

Abstract
The signaling lipid phosphatidylinositol-4,5-bisphosphate (PIP2) regulates many ion channels. It inhibits eukaryotic cyclic nucleotide-gated (CNG) channels while activating their relatives, the hyperpolarization-activated and cyclic nucleotide-modulated (HCN) channels. The prokaryotic SthK channel from Spirochaeta thermophila shares features with CNG and HCN channels and is an established model for this channel family. Here, we show SthK activity is inhibited by PIP2. A cryo-EM structure of SthK in nanodiscs reveals a PIP2-fitting density coordinated by arginine and lysine residues from the S4 helix and the C-linker, located between voltage-sensing and pore domains of adjacent subunits. Mutation of two arginine residues weakens PIP2 inhibition with the double mutant displaying insensitivity to PIP2. We propose that PIP2 inhibits SthK by gluing S4 and S6 together, stabilizing a resting channel conformation. The PIP2 binding site is partially conserved in CNG channels suggesting the possibility of a similar inhibition mechanism in the eukaryotic homologs.
Similar content being viewed by others


Introduction
Each cell is confined by a membrane, largely composed of lipids that form a bilayer separating the intra- from the extra-cellular space. In addition to simply forming a barrier, lipids have diverse physicochemical properties and are involved in cellular signaling pathways, cellular trafficking, and modulation of protein function, among others1,2,3. Membrane proteins greatly rely on their surrounding lipid environment and protein-lipid interactions influence protein structure as well as function4. Regarding the modulation of protein structure and function, the best studied lipids are the signaling lipids phosphatidylinositol-4,5-bisphosphate (PIP2)5 and phosphatidic acid (PA)6.
PIP2 is the most abundant of the seven phosphorylated phosphatidylinositides (PI) in the cell and is mainly located in the inner leaflet of the plasma membrane7. It is produced from PI(4)P and PI(3,4,5)P3 by specific kinases and phosphatases (reviewed in Mandal5) and can exert a large influence on integral and peripheral membrane proteins even though it is only present in low abundance, usually accounting for less than 1.5 % of all cellular lipids8,9,10. Signaling via PIP2 is mainly facilitated by decreasing its concentration through receptor-mediated hydrolysis by phospholipase C (PLC) subtypes to diacylglycerol (DAG) and soluble inositol triphosphate (IP3). PLCs are activated by G-protein coupled receptor and receptor tyrosine kinase signaling pathways and can nearly deplete the cellular PIP2 pool in a matter of seconds11,12 allowing for fast regulation of PIP2-sensitive proteins.
For ion channels, inhibition as well as potentiation by PIP2 has been observed. The first examples of PIP2 regulation of ion channel activity include KATP and a Na+-Ca2+-exchanger where PIP2-dependent current rundown was observed in electrophysiological recordings13. Since then, over 80 channels11, including voltage-gated calcium and potassium channels14, TRP channels15,16, cyclic nucleotide-modulated channels17,18, and two-pore domain potassium channels19, as well as several transporters were found to be regulated by PIP2. This regulation is often conserved within channel families, but the family of cyclic nucleotide-modulated channels presents an exception. Here, the two subfamilies of cyclic nucleotide-gated (CNG) channels and hyperpolarization-activated and cyclic nucleotide-modulated (HCN) channels are oppositely affected by PIP2. CNG channels are strictly inhibited by PIP217 while HCN channels show a potentiating shift in their voltage dependence (towards less negative potentials) in the presence of PIP220,21. As such, PIP2 may take up important regulatory roles in phototransduction and pacemaking cascades.
SthK, a cyclic nucleotide-modulated ion channel from Spirochaeta thermophila22, shares functional and structural characteristics with both the CNG and HCN subfamilies23,24,25,26,27,28. The function of SthK is strongly potentiated by anionic lipids via a mechanism where lipids unlock a salt bridge at the bundle crossing shared with HCN channels29. However, it is not known whether PIP2 also modulates SthK and, if so, whether it leads to inhibition as for CNG or potentiation as for HCN channels. Thus, investigating how PIP2 affects SthK channels, may provide insights into the modulation of cyclic nucleotide-modulated channels, either CNG or HCN, by PIP2. Furthermore, although PIP2 is mainly found in eukaryotes, the presence of phosphatidylinositide lipids has been reported for Treponema pallidum, another member of the Spirochaetaceae family suggesting the possibility of a physiological regulation by such lipids in the host bacteria30 (the lipidome of S. thermophila is unknown at this time).
Here, we show inhibition of purified and liposome-reconstituted SthK ion channels when PIP2 is present in the liposomal membranes, using flux assays and single-channel recordings. The cryo-EM structure of SthK reconstituted in lipid nanodiscs reveals a density that fits PIP2 and that has not been observed for SthK in other lipid compositions. This density occupies a pocket formed by multiple Arg and Lys residues, two of which belong to the voltage sensing S4 helix. Removal of these two S4 helix arginine residues results in a PIP2-insensitive SthK channel. We propose a mechanism where PIP2 glues together the S4-S6 helices in a resting state position, which prevents the outward movement of the S4-S5-S6 helices and the upwards movement of the CNBD required to open the SthK pore. An equivalent binding site exists in closed CNG, but not closed HCN channel structures, possibly providing clues into the differential effect of PIP2 on the two channel subfamilies.
Results
PIP2 inhibits SthK channel activity
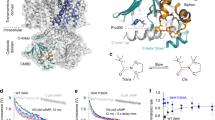
To investigate whether SthK is affected by the signaling lipid PIP2, we first monitored channel activity using a stopped-flow fluorescence flux assay. This ensemble method measures the flux of ions through reconstituted channels and allows for screening of channel activity in various lipid compositions. We chose a reference lipid composition of 3:1 DOPC:POPG, in which SthK shows intermediate activity29, allowing observation of both an increase and a decrease in activity upon PIP2 addition. The quenching kinetics in the presence of PIP2 were distinctively slower than in the absence of PIP2 at all concentrations tested (Fig. 1a, b, Supplementary Fig. 1e). This indicates that, similar to its action on eukaryotic CNG channels, PIP2 inhibits SthK.

a Representative quenching kinetics from a Tl+ flux assay measuring SthK activity in proteo-liposomes with a lipid composition of 3:1 DOPC:POPG (shades of grey and black) and 3:1 DOPC:POPG + 2.5 % PIP2 (shades of blue). Individual traces are: without quencher and without cAMP (lightest grey and lightest blue, no signal change observed), with quencher but without cAMP (medium light grey and medium light blue, slow, minimal, linear fluorescence decline due to leakage of Tl+ across liposomal membrane) and with quencher and 200 µM cAMP (black and dark blue, exponential quenching kinetics reflecting channel activity upon activation). b Initial Tl+ flux rates (Eqs. (1) and (2)) from experiments as in (a), averaged values ± SEM are normalized to the flux rate without PIP2 (n = 3 independent repeats), individual data points are in grey. See “methods” for normalization procedure. c Representative single-channel recordings of SthK in the presence of 300 µM cAMP reconstituted into 3:1 DOPC:POPG (top) and 3:1 DOPC:POPG + 5 % PIP2 (bottom) at +100 mV and -100 mV. Closed levels are indicated by dashed lines. d Normalized all-amplitude histograms from single-channel recordings as in (c) (see “methods”), with PIP2 (blue) and without PIP2 (black), at +100 mV. e Single-channel current-voltage (I/V) relation and (f) single-channel open probability (Po) obtained from recordings as in (c). Individual data points (light colors) and averaged values ± s.d. (dark colors) are depicted (number of independent repeats n is specified next to each data point in the corresponding color).
To describe the observed PIP2 inhibition in more detail, we next investigated SthK activity at the single-molecule level with electrophysiological recordings in planar lipid bilayers. In a 3:1 DOPC:POPG lipid bilayer, SthK showed the characteristic, asymmetric voltage-dependent behavior with smaller single-channel amplitude and higher open probability (Po) at positive voltages and larger amplitudes but a lower Po at negative voltages (Fig. 1c, f, Po = 0.25 ± 0.006 at + 100 mV, similar to previous reports29). Analysis of recordings for WT SthK in the presence of 5 % PIP2 revealed that the Po at + 100 mV is decreased about fourfold (Po = 0.058 ± 0.019) compared to recordings without PIP2 (Po = 0.25 ± 0.006, P < 0.001, Fig. 1f), while the single-channel current amplitude-voltage relationship remained unchanged (Fig. 1d, e).
Structural identification of the PIP2 binding site
We next sought to identify the PIP2 binding site on SthK to better describe the mechanism of its inhibitory action. We reconstituted SthK into nanodiscs containing 3:1 DOPC:POPG with additional 10 % PIP2. According to our stopped-flow results, 2.5 % PIP2 almost completely abolishes channel activity (Fig. 1b) and thus, 10 % PIP2 in the nanodiscs should be sufficient to saturate all PIP2 binding sites, potentially allowing for structural identification of the lipid.
The resulting cryo-EM density shows the tetrameric assembly of SthK where each subunit consists of a voltage sensing domain (S1–S4), a pore domain (S5–S6) and a cytosolic cyclic nucleotide-binding domain (CNBD) that is linked to the pore via a helical C-linker (Fig. 2a). Data were collected in the presence of saturating concentrations of cAMP and only one conformation, identical to previous closed state structures of SthK24, was identified (RMSD 0.915 Å, Supplementary Fig. 1, 2 and 7a).

a Density map of SthK in 3:1 DOPC:POPG + 10 % PIP2 nanodiscs. Protein density is shown in grey, lipid densities in yellow and PIP2 density in blue. b Zoom of inner leaflet lipids shown in (a). A PIP2 molecule is modeled into the lipid density (blue mesh) in the inner leaflet. Yellow mesh is an additional lipid binding along the S6 helix, previously found to modulate SthK activity. c Model of the PIP2 binding site highlighting residues coordinating PIP2. d Electrostatic surface potential of the binding pocket with blue representing positive and red negative potential. e The modeled PIP2 density (left) is absent from other SthK structures in different lipid compositions at similar resolution and threshold (middle: EMD-25916, right: EMD-24670).
The structure was refined to a resolution of 2.9 Å, which revealed several densities in the transmembrane region, that are distinct from the protein and are lipid-shaped (Fig. 2a, Supplementary Fig. 3). In search of potential densities for PIP2, we focused on the inner leaflet of the membrane, as this is the predominant location of PIP231, taking into account the following: i) inositol-4,5-bisphosphate is a large head group which should lead to a large volume of density in addition to the lipid tails (Fig. 2a, b, Supplementary Fig. 3), ii) PIP2 binding sites display characteristic physicochemical properties, with several positively charged residues forming a binding pocket that can accommodate the negatively charged PIP2 head group (Fig. 2c, d), and iii) the unique size of PIP headgroups allows them to reach out of the membrane by up to 17 Å31,32. We identified one density that satisfies these criteria, fits the model of PIP2 well (Fig. 2a–d, Supplementary Fig. 3), and, importantly, is absent from SthK maps without PIP2 at similar resolutions (Fig. 2e). In addition, the S2-S3 linker was not visible in previous structures but is now resolved, potentially due to its proximity to the PIP2 molecule (Supplementary Figs. 3b, c).
At this location, the phosphatidylinositol head group of PIP2 binds close to the intracellular ends of the S4 and S5 helices of one subunit and the C-linker of the adjacent subunit, with four positively charged amino acid residues forming the binding site (Fig. 2c, d). Arg120 and Arg124 from the S4 helix form salt bridges with the 3-phosphate of the lipid backbone as well as one phosphate from the lipid head group, respectively. The two residues from the C-linker, Lys229 and Arg268, both coordinate the second phosphate of the PIP2 head group. This PIP2-binding pocket was previously identified as a nexus for channel activation, ideally positioning PIP2 to modulate channel gating28,29.
Arginine residues on the S4 helix contribute to PIP2 binding
To functionally validate the structure-identified PIP2 binding site, we mutated the PIP2-coordinating arginine residues on the S4 helix (Fig. 3). SthK R120A was previously investigated due to its role in voltage sensing, where this mutation was reported to lead to a more active channel and cryo-EM analysis revealed a closed state and three different open states28. Stopped-flow measurements in the presence of PIP2 showed that while the inhibitory effect is reduced, higher PIP2 concentrations still lead to a decrease in activity in SthK R120A (Fig. 3g). The same was observed in single-channel recordings, where the Po at +100 mV is reduced by about 75 % in the presence of 5 % PIP2 (in the absence of PIP2 Po = 0.46 ± 0.07, in the presence of 5 % PIP2 Po = 0.11 ± 0.05, P < 0.0001, Fig. 3a, d and h, Supplementary Fig. 4a, d and g). This is consistent with a lower apparent affinity for PIP2 in SthK R120A, due to modifications in the PIP2-binding site.

Representative single-channel recordings for (a) SthK R120A, (b) SthK R124A, and (c) SthK R120A/R124A in the presence of 300 µM cAMP reconstituted into 3:1 DOPC:POPG (top traces) and 3:1 DOPC:POPG + 5 % PIP2 (bottom traces). Recordings at −100 mV and +100 mV are shown. Average Po ± s.d. of (d) SthK R120A, (e) SthK R124A and (f) SthK R120A/R124A at different voltages without PIP2 (squares) and in the presence of 5 % PIP2 (circles). The number of independent repeats, n, is indicated for each voltage in the plots. Individual data points are shown in grey. g Comparison of initial Tl+ flux rates of the different SthK variants. Averaged values ± SEM (n = 3 independent repeats) of measurements with different lipid compositions are normalized to the rate without PIP2 of the respective variant. Individual data points are shown in grey. h Normalized Po of single channel recordings at + 100 mV of different SthK variants in bilayers with or without 5 % PIP2. Averaged values ± s.d. are shown (number of independent repeats, n, for each condition is shown in the corresponding bars) as well as individual data points (grey). Statistical significance was assessed with a two-tailed unpaired t-test with a confidence level of 95 % resulting in P = 4 × 10−8 for WT SthK and P = 5 × 10−8 for SthK R120A. *** P < 0.001.
The cryo-EM structure of SthK R120A in the presence of 10 % PIP2 revealed a single closed state, in contrast to the multiple open states observed in the absence of PIP228, suggesting that PIP2 stabilizes the closed state, consistent with PIP2 inhibiting the channel (Fig. 4a). Furthermore, the cryo-EM map of SthK R120A revealed the presence of the same density in the PIP2 binding site, albeit weaker than in WT SthK (Fig. 4a, c, Supplementary Fig. 8). This suggests a lower PIP2 occupancy or a less well-defined positioning of PIP2, likely due to losing one of the interactions in the binding site.

a Density map of SthK R120A in 3:1 DOPC:POPG + 10 % PIP2 nanodiscs. Protein density is shown in light blue, lipid densities in yellow and PIP2 density in blue. Zoom focuses on the lipid densities. b Density map of SthK R120A/R124A in 3:1 DOPC:POPG + 10 % PIP2 nanodiscs. Protein density is shown in light green and lipid densities in yellow. Zoom focuses on the lipid densities. c Comparison of lipid densities in cryo-EM maps of WT SthK (top), SthK R120A (middle), and SthK R120A/R124A (bottom). Density of PIP2 is shown in blue and of the additional lipid bound along the S6 helix in yellow.
We next investigated the importance of Arg124, the second PIP2-interacting residue on the S4 helix. SthK R124A was still inhibited by PIP2, albeit much more weakly, when analyzed using the ensemble stopped-flow flux assay (Fig. 3g, Supplementary Figs. 4e, g). However, single-channel electrophysiology recordings revealed a nearly complete loss of inhibition by PIP2 at the tested concentration (Fig. 3b, e, h, Supplementary Fig. 4, in the absence of PIP2 Po = 0.32 ± 0.12, in the presence of 5 % PIP2 Po = 0.25 ± 0.1; differences are statistically insignificant with P = 0.4). In summary, removal of the positive charge at position 124 in the voltage sensing S4 helix considerably lowered the sensitivity of SthK R124A towards PIP2.
SthK R120A/R124A is insensitive to PIP2
Single arginine mutations in the PIP2-binding pocket led to partial reductions in PIP2 inhibition. We next asked whether the removal of both arginines would completely abolish the inhibitory effect of PIP2 on SthK channel activity within the experimentally accessible PIP2-concentration range. SthK R120A/R124A showed high activity in the stopped-flow assay, even in the presence of 2.5 % PIP2 in the liposomes (Fig. 3g, Supplementary Fig. 4). Similarly, the single channel activity of SthK R120A/R124A was virtually identical in the absence and presence of PIP2 (Fig. 3c, f, in the absence of PIP2 Po = 0.21 ± 0.04, in the presence of 5 % PIP2 Po = 0.25 ± 0.08 at +100 mV; differences are statistically insignificant with P = 0.4). Together, our functional results indicate that the structurally identified binding pocket is the PIP2 inhibitory site in SthK channels.
To further corroborate our functional results, we solved the cryo-EM structure of SthK R120A/R124A reconstituted into nanodiscs containing 10 % PIP2 (same conditions as used before for WT and for SthK R120A). In the resulting cryo-EM structure of SthK R120A/R124A we only identified a closed conformation similar to WT and SthK R120A in the presence of PIP2 (Po at 0 mV ~0.08, RMSD to WT SthK (PDB: 6CJU) 0.676 Å). However, despite PIP2 availability in the sample, this structure did not feature a density for the PIP2 headgroup and the S2-S3 linker could no longer be resolved, indicating that PIP2 binding is disrupted by the two arginine to alanine substitutions in the S4 helix (Fig. 4b, c, Supplementary Figs. 6, 7, and 8).
Discussion
Eukaryotic HCN and CNG channels are both regulated by the signaling lipid PIP217,20,21. Here we employed the bacterial homolog SthK to investigate PIP2-regulation in this channel family in molecular detail with biophysical and structural approaches using purified SthK protein in liposomes with controlled amounts of PIP2. We showed that PIP2 inhibits SthK function, similar to reports for CNG channels17. Functional experiments revealed that low concentrations of PIP2, that are in the range of the physiological abundance of this lipid (typically 0.5−1.5 % in the mammalian plasma membrane8,9,10), are sufficient to almost completely abolish channel function in vitro. Combining structural, functional, and mutational studies, we then identified and validated the binding site for PIP2 on SthK allowing us to propose a model by which PIP2 inhibits activity in this channel.
In SthK, PIP2 is positioned between the intracellular end of the S4 helix and the C-linker of an adjacent subunit. This puts the signaling lipid in a core region of conformational changes during channel opening. Upwards movement of the C-linker closer to the membrane and its simultaneous outwards rotation are required to generate tension on the intracellular end of the pore lining S6 helix, which is relieved by opening of the pore at the bundle crossing by rotation and dilation of S6 accompanied by small S4 and S5 movements28,29. The identified PIP2 binding pocket is positioned at the center of these conformational rearrangements and is comprised of the residues Arg120 and Arg124 of the S4 helix and Lys229 and Arg268 of the C-linker. In the presence of PIP2, the lipid holds both elements in place by gluing the voltage-sensing S4 helix and the C-linker together and restricting domain movements. The interactions with Arg residues on S4 hold the VSD in place and restrict movement of the S4-S5 linker. At the same time, the position of PIP2 inhibits the outward rotation of the S6 helix as well as upwards movement of the C-linker as it would lead to clashes with these elements in the open state (Fig. 5). The coordination of PIP2 as revealed in this study suggests a stabilization of a closed, resting state, resulting in the observed decrease in channel activity.

a Alignment of the PIP2 inhibited structure (grey) with SthK in the open conformation (blue, PDB: 7TJ6). Movements of the helices associated with channel opening are depicted by yellow arrows. The C-linker undergoes an upward movement leading to an outward rotation of the S6 helix that can only be facilitated by outward movements of the lower S4-S5 helices. b PIP2 binding in the closed state (top) and superimposed on the open state (bottom). In the closed state, Arg residues on S4 electrostatically coordinate PIP2 (green dashes) while in the open state the distance is too far. In addition, steric clashes are observed between PIP2 and the C-linker (red traffic circles). c Alignment of the SthK PIP2 binding site with the structure of human CNGA1 (PDB: 7LFT). Positively charged residues responsible for PIP2 binding in SthK are labelled and three of them are conserved in CNGA1.
Recently, PIP2 binding and regulation of channel function via the S4-S5 linker was observed for voltage-gated potassium channels of the Kv7 family33,34. PIP2 activates Kv7 and is necessary for channel opening. This is different from the inhibitory effect observed here and might be due to different channel architectures. The VSDs in Kv7 channels are domain-swapped while SthK is a nondomain-swapped channel, leading to different roles of the S4-S5 linker for channel activation. Further studies will be needed to reveal whether differences in VSD organization directly correlate to different functional effects of PIP2 on ion channel activity.
Lipid modulation of membrane proteins is not limited to interactions with VSDs. Previously, a broader analysis of lipid regulation of SthK revealed that anionic lipids bind with high affinity to SthK and potentiate channel function with the efficacy of activation correlating to the electronegativity of the lipid head group29. The mechanism of activation relies on binding of lipids along the pore-lining S6 helices resulting in the positioning of their head groups near a salt bridge at the bundle crossing. Anionic lipids preferentially bind in this location and electrostatically destabilize this salt bridge, making it easier for the channel to open. In contrast, although PIP2 also has a highly negatively charged head group, it exerts the opposite effect on SthK function and efficiently inhibits channel activity. In all SthK structures a lipid binds along the S6 helix, independent of the lipid composition, including the structures solved in this study. Our data show that the S6 lipid and PIP2 bind at distinct sites, which can explain the different functional effects of anionic lipids on SthK function. The different binding locations can be explained by the different chemical structures of the two lipids. PA lacks a head group and thus has minimal steric requirements which allows binding in an ideal distance to the salt bridge at the intracellular end of S629, while the doubly phosphorylated inositol of PIP2 is a considerably larger head group which cannot fit in a similar position as PA. Together, SthK provides detailed insights into how different signaling lipids can regulate the same protein in opposite ways.
Similar to our findings for SthK, eukaryotic CNG channels are inhibited by PIP217. Given the high sequence and structure homology between these proteins22,23,24, it is possible that a similar mechanism as identified for PIP2 modulation of SthK may also apply to eukaryotic CNG channels. Three of the four residues that form the PIP2 binding pocket in SthK are conserved in CNG channels (Fig. 5c and Supplementary Fig. 9) and are similarly positioned in available structures. This pocket in CNGA1 closely resembles the PIP2 binding site of SthK R120A (Fig. 5c). The fourth residue is a glutamate in CNG channels. However, a structural alignment between SthK and CNGA1 shows that the glutamate residue in CNGA1 points away from the potential PIP2 binding site and is in salt bridge forming distance to Arg218 on the S2 helix (Supplementary Fig. 10a). As such, the environment in CNGA1 is similar to the environment in SthK R120A, which is efficiently inhibited by PIP2, and it is conceivable that a similar mechanism of inhibition applies to SthK and CNG channels. This may add another layer of CNG channel regulation by phosphoinositides to the previously identified N- and C-terminal interactions in CNGA3 channels that influence cyclic nucleotide affinity and efficacy18,35. In the plasma membrane of the photoreceptor outer segment, CNG channels are responsible for depolarization during darkness (i.e. dark current). High concentrations of cGMP activate the channel resulting in influx of cations. After light stimulation, a G-protein coupled signaling cascade is initiated that results in activation of a cGMP phosphodiesterase (PDE6)36. The subsequent cGMP hydrolysis leads to closing of CNG channels. It was shown that PDE6 activity is strongly stimulated by PIP2 in the membrane, leading to a faster degradation of the cGMP pool37. By stimulating the hydrolysis of cGMP and, simultaneously, directly inhibiting CNG channels, PIP2 could provide a fast response to changing light levels.
On the other hand, HCN channels are activated by PIP220,21, and only two of the four PIP2 binding residues are structurally conserved. While the two positively charged residues of the C-linker in HCN channels are similarly positioned, the lower part of the HCN S4 helix does not contain any Arg or Lys residues in the depolarized state, thus closely resembling SthK R120A/R124A (Supplementary Fig. 9 and 10b). This changes upon hyperpolarization when the S4 helix of HCN1 moves downwards resulting in a helix break38 and a shift of Arg276 to a position similar to Arg124 of SthK which is involved in coordinating the PIP2 headgroup (Supplementary Fig. 10c). Thus, it is possible that PIP2 binds in a similar overall location but in activated rather than closed HCN channels, stabilizing the activated conformation of the voltage sensor in HCN channels and thus the open state.
In summary, we here propose a model of PIP2 modulation for an ion channel in the family of cyclic nucleotide-gated channels. We show that SthK is strongly inhibited when reconstituted in a lipid composition containing PIP2 and identified a binding site comprised of residues of the lower S4 helix and the C-linker by cryo-EM. Substitutions of the S4 residues involved in PIP2 coordination weakened or eliminated the inhibitory effect by PIP2, validating the PIP2 binding site. Together, our results suggest a mechanism in which movements of the S4-S5 linker as well as upwards movement of the C-linker are inhibited by PIP2 binding and, thus, signal transmission from cAMP binding and pore opening is efficiently blocked. The results presented here add to our understanding of the interplay between protein structure and the surrounding lipid environment.
Methods
Expression and Purification of SthK
Expression and purification of SthK were performed as previously described23. In brief, SthK was expressed in E. coli C41 (DE3) cells (Lucigen) grown in 4 L LB media (with 100 µg/ml ampicillin) at 37 °C to a density of OD600 = 0.4, after which temperature was decreased to 20 °C. At OD600 = 0.8 protein expression was induced with 0.5 mM IPTG. 12 h after induction the cells were harvested by centrifugation (5000 g, 10 min, 4 °C) and resuspended in 50 ml breaking buffer (50 mM Tris, 100 mM KCl, pH 8) supplemented with 85 µg/ml PMSF (Roche), 0.95/1.4 µg/ml Leupeptin/Pepstatin (Roche), 1 mg DNaseI (Sigma-Aldrich), 1 mg lysozyme (Sigma-Aldrich), ultra cOmplete protease inhibitor (Roche) and 200 µM cAMP. The cells were broken by sonication (Sonic Desmembrator 500, Fisher Scientific) and the resulting lysate was solubilized with 30 mM β-DDM (n-dodecyl-β-d-maltopyranoside, Anatrace) for 1.5 h at 4 °C under constant agitation. Insoluble fragments and cell debris were separated by centrifugation (38000 g, 45 min, 4 °C) and the supernatant was filtered through a 0.22 µm membrane. A 5 ml HiTrap chelating HP column (GE Life Sciences) was charged with Co2+ and equilibrated with running buffer (20 mM HEPES, 100 mM KCl, 1 mM β-DDM, 200 μM cAMP, 40 mM imidazole, pH 7.8). The filtered lysate was loaded onto the column and washed with 75 ml running buffer before elution of the protein with elution buffer (20 mM HEPES, 100 mM KCl, 1 mM β-DDM, 200 μM cAMP, 300 mM imidazole, pH 7.8). The protein eluate was concentrated to about 10 mg/ml using a concentrator with a cut-off molecular weight of 100 kDa (Amicon Ultra, Millipore). Purification was finalized by gel filtration over a Superdex200 10/300 column (GE Lifescience, 10 mM HEPES, 100 mM KCl, 0.5 mM β-DDM, pH 7.4) at 4 °C. SthK from the peak fractions was collected, concentrated to 10 mg/ml, flash frozen in liquid nitrogen and stored at −80 °C.
The R120A mutation in the SthK gene was previously described28 and R124A as well as the double mutation R120A/R124A were introduced by Quickchange PCR (Q5 Polymerase, NEB). All SthK variants were expressed and purified using the same protocol and resulted in similar yields and purity (Supplementary Fig. 1).
Reconstitution of SthK into liposomes for the stopped-flow flux assay
All lipids are from Avanti and we used brain phosphatidylinositol-4,5-bisphosphate (PIP2).
To perform a stopped-flow Tl+ flux assay, SthK was reconstituted into large unilamellar vesicles (LUVs) containing the fluorophore ANTS (8-Aminonaphthalene-1,3,6-trisulfonic acid, Life Technologies) as previously described23,39. A total of 7.5 mg lipids (3:1 DOPC:POPG alone or with PIP2 added) in chloroform were dried in a glass tube under constant N2 gas stream to a thin film and subsequently further dried under vacuum overnight. Rehydration of the lipids was performed with reconstitution buffer (15 mM HEPES, 150 mM KNO3, pH 7.4) before solubilization with CHAPS (33 mM) by sonication. Purified protein (30 µg/mg lipid) and 280 μl ANTS (75 mM stock solution in ddH2O, pH 7.4) were added to the solubilized lipids and incubated for 10 min at room temperature after which another 250 μl ANTS were added to reach a final concentration of 25 mM. Detergent was removed with BioBeads (SM-2, BioRad). 750 mg BioBeads from a 50 % slurry in pre-mix buffer (10 mM HEPES, 140 mM KNO3, pH 7.4) were added and incubated for 3 h at 21 °C under constant agitation. Overnight, the supernatant containing the liposomes was stored in a new glass tube at 13 °C. Stored liposomes were briefly sonicated in a water bath sonicator before extrusion through a 0.1 µm filter (mini extruder, Avanti Polar Lipids). The liposomes were then passed over a PD-10 desalting column (GE Lifesciences) to remove extra-vesicular ANTS. For a good signal-to-noise ratio the liposomes were diluted 5-fold in pre-mix buffer right before measurements.
Stopped-flow flux assay
All experiments were performed with a SX.20 LED stopped-flow spectrophotometer (Applied Photophysics, ProDataSX software, Leatherhead, UK) in sequential mixing mode with fluorescence excitation at 360 nm and detection above 420 nm at 25 °C. In a first activation step, LUVs were mixed 1:1 with pre-mix buffer (10 mM HEPES, 140 mM KNO3, pH 7.4) containing 400 µM cAMP and incubated for different amounts of time (12 ms to 10 s). Following, a second 1:1 mixing step with quenching buffer (10 mM HEPES, 90 mM KNO3, 50 mM TlNO3, 200 µM cAMP, pH 7.4) was performed and ANTS fluorescence quenching (excitation 360 nm, 420 nm high-pass filter) was recorded for 1 s with 5000 data points. For each delay time at least 8 technical repeats were performed. Control measurements were conducted with recording buffer without quencher and without the activating ligand cAMP. Every experiment was repeated at least three times with samples from independent reconstitutions.
Stopped-flow data were analyzed as described using Matlab40. The first 100 ms of the fluorescence decay were fitted with a stretched exponential function (Eq. 1) due to different liposome size distribution and varying number of active channels per liposome. Determined parameters of the stretched exponential function were then used to calculate the rate of Tl+ influx at 2 ms (Eq.2).
With Ft, F0 and F∞ denoting the fluorescence at timepoint t, the initial and final fluorescence, respectively. τ is the time constant of the quenching reaction (in s), β the stretched exponential coefficient and kt the Tl+ influx rate (in s−1) at 2 ms. For each experimental repeat measurements in the presence and absence of PIP2 were performed side by side, using samples prepared from the same stock solutions. The obtained flux rates in the presence of PIP2 then were normalized to the respective measurement in the absence of PIP2 for each independent repeat separately.
Electrophysiology
Electrophysiologic single-channel recordings were performed in a horizontal lipid bilayer setup at room temperature. Liposomes for bilayer recordings were prepared by drying 5 mg of lipids (3:1 DOPC:POPG alone or with PIP2 added) under constant N2 gas stream in a glass tube to a thin film. Lipids were then rehydrated with reconstitution buffer (10 mM HEPES, 400 mM KCl, 5 mM NMDG (n-methyl-D-glucamine), pH 7.6) and solubilized with 33 mM CHAPS by sonication in a water bath sonicator. Purified SthK (50 µg/mg lipid) was added to the lipid mixture and incubated for 20 min. Liposomes were formed by detergent removal via gravity flow over an 18 ml G-50 (Sephadex G-50 fine beads, GE Life Sciences) column and collected in 500 µl fractions. Liposome-containing fractions were identified by turbidity and pooled, before preparation of 50 µl aliquots that were flash frozen in liquid nitrogen and stored at −80 °C.
In the horizontal bilayer setup, two chambers were separated by a partition with a 100 µm aperture. A small air bubble on the tip of a 10 µl pipette tip was used to paint a bilayer of DPhPC (1,2-diphytanoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids, 8 mg/ml in decane) over the aperture. Bilayer formation was monitored by capacitance measurements and good bilayers were selected to be between 35–60 pF. The two chambers of the horizontal setup were filled with filtered recording buffer (100 mM KCl, 10 mM HEPES, pH 7.4) with addition of 300 µM cAMP only to the bottom chamber. This ensures that only activity from channels oriented with the CNBD facing the bottom chamber is recorded.
The software Clampex 10.7.0.3 (Molecular Devices) was used for data acquisition in combination with an Axopatch 200B amplifier (Molecular Devices). Recordings were filtered online at 2 kHz with an eight-pole, low-pass Bessel filter and digitized at 20 kHz (Digidata 1440 A, Molecular Devices). Recorded single-channel traces were analyzed with Clampfit 10.7.0.3 (Molecular Devices). Statistical significance was tested with a two-tailed unpaired t-test with a confidence level of 95 % and P-values are indicated as follows: *** = P-value < 0.001, ** = P-value < 0.01, *= P-value < 0.05. The all-amplitude histograms were normalized to the total number of events, and the peak heights sum up to 1.
For single channel recordings of WT SthK in the presence of PIP2, we observed two different behaviors. The large majority of recordings (~90 %) had SthK-characteristic voltage dependence and very low open probability, in agreement with the phenotype observed in our ensemble assay (Fig. 1c, d). A few recordings (~10 %) did not show SthK-characteristic features, were neither voltage nor cAMP dependent, and had larger single-channel conductance and large open probability (~ 0.8 − 0.9). For these reasons we did not include them in our analysis.
Cryo-EM sample preparation and data collection
For structural studies with cryo-EM, SthK was reconstituted into nanodiscs. A lipid stock of 125 mM was prepared by drying a lipid mixture of 3:1 DOPC:POPG + 10 % PIP2 (65 % DOPC, 25 % POPG, 10 % PIP2) under a constant N2 gas stream to a thin film and further drying under vacuum overnight. Lipids were rehydrated with reconstitution buffer (10 mM HEPES, 100 mM KCl, pH 7.4) and solubilized with 250 mM Na-Cholate. The lipid stock was flash-frozen in liquid nitrogen and stored at −80 °C. The membrane scaffolding protein MSP1E3 was prepared as described41 and concentrated to 10 mg/ml in reconstitution buffer. Purified SthK prepared as above was concentrated to 25 mg/ml after gel filtration. Five identical reconstitutions were prepared simultaneously, each with a total volume of 50 µl and an SthK concentration of 100 µM. SthK, MSP1E3 and lipids were mixed 1:1.1:70 (molar ratio) in reconstitution buffer and incubated for 20 min at room temperature. Detergent was removed by addition of 20 mg SM-2 BioBeads (BioRad, pre-washed in reconstitution buffer) to each sample and incubated for 2 h at room temperature under gentle agitation. The supernatant was transferred to new reaction tubes with fresh BioBeads and detergent removal was continued overnight. All five samples were pooled and the BioBeads were washed with 250 µl reconstitution buffer, resulting in a final volume of 500 µl. The sample was filtered through a Spin-X column and nanodiscs with tetrameric SthK were separated by gel filtration using a Superose6 10/300 column (GE Life Sciences) equilibrated in reconstitution buffer. The reconstitution buffer for gel filtration was supplemented with 200 µM cAMP. The peak fraction of SthK nanodiscs was collected and concentrated to 30 mg/ml with 1 A280 = 1 mg. For cryo-EM grid preparation the final sample consisted of 7.5 mg/ml protein, 5 mM cAMP, 3 mM fluorinated Foscoline-8 (Anatrace) in reconstitution buffer.
Grids were prepared by plunge-freezing in liquid ethane using a Vitrobot Mark IV (FEI, Thermo Fischer). Gold grids (UltrAuFoil, R1.2/1.3, 300 mesh, Quantifoil) were glow-discharged (PELCO easiGlow Glow Discharge Cleaning System, 80 s, −25 mA). 3.5 µl of the final sample were applied onto the grid, incubated for 30 s at 25 °C and 100 % humidity and blotted for 2 s with a blot force of −4 before plunge freezing.
For WT SthK and SthK R120A, data were collected at a 200 kV Talos Arctica TEM (FEI, Thermo Fisher) in super resolution with a GatanK3 direct electron detector at 36,000x nominal magnification and a pixel size of 0.548 Å. Movies were recorded with an exposure rate of 21.26 e−/Å2/s and a nominal defocus range of 0.9–2.9 µm using Leginon42. A total of 48 frames per micrograph with 50 ms exposure per frame for a total exposure time of 2400 ms were collected.
For SthK R120A/R124A, data were collected at a Titan Krios G3i operated at 300 kV equipped with a GatanK3 direct electron detector and a Bioquantum energy filter (20 eV) using Leginon42. Movies were recorded in super resolution (105,000x nominal magnification, pixel size 0.4125 Å) at a frame rate of 40 ms. A total of 50 frames per movie were recorded at an exposure rate of 28.32 e−/Å2/s leading to an electron exposure of 56.64 e−/Å2.
Cryo-EM data processing
All datasets were analyzed in RELION4_beta43 following a general processing pipeline as described here. Detailed processing schemes for each protein analyzed in this study are included in the Supplementary Information (Supplementary Figs. 2, 5 and 6). Data collection parameters and structure validation are presented in Supplementary Table 1.
Movies were motion corrected with RELION’s own implementation of MotionCor2 and binned. CTF estimation without dose-weighting of the micrographs was performed with CTFFIND 4.144. Particles were picked using the software crYOLO45 with a general model without training and extracted with a 256 px box size. Junk particles were sorted out by 2D classification and a 3D refinement with an ab initio SthK model as reference in C1 was performed before further sorting particles by 3D classification without alignment. The selected particles were subjected to multiple rounds of CTF-refinement, Bayesian polishing and 3D refinement in C4, as no asymmetry was observed. After the resolution converged, the particles were transferred to CryoSPARC46 for non-uniform refinement, ab initio reconstructions and heterogeneous refinements. Particles were transferred back to RELION by means of csparc2star.py (part of UCSF pyem47). For final processing, iterative CTF-refinement, Bayesian polishing, and 3D refinement were performed. To account for overfitting, reconstruction with sidesplitter48 was integrated into the last round of 3D refinement.
This resulted in a final resolution of 2.9 Å for WT SthK with PIP2 from 1,059,574 particles following the gold standard FSC cut-off at 0.143. Similarly, SthK R120A with PIP2 led to a cryo-EM density with 3.0 Å global resolution (228,035 particles), and SthK R120A/R124A with PIP2 was resolved at 2.5 Å from 431,535 particles. Model building was performed based on densities after sharpening of unfiltered maps of the final 3D refinement with deepEMhancer49 while applying the same masks as used for 3D refinement.
Model building and validation
For model building a previously published SthK structure (PDB-ID: 6CJQ) was placed into the density using UCSF Chimera50 and manually adjusted in COOT51. Newly resolved residues of the S2-S3 linker were added for WT SthK and SthK R120A. Models were real-space refined in Phenix52,53 and further improved using ISOLDE54. cAMP and lipids were manually added to each model in COOT and outliers were fixed. Quality of the final models was validated with MolProbity55 against unfiltered half maps and the unsharpened full map. All structural figures were prepared using Chimera50 and ChimeraX56.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. The maps and models produced in this study have been deposited to the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB). Accession codes are: WT SthK in the presence of cAMP and PIP2 (EMD-43520, PDB: 8VT9); SthK R120A in the presence of cAMP and PIP2 (EMD-43521, PDB: 8VTA); SthK R120A/R124A in the presence of cAMP and PIP2 (EMD-43522, PDB: 8VTB). This study refers to the previously published cryo-EM maps of SthK with the accession codes EMD-7482, EMD-25916 and EMD-24670 as well as the previously published models of SthK (PDB: 6CJU, PDB: 6CJQ and PDB: 7TJ6), CNGA1 (PDB: 7LFT) and HCN1 (PDB: 5U6O and PDB: 6UQF). Raw traces of all functional recordings presented here are available from the corresponding author upon request. The source data underlying Figs. 1b, e, f, 3d–h and Supplementary Figs. 1a, d and e and 4a–c and g are provided as a Source Data file. Source data are provided in this paper.
References
van Meer, G., Voelker, D. R. & Feigenson, G. W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 (2008).
Fernandis, A. Z. & Wenk, M. R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 18, 121 (2007).
Manna, M., Nieminen, T. & Vattulainen, I. Understanding the role of lipids in signaling through atomistic and multiscale simulations of cell membranes. Annu. Rev. Biophysics 48, 421–439 (2019).
Levental, I. & Lyman, E. Regulation of membrane protein structure and function by their lipid nano-environment. Nat. Rev. Mol. Cell Biol. 24, 107–122 (2023).
Mandal, K. Review of PIP2 in cellular signaling, functions and diseases. Int J. Mol. Sci. 21, 8342 (2020).
Wang, X., Devaiah, S. P., Zhang, W. & Welti, R. Signaling functions of phosphatidic acid. Prog. Lipid Res 45, 250–278 (2006).
Dickson, E. J. & Hille, B. Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1–23 (2019).
McLaughlin, S., Wang, J., Gambhir, A. & Murray, D. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 (2002).
Finkelstein, S. et al. Phosphoinositide profile of the mouse retina. Cells 9, 1417 (2020).
Nasuhoglu, C. et al. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal. Biochem. 301, 243–254 (2002).
Jensen, J. B. et al. Biophysical physiology of phosphoinositide rapid dynamics and regulation in living cells. J. Gen. Physiol. 154, e202113074 (2022).
Falzone, M. E. & MacKinnon, R. Gβγ activates PIP2 hydrolysis by recruiting and orienting PLCβ on the membrane surface. Proc. Natl Acad. Sci. USA 120, e2301121120 (2023).
Hilgemann, D. W. & Ball, R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273, 956–959 (1996).
Whorton, M. R. & MacKinnon, R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 (2011).
Rohacs, T. Phosphoinositide regulation of TRPV1 revisited. Pflug. Arch. - Eur. J. Physiol. 467, 1851–1869 (2015).
Large, W. A., Saleh, S. N. & Albert, A. P. Role of phosphoinositol 4,5-bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium 45, 574–582 (2009).
Womack, K. B. et al. Do phosphatidylinositides modulate vertebrate phototransduction? J. Neurosci. 20, 2792–2799 (2000).
Dai, G. & Varnum, M. D. CNGA3 achromatopsia-associated mutation potentiates the phosphoinositide sensitivity of cone photoreceptor CNG channels by altering intersubunit interactions. Am. J. Physiol. Cell Physiol. 305, C147–C159 (2013).
Riel, E. B. et al. The versatile regulation of K2P channels by polyanionic lipids of the phosphoinositide and fatty acid metabolism. J. Gen. Physiol. 154, e202112989 (2021).
Pian, P., Bucchi, A., Robinson, R. B. & Siegelbaum, S. A. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 128, 593–604 (2006).
Zolles, G. et al. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron 52, 1027–1036 (2006).
Brams, M., Kusch, J., Spurny, R., Benndorf, K. & Ulens, C. Family of prokaryote cyclic nucleotide-modulated ion channels. Proc. Natl Acad. Sci. Usa. 111, 7855–7860 (2014).
Schmidpeter, P. A. M., Gao, X., Uphadyay, V., Rheinberger, J. & Nimigean, C. M. Ligand binding and activation properties of the purified bacterial cyclic nucleotide-gated channel SthK. J. Gen. Physiol. 150, 821–834 (2018).
Rheinberger, J., Gao, X., am Schmidpeter, P. & Nimigean, C. M. Ligand discrimination and gating in cyclic nucleotide-gated ion channels from apo and partial agonist-bound cryo-EM structures. eLife 7, e39775 (2018).
Kesters, D. et al. Structure of the SthK carboxy-terminal region reveals a gating mechanism for cyclic nucleotide-modulated ion channels. PloS one 10, e0116369 (2015).
Morgan, J. L. W., Evans, E. G. B. & Zagotta, W. N. Functional characterization and optimization of a bacterial cyclic nucleotide-gated channel. J. Biol. Chem. 294, 7503–7515 (2019).
Evans, E. G. B., Morgan, J. L. W., DiMaio, F., Zagotta, W. N. & Stoll, S. Allosteric conformational change of a cyclic nucleotide-gated ion channel revealed by DEER spectroscopy. Proc. Natl Acad. Sci. 117, 10839–10847 (2020).
Gao, X. et al. Gating intermediates reveal inhibitory role of the voltage sensor in a cyclic nucleotide-modulated ion channel. Nat. Commun. 13, 6919 (2022).
Schmidpeter, P. A. M. et al. Anionic lipids unlock the gates of select ion channels in the pacemaker family. Nat. Struct. Mol. Biol. 29, 1092–1100 (2022).
Belisle, J. T., Brandt, M. E., Radolf, J. D. & Norgard, M. V. Fatty acids of Treponema pallidum and Borrelia burgdorferi lipoproteins. J. Bacteriol. 176, 2151–2157 (1994).
Suh, B.-C. & Hille, B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev. Biophys. 37, 175–195 (2008).
Sansom, M. S. P. et al. Molecular simulations and lipid–protein interactions: potassium channels and other membrane proteins. Biochem. Soc. Trans. 33, 916 (2005).
Mandala, V. S. & MacKinnon, R. The membrane electric field regulates the PIP2-binding site to gate the KCNQ1 channel. Proc. Natl Acad. Sci. 120, e2301985120 (2023).
Pant, S. et al. PIP2-dependent coupling of voltage sensor and pore domains in Kv7.2 channel. Commun. Biol. 4, 1–14 (2021).
Dai, G., Peng, C., Liu, C. & Varnum, M. D. Two structural components in CNGA3 support regulation of cone CNG channels by phosphoinositides. J. Gen. Physiol. 141, 413–430 (2013).
Stryer, L. Cyclic GMP cascade of vision. Annu Rev. Neurosci. 9, 87–119 (1986).
He, F., Mao, M. & Wensel, T. G. Enhancement of phototransduction G protein-effector interactions by phosphoinositides. J. Biol. Chem. 279, 8986–8990 (2004).
Lee, C.-H. & MacKinnon, R. Voltage sensor movements during hyperpolarization in the HCN channel. Cell 179, 1582–1589.e7 (2019).
Posson, D. J., Rusinova, R., Andersen, O. S. & Nimigean, C. M. Stopped-flow fluorometric ion flux assay for ligand-gated ion channel studies. Methods Mol. Biol. 1684, 223–235 (2018).
Ingólfsson, H. I. & Andersen, O. S. Screening for small molecules’ bilayer-modifying potential using a gramicidin-based fluorescence assay. Assay. Drug Dev. Technol. 8, 427–436 (2010).
Ritchie, T. K. et al. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem J. 478, 4169–4185 (2021).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Wagner, T. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Asarnow, D., Palovcak, E. & Cheng, Y. asarnow/pyem: UCSF pyem v0.5. Zenodo. https://doi.org/10.5281/ZENODO.3576630 (2019).
Ramlaul, K., Palmer, C. M., Nakane, T. & Aylett, C. H. S. Mitigating local over-fitting during single particle reconstruction with SIDESPLITTER. J. Struct. Biol. 211, 107545 (2020).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Compt. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D: Struct. Biol. 74, 531–544 (2018).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Cryst. D. 74, 519–530 (2018).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
Cryo-EM data collection was performed at NYU Langone Health’s Cryo-Electron Microscopy Laboratory (RRID: SCR_019202) with assistance from William Rice, Bing Wang, and Alice Paquette. The work presented here was sponsored in part by the National Institutes of Health (NIH, GM124451 to CN), and the German Academic Exchange Service (DAAD, to OT).
Author information
Authors and Affiliations
Contributions
P.S., C.N., and O.T. designed research. O.T., P.S., and Z.W. performed all experiments and analyzed the data. P.S., O.T., and C.N. interpreted the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Thon, O., Wang, Z., Schmidpeter, P.A.M. et al. PIP2 inhibits pore opening of the cyclic nucleotide-gated channel SthK. Nat Commun 15, 8230 (2024). https://doi.org/10.1038/s41467-024-52469-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52469-1
- Springer Nature Limited