration of agonists, Imax is the maximum normalized current, EC50 is the concentration of agonists yielding one half of maximal currents, and n is the Hill coefficient.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The complete data generated in this study are described and provided in this document and its associated Supplementary Information and the Source data file. Previously solved structures were accessed from the PDB with accession codes: 5SVJ, 5SVK, 6U9W and 6U9V. Data for a previously released and re-analyzed structure of the P2X3 receptor in complex with AF-219 (PDB 5YVE) (map/data table) and Membrane ANM analysis data have been deposited to figshare [https://doi.org/10.6084/m9.figshare.26056909]. Source data are provided with this paper.
References
Verkest, C. et al. Intrinsically disordered intracellular domains control key features of the mechanically-gated ion channel PIEZO2. Nat. Commun. 13, 1365 (2022).
Codding, S. J., Johnson, A. A. & Trudeau, M. C. Gating and regulation of KCNH (ERG, EAG, and ELK) channels by intracellular domains. Channels 14, 294–309 (2020).
Li, M. et al. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 542, 60–65 (2017).
Wang, L. et al. Structures and gating mechanism of human TRPM2. Science 362, eaav4809 (2018).
Huang, Y., Winkler, P. A., Sun, W., Lü, W. & Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 562, 145–149 (2018).
Macpherson, L. J. et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545 (2007).
Yan, J., Bengtson, C. P., Buchthal, B., Hagenston, A. M. & Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 370, eaay3302 (2020).
Duan, J. et al. Structure of full-length human TRPM4. Proc. Natl Acad. Sci. 115, 2377–2382 (2018).
Sahni, J. & Scharenberg, A. M. TRPM7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 8, 84–93 (2008).
Qiao, W. et al. TRPM7 kinase-mediated immunomodulation in macrophage plays a central role in magnesium ion-induced bone regeneration. Nat. Commun. 12, 2885 (2021).
Wang, J.-J. et al. Disruption of auto-inhibition underlies conformational signaling of ASIC1a to induce neuronal necroptosis. Nat. Commun. 11, 475 (2020).
Wang, Y. Z. et al. Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. Elife 4, e05682 (2015).
Schmid, R. & Evans, R. J. ATP-gated P2X receptor channels: molecular insights into functional roles. Annu. Rev. Physiol. 81, 43–62 (2019).
Alexander, S. P. H. et al. The concise guide to PHARMACOLOGY 2023/24: ion channels. Br. J. Pharm. 180, S145–S222 (2023).
North, R. A. P2X receptors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150427 (2016).
Khakh, B. S. & North, R. A. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron 76, 51–69 (2012).
Yin, Y. et al. Leveraging the ATP‐P2X7 receptor signalling axis to alleviate traumatic CNS damage and related complications. Med. Res. Rev. 43, 1346–1373 (2023).
Schneider, R. et al. A novel P2X2‐dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol. Med. 13, e12724 (2020).
Bernier, L. P., Ase, A. R. & Seguela, P. P2X receptor channels in chronic pain pathways. Br. J. Pharm. 175, 2219–2230 (2018).
Beggs, S., Trang, T. & Salter, M. W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 15, 1068–1073 (2012).
McGarvey, L. P. et al. Efficacy and safety of gefapixant, a P2X3 receptor antagonist, in refractory chronic cough and unexplained chronic cough (COUGH-1 and COUGH-2): results from two double-blind, randomised, parallel-group, placebo-controlled, phase 3 trials. Lancet 399, 909–923 (2022).
Pijacka, W. et al. Purinergic receptors in the carotid body as a new drug target for controlling hypertension. Nat. Med. 22, 1151–1159 (2016).
Shokoples, B. G., Paradis, P. & Schiffrin, E. L. P2X7 receptors–mediated inflammation in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 41, 186–199 (2021).
Yamamoto, K. et al. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat. Med. 12, 133–137 (2005).
Di Virgilio, F., Sarti, A. C., Falzoni, S., De Marchi, E. & Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 18, 601–618 (2018).
Romagnani, A. et al. P2X7 receptor activity limits accumulation of T cells within tumors. Cancer Res. 80, 3906–3919 (2020).
Csóka, B. et al. Macrophage P2X4 receptors augment bacterial killing and protect against sepsis. JCI Insight 3, e99431 (2018).
Zhang, X. et al. Activation of ATF3/AP-1 signaling pathway is required for P2X3-induced endometriosis pain. Hum. Reprod. 35, 1130–1144 (2020).
Xia, L.-P. et al. GPR151 in nociceptors modulates neuropathic pain via regulating P2X3 function and microglial activation. Brain 144, 3405–3420 (2021).
Richards, D., Gever, J. R., Ford, A. P. & Fountain, S. J. Action of MK‐7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation. Br. J. Pharmacol. 176, 2279–2291 (2019).
Smith, J. A. The therapeutic landscape in chronic cough. Lung 202, 5–16 (2023).
Guo, C.-R. et al. Chronic cough relief by allosteric modulation of P2X3 without taste disturbance. Nat. Commun. 14, 5844 (2023).
Yardley, M. M. et al. Ivermectin reduces alcohol intake and preference in mice. Neuropharmacology 63, 190–201 (2012).
Popova, M. et al. Residues in transmembrane segments of the P2X4 receptor contribute to channel function and ethanol sensitivity. Int. J. Mol. Sci. 21, 2471 (2020).
Teng, Y. et al. Intrathecal injection of bone marrow stromal cells attenuates neuropathic pain via inhibition of P2X4R in spinal cord microglia. J. Neuroinflamm. 16, 271 (2019).
Alqallaf, S. M., Evans, B. A. J. & Kidd, E. J. Atypical P2X7 receptor pharmacology in two human osteoblast‐like cell lines. Br. J. Pharmacol. 156, 1124–1135 (2009).
Cao, F. et al. P2X7 receptor: a potential therapeutic target for autoimmune diseases. Autoimmun. Rev. 18, 767–777 (2019).
Yin, C. et al. Inhibitory effects of palmatine on P2X7 receptor expression in trigeminal ganglion and facial pain in trigeminal neuralgia rats. Front. Cell. Neurosci. 15, 672022 (2021).
Bolaños, J. P. et al. Astrocyte adaptation in Alzheimer’s disease: a focus on astrocytic P2X7R. Essays Biochem. 67, 119–130 (2023).
Calzaferri, F. et al. The purinergic P2X7 receptor as a potential drug target to combat neuroinflammation in neurodegenerative diseases. Med. Res. Rev. 40, 2427–2465 (2020).
Sheng, D. & Hattori, M. Recent progress in the structural biology of P2X receptors. Proteins 90, 1779–1785 (2022).
Kawate, T., Michel, J. C., Birdsong, W. T. & Gouaux, E. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature 460, 592–598 (2009).
Illes, P. et al. Update of P2X receptor properties and their pharmacology: IUPHAR review 30. Br. J. Pharmacol. 178, 489–514 (2020).
Mansoor, S. E. et al. X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 538, 66–71 (2016).
McCarthy, A. E., Yoshioka, C. & Mansoor, S. E. Full-length P2X7 structures reveal how palmitoylation prevents channel desensitization. Cell 179, 659–670.e13 (2019).
Durner, A., Durner, E. & Nicke, A. Improved ANAP incorporation and VCF analysis reveal details of P2X7 current facilitation and a limited conformational interplay between ATP binding and the intracellular ballast domain. eLife 12, e82479 (2022).
Fisher, J. A., Girdler, G. & Khakh, B. S. Time-resolved measurement of state-specific P2X2 ion channel cytosolic gating motions. J. Neurosci. 24, 10475–10487 (2004).
Bacart, J., Corbel, C., Jockers, R., Bach, S. & Couturier, C. The BRET technology and its application to screening assays. Biotechnol. J. 3, 311–324 (2008).
Pfleger, K. D. G. & Eidne, K. A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174 (2006).
Yang, F. et al. The conformational wave in capsaicin activation of transient receptor potential vanilloid 1 ion channel. Nat. Commun. 9, 2879 (2018).
Lörinczi, É. et al. Involvement of the cysteine-rich head domain in activation and desensitization of the P2X1 receptor. Proc. Natl. Acad. Sci. 109, 11396–11401 (2012).
Andriani, R. T. & Kubo, Y. Voltage-clamp fluorometry analysis of structural rearrangements of ATP-gated channel P2X2 upon hyperpolarization. eLife 10, e65822 (2021).
Lee, H. S., Guo, J., Lemke, E. A., Dimla, R. D. & Schultz, P. G. Genetic incorporation of a small, environmentally sensitive, fluorescent probe into proteins in Saccharomyces cerevisiae. J. Am. Chem. Soc. 131, 12921–12923 (2009).
Pippel, A. et al. Localization of the gate and selectivity filter of the full-length P2X7 receptor. Proc. Natl. Acad. Sci. 114, E2156–E2165 (2017).
Dombkowski, A. A., Sultana, K. Z. & Craig, D. B. Protein disulfide engineering. FEBS Lett. 588, 206–212 (2013).
Hausmann, R. et al. A hydrophobic residue in position 15 of the rP2X3 receptor slows desensitization and reveals properties beneficial for pharmacological analysis and high-throughput screening. Neuropharmacology 79, 603–615 (2014).
Allsopp, R. C. & Evans, R. J. The intracellular amino terminus plays a dominant role in desensitization of ATP-gated P2X receptor ion channels. J. Biol. Chem. 286, 44691–44701 (2011).
Fountain, S. J. & North, R. A. A C-terminal lysine that controls human P2X4 receptor desensitization. J. Biol. Chem. 281, 15044–15049 (2006).
Hattori, M. & Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485, 207–212 (2012).
Browne, L. E. et al. P2X receptor channels show threefold symmetry in ionic charge selectivity and unitary conductance. Nat. Neurosci. 14, 17–18 (2010).
Stelmashenko, O. et al. Activation of trimeric P2X2 receptors by fewer than three ATP molecules. Mol. Pharmacol. 82, 760–766 (2012).
Kunori, S. et al. Involvement of prostaglandin F2α receptor in ATP-induced mechanical allodynia. Neuroscience 163, 362–371 (2009).
Nakagawa, T. et al. Intrathecal administration of ATP produces long-lasting allodynia in rats: differential mechanisms in the phase of the induction and maintenance. Neuroscience 147, 445–455 (2007).
Wang, W. et al. Exchange factor directly activated by cAMP–PKCε signalling mediates chronic morphine‐induced expression of purine P2X3 receptor in rat dorsal root ganglia. Br. J. Pharmacol. 175, 1760–1769 (2018).
Gever, J. R. et al. AF‐353, a novel, potent and orally bioavailable P2X3/P2X2/3 receptor antagonist. Br. J. Pharmacol. 160, 1387–1398 (2010).
Inoue, K. Nociceptive signaling of P2X receptors in chronic pain states. Purinergic Signal. 17, 41–47 (2020).
Li, H., Wei, W. & Xu, H. Drug discovery is an eternal challenge for the biomedical sciences. Acta Mater. Med. 1, 1–3 (2022).
Boué-Grabot, É., Archambault, V. & Séguéla, P. A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X2 ATP-gated channels. J. Biol. Chem. 275, 10190–10195 (2000).
Wang, J. et al. Druggable negative allosteric site of P2X3 receptors. Proc. Natl Acad. Sci. 115, 4939–4944 (2018).
Ma, X.-F. et al. The long β2,3-sheets encoded by redundant sequences play an integral role in the channel function of P2X7 receptors. J. Biol. Chem. 298, 102002 (2022).
Bond, P. S., Wilson, K. S. & Cowtan, K. D. Predicting protein model correctness in Coot using machine learning. Acta Crystallogr. Sect. D. Struct. Biol. 76, 713–723 (2020).
Liebschner, D., Afonine, P. V., Poon, B. K., Moriarty, N. W. & Adams, P. D. Improved joint X-ray and neutron refinement procedure in Phenix. Acta Crystallogr. Sect. D. Struct. Biol. 79, 1079–1093 (2023).
Lezon, T. R. & Bahar, I. Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys. J. 102, 1331–1340 (2012).
Zhang, Y., Zhang, S., Xing, J. & Bahar, I. Normal mode analysis of membrane protein dynamics using the vibrational subsystem analysis. J. Chem. Phys. 154, 195102 (2021).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Bakan, A., Meireles, L. M. & Bahar, I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 (2011).
Acknowledgements
This study was supported by funds from Hunan “Huxiang” High-level Talent Program (2021RC5013 to Y.Y.), Changsha “Jie Bang Gua Shuai” Major Science and Technology Programs (KQ2301004 to Y.Y.), National Natural Science Foundation of China (no. 32371289 to Y.Y. and no. 32000869 to J.W.), Innovation and Entrepreneurship (Shuangchuang) Program of Jiangsu Province (2020 and 2023 to Y.Y.), Natural Science Foundation of Jiangsu Province (BK20202002 to Y.Y.), “Xing Yao” Leading Scholars of China Pharmaceutical University (2021, Y.Y.), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-074, Y.Y.), the Medical Innovation and Development Project of Lanzhou University (lzuyxcx-2022-156, Y.Y.), and the Fundamental Research Funds for the Central Universities (2632024ZD10 to J. W).
Author information
Authors and Affiliations
Contributions
Y. Yu initiated the project. J. Wang and Y. Yu designed the research. Y.Y. Lin, Y. Lu, C.Y. Li, X.F. Ma, M.Q. Shao, Y.H. Gao, Y.Q. Zhang, H.N. Jiang, Y. Liu, Y. Yang, L.D. Huang, P. Cao, and J. Wang performed experiments. Y.Y. Lin, H.S. Wang, J. Wang. and Y. Yu wrote the manuscript. All authors discussed the results and commented on the manuscript. All authors declare no competing financial interests.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Samuel Fountain and Yoshihiro Kubo for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, YY., Lu, Y., Li, CY. et al. Finely ordered intracellular domain harbors an allosteric site to modulate physiopathological function of P2X3 receptors. Nat Commun 15, 7652 (2024). https://doi.org/10.1038/s41467-024-51815-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51815-7
- Springer Nature Limited