
Abstract
Palbociclib combined with endocrine therapy is approved for treating patients with hormone-receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2−) advanced breast cancer; however, data on palbociclib combined with tamoxifen are limited. We investigated the efficacy and safety of palbociclib–tamoxifen in patients with HR+/HER2− advanced breast cancer. This double-blind phase 3 study included 184 women who were randomly assigned 1:1 to receive palbociclib–tamoxifen or placebo–tamoxifen. Pre/perimenopausal women also received goserelin. The primary endpoint was investigator-assessed progression-free survival (PFS). Secondary endpoints included overall survival (OS) and safety. Median PFS was 24.4 months (95% confidence interval [CI], 13.1–32.4) with palbociclib–tamoxifen and 11.1 months (95% CI, 7.4–14.6) with placebo–tamoxifen (hazard ratio [HR], 0.60; 95% CI, 0.43–0.85; P = 0.002). Palbociclib–tamoxifen improved PFS in patients who were treated with first-line or second-line endocrine therapy and pre-, peri-, and postmenopausal patients. Though OS data are still immature (median not reached in both groups), an overall risk reduction of 27% (HR, 0.73; 95% CI, 0.44–1.21) with palbociclib–tamoxifen was observed at the time of PFS analysis. The most common grade 3/4 adverse event with palbociclib–tamoxifen was neutropenia (89.0% [none were febrile] versus 1.1% with placebo–tamoxifen). There were no deaths owing to adverse events in either group. Among patients with HR+/HER2− advanced breast cancer, palbociclib–tamoxifen resulted in significantly longer PFS than tamoxifen alone. Early OS data showed a trend favoring palbociclib–tamoxifen. Trial registration: ClinicalTrials.gov number, NCT03423199. Study registration date: February 06, 2018.
Similar content being viewed by others


Introduction
The tumor characteristics of breast cancer in premenopausal women differ from those of postmenopausal women. Premenopausal patients tend to be diagnosed with more advanced cancer that is associated with worse clinical outcomes1. In Asian countries, the incidence of breast cancer is increasing, with a higher proportion of pre/perimenopausal cases than in Western countries2,3. However, treatment options for pre/perimenopausal women with breast cancer remain limited. When this study began in 2018, the combination of tamoxifen and luteinizing hormone-releasing hormone agonists (such as goserelin) for ovarian function suppression was one of the treatment options for first-line endocrine therapy (ET) for pre/perimenopausal patients with hormone receptor-positive (HR+)/human epidermal growth factor receptor 2–negative (HER2−) advanced breast cancer4. Tamoxifen was also a treatment option for postmenopausal patients with advanced breast cancer after treatment with aromatase inhibitors (AIs) or when AIs were intolerable5. Though ET remains the mainstay of treatment, there is inevitable resistance after a period of time, which has led to the development of targeted therapies6.
The cyclin-dependent kinase 4/6 (CDK4/6)-cyclin D axis is hyperactive in HR+/HER2− breast cancer7. Two pivotal studies, PALOMA-2 and PALOMA-3, demonstrated that adding the CDK4/6 inhibitor palbociclib to ET resulted in prolonged progression-free survival (PFS) over ET alone in patients with HR+/HER2− breast cancer8,9. CDK4/6 inhibitors, such as palbociclib, in combination with ET have become an established therapeutic approach for HR+/HER2− breast cancer7. Despite significant progress with CDK4/6 inhibitors, no phase 3 studies have evaluated the efficacy and safety of palbociclib in combination with tamoxifen in patients with HR+/HER2− advanced breast cancer regardless of menopausal status.
Here we report results from the PATHWAY trial (NCCH1607), which has investigated the benefit of adding palbociclib to tamoxifen in patients with HR+/HER2− advanced breast cancer, in both pre-, peri-, and postmenopausal patients versus tamoxifen alone.
Results
Patient characteristics
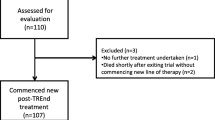
From February 15, 2018, to July 30, 2019, 184 patients were randomly assigned to receive palbociclib–tamoxifen (91 patients) or placebo–tamoxifen (93 patients) at 22 sites in 4 countries. All randomly assigned patients were treated (Fig. 1). The baseline demographic and clinical characteristics were balanced between the 2 treatment groups (Table 1). Overall, approximately 72% of the patients were postmenopausal and 61% received study treatment as first-line therapy.

ITT intent-to-treat; BICR blinded independent central review.
Progression-free survival
At the data cut-off date (September 15, 2022), 138 PFS events had occurred after a median duration of follow-up of 40.9 months for censored patients. The median PFS was 24.4 months (95% confidence interval [CI], 13.1–32.4) with palbociclib–tamoxifen, compared with 11.1 months (95% CI, 7.4–14.6) with placebo–tamoxifen (hazard ratio [HR], 0.60 [95% CI 0.43–0.85]; P = 0.002; Fig. 2a). The median PFS as assessed by blinded independent central review (BICR) was 35.0 months (95% CI, 14.9–not estimable [NE]) with palbociclib–tamoxifen, compared with 12.9 months (95% CI, 7.4–16.6) with placebo–tamoxifen (HR, 0.44 [95% CI 0.23–0.87]; P = 0.007; Fig. 2b).

a Progression-free survival by investigator assessment. b Progression-free survival by blinded independent central review. The tick marks indicate censored data. CI confidence interval; HR hazard ratio; NE not estimable; PFS progression-free survival.
In subgroup analyses, PFS favored palbociclib–tamoxifen over placebo–tamoxifen treatment across all subgroups, except for patients with bone-only metastasis. No treatment interactions were observed in all subgroups (Fig. 3). The small number of patients with bone-only metastasis (palbociclib–tamoxifen: 9 and placebo–tamoxifen: 7) made results difficult to interpret. In the pre/perimenopausal subgroup, median PFS was longer with palbociclib–tamoxifen (29.5 months; 95% CI, 16.2–NE) compared with placebo–tamoxifen (11.1 months; 95% CI, 3.9–18.4; HR, 0.38; 95% CI, 0.19–0.74) (Fig. 4a). Similarly, in the postmenopausal subgroup, median PFS was longer with palbociclib–tamoxifen (24.0 months; 95% CI, 11.2–32.3) than with placebo–tamoxifen (11.0 months; 95% CI, 5.7–14.9; HR, 0.68; 95% CI, 0.46–1.01) (Fig. 4b).

aBased on the registration system. bBrookmeyer and Crowley method. cHR and the corresponding 2-sided 95% CI for the palbociclib group relative to the placebo group were calculated by unstratified Cox proportional hazards model. dCategorized by IHC. Tests by IHC were not conducted in 4 patients in the Palbociclib-Tamoxifen group. eAll patients with HER2 2+ were negative by in situ hybridization. BRCA1/2 breast cancer 1 or 2 gene; CI confidence interval; ECOG Eastern Cooperative Oncology Group; ER estrogen receptor; ESR1 estrogen receptor 1 gene; ET endocrine therapy; HER2 human epidermal growth factor receptor 2-negative; HR hazard ratio; IHC immunohistochemistry; NE not estimable; PFS progression-free survival; PIK3CA phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha gene; PR progesterone receptor.

a Progression-free survival in pre/perimenopausal patients. b Progression-free survival in postmenopausal patients. c Progression-free survival in patients treated with first-line endocrine therapy. d Progression-free survival in patients treated with second-line endocrine therapy. The tick marks indicate censored data. CI confidence interval; HR hazard ratio; NE not estimable; PFS progression-free survival.
The first-line ET subgroup had a HR of 0.52 (95% CI, 0.33–0.82) with a median PFS of 29.5 months (95% CI, 16.8–38.0) with palbociclib–tamoxifen and 12.7 months (95% CI, 5.7–20.3) with placebo–tamoxifen (Fig. 4c). The second-line ET subgroup had a HR of 0.71 (95% CI, 0.42–1.19), median PFS was 11.2 months (95% CI, 5.8–29.3) with palbociclib–tamoxifen and 10.9 months (95% CI, 5.4–13.1) with placebo–tamoxifen (Fig. 4d).
Overall survival
The median overall survival (OS) was not reached in either the palbociclib–tamoxifen (95% CI, 47.2–NE) or the placebo–tamoxifen group (95% CI, 46.2–NE), with a HR of 0.73 (95% CI, 0.44–1.21; Fig. 5). The estimated survival rates (95% CI) in the palbociclib–tamoxifen and placebo–tamoxifen groups were 98.8% (92.0–99.8) and 93.5% (86.2–97.0) at 1 year, 93.0% (85.1–96.8) and 78.3% (68.4–85.4) at 2 years, and 79.1% (68.9–86.3) and 66.8% (56.1–75.5) at 3 years. Double-blinding of the study will be maintained until the final OS analysis.

Tick marks represent data censored at the last time the patient was known to be alive. CI confidence interval; HR hazard ratio; NE not estimable; NR not reached.
Objective response
The objective response rate in the intent-to-treat population was higher in the palbociclib–tamoxifen group at 44.0% (95% CI, 33.6–54.8) compared with 28.0% (95% CI, 19.1–38.2) in the placebo–tamoxifen group (Supplementary Table 1). Consistent with these findings, patients who had measurable disease had a more favorable response rate with palbociclib–tamoxifen at 53.4% (95% CI, 41.4–65.2) than with placebo–tamoxifen (34.2%; 95% CI, 23.7–46.0).
Clinical benefit
The clinical benefit rate was greater with palbociclib–tamoxifen treatment (75.8%; 95% CI, 65.7–84.2) versus placebo–tamoxifen (61.3%; 95% CI, 50.6–71.2).
Exposure and safety
The median (range) duration of treatment with palbociclib was 19.9 months (0.2–52.2), whereas placebo was administered for a median of 10.8 months (0.7–50.9). The median (range) duration of treatment with tamoxifen was 20.2 months (0.2–52.4) in the palbociclib–tamoxifen group and 11.0 months (1.0–50.9) in the placebo–tamoxifen group. There were 61 (67.0%) patients who needed at least 1 dose reduction of palbociclib. Of those patients, 38 (41.8%) had 2 dose reductions from 125 mg to 75 mg. The median relative dose intensity of palbociclib was 68.8% in the palbociclib–tamoxifen group and 98.7% for placebo in the placebo–tamoxifen group. The median relative dose intensities of tamoxifen were 99.2% and 99.8% in the palbociclib–tamoxifen and placebo–tamoxifen groups, respectively.
Adverse events (AEs) were reported in 89 patients in the palbociclib–tamoxifen group (97.8%) and 81 patients in the placebo–tamoxifen group (87.1%; Table 2), grade 3/4 AEs were reported in 85 (93.4%) and 19 (20.4%) patients. The most common grade 3/4 AEs were neutropenia (89.0%) and leukopenia (28.6%; none were grade 4 AEs) in the palbociclib–tamoxifen group. Febrile neutropenia was not reported in either treatment group. Grade 5 AEs (deaths) were not reported in either treatment group.
Serious AEs were reported in 17.6% of patients treated with palbociclib–tamoxifen and in 15.1% of patients treated with placebo–tamoxifen; 2.2% and 3.2% were reported as treatment-related serious AEs (Supplementary Table 2). AEs leading to treatment discontinuation were reported in 3.3% and 2.2% of patients in the palbociclib–tamoxifen and placebo–tamoxifen groups, respectively (Supplementary Table 3). One patient in each treatment group experienced a greater than 60 msec QTcF prolongation from baseline; there were no patients with postbaseline QTcF of more than 480 msec in either treatment group.
Subsequent line of therapy
At the data cut-off date, 70 and 84 patients had discontinued treatment with palbociclib–tamoxifen and placebo–tamoxifen, respectively. Subsequent anticancer therapy was reported in 64 (70.3%) and 80 (86.0%) patients in the palbociclib–tamoxifen and placebo–tamoxifen groups, respectively (Supplementary Table 4). CDK4/6 inhibitors as first subsequent therapy were received by 11 (12.1%) and 34 patients (36.6%), respectively.
Biomarkers
PFS was compared by baseline mutational status for specific genes in both treatment groups (Fig. 3). Regardless of PIK3CA mutation status, palbociclib–tamoxifen treatment trended in favor of improved PFS over placebo–tamoxifen. The small number of patients with ESR1 (palbociclib–tamoxifen: 12, placebo–tamoxifen: 9) or BRCA1/2 mutations (palbociclib–tamoxifen: 5, placebo–tamoxifen: 3) precluded comparison of PFS between the 2 treatment groups.
Discussion
PATHWAY is a phase 3 trial evaluating efficacy and safety of palbociclib in combination with tamoxifen. This trial achieved its primary endpoint, demonstrating both statistically significant and clinically meaningful improvements in PFS for patients with HR+/HER2− advanced breast cancer treated with palbociclib–tamoxifen compared with placebo–tamoxifen. A clinical benefit in PFS with palbociclib–tamoxifen treatment was observed both in patients who were treated with first- and second-line ET and regardless of menopausal status.
This study included a heterogeneous population of pre- and postmenopausal patients receiving first-line ET as well as those resistant to AIs and receiving second-line ET. Although the number of pre/perimenopausal patients in this study is small, there was a 62% lower relative risk of progression or death with palbociclib–tamoxifen in this population compared with those receiving placebo–tamoxifen (HR, 0.38 [95% CI, 0.19–0.74]). In MONALEESA-7, a phase 3, placebo-controlled trial which included 672 premenopausal women with advanced, HR-positive breast cancer, the HR of PFS in the subgroup of ribociclib in combination with tamoxifen was 0.59 (95% CI, 0.39–0.88)10. However, cross-trial comparisons should be interpreted with caution because of differences in study designs and patient populations. Ribociclib is approved in the Republic of Korea, Taiwan, and Singapore, but its development has been halted in Japan due to dose-limiting toxicities that led to a different recommended phase 2 dose11. Since tamoxifen remains a valid treatment option for both pre-, peri-, and postmenopausal women with HR+/HER2− advanced breast cancer, given the higher risk of prolonged QTcF with tamoxifen–ribociclib and higher risk of venous thromboembolic events with tamoxifen–abemaciclib10,12, new treatment options that reduce the risk of serious AEs would be of clinical importance. Beyond clinical trials, the incidence of thromboembolic events with CDK4/6 inhibitors remains a topic of research interest. In a real-world study of 266 patients with breast cancer receiving CDK4/6 inhibitors, thromboembolic events including arterial and venous events were more frequent with palbociclib and ribociclib than with abemaciclib; however, palbociclib comprised the vast majority of CDK 4/6 inhibitors in the study, making comparisons between the agents challenging13. Furthermore, the incidence of venous thromboembolic events reported in this study was higher than that observed in a meta-analysis of randomized controlled trials in patients treated with CDK4/6 inhibitors14. Therefore, this finding needs to be validated by other real-world data; nevertheless, physicians monitor thromboembolism in every patient who receives a CDK4/6 inhibitor.
Though, in postmenopausal patients with advanced breast cancer, the use of tamoxifen as the first-line ET backbone is not common and limited, the results of the SONIA trial suggest that an AI alone may remain a treatment option for first-line treatment15. Therefore, for those who have failed AI, or who are unable to maintain adherence due to side effects such as arthralgia and osteoporosis, this study showed the efficacy of palbociclib plus tamoxifen in postmenopausal breast cancer and supports the use of this combination therapy as one of the treatment options.
The PATHWAY study had limited enrollment of premenopausal patients who have received AI plus ovarian function suppression, either as adjuvant ET or as first-line ET in advanced setting. However, following the results of SOFT and TEXT trials16,17, the use of AI plus ovarian function suppression as adjuvant ET in premenopausal patients has been increasing, particularly for those with high risk of recurrence. Since incomplete ovarian function suppression remains a concern for some patients18, tamoxifen plus ovarian function suppression plus palbociclib is a reasonable choice once patients develop disease recurrence with AI plus ovarian function suppression. In addition, given that palbociclib and tamoxifen appear to be a safe combination therapy, it could be used as a bridge when starting ovarian suppression and waiting for the ovaries to be suppressed (prior to being able to initiate treatment with an AI).
Although fulvestrant was a strongly recommended treatment for patients previously treated with ET, tamoxifen, too, remained a treatment option based on patient preference. Hence, patients who were being considered for tamoxifen were included in this study. In addition, several agents such as alpelisib or capivasertib are now being available for use in combination with fulvestrant in the subsequent line after treatment with ET or ET plus CDK4/6 inhibitor19,20,21. The combination of palbociclib and tamoxifen provides value in expanding the treatment options for HR+/HER2− advanced breast cancer beyond palbociclib combinations with AI or fulvestrant.
The OS data had not matured, and median OS was not reached in either treatment group at the time of the PFS analysis; however, a trend favoring the palbociclib–tamoxifen group was observed. The clinical benefit of palbociclib in combination with tamoxifen was also maintained across all secondary endpoints evaluated including objective response and clinical benefit response, confirming the robustness of the results.
Attributable toxicities with palbociclib in combination with tamoxifen were manageable with dosing interruptions and/or dose reduction of palbociclib. The AEs reported were generally consistent with the known safety profile of palbociclib in combination with other ET. There were no unexpected major safety findings in this study population. Neutropenia was the most common AE among patients receiving palbociclib in this study. The incidence of neutropenia in this study were similar to those reported in the Asian-race subgroup analysis from PALOMA-2 and PALOMA-322,23. QTcF interval prolongation with palbociclib–tamoxifen was not frequent.
In PALOMA-3, a clinical benefit was reported with palbociclib–fulvestrant regardless of PIK3CA- and ESR1-mutation status24,25. Although a clinical benefit was observed with palbociclib–tamoxifen treatment regardless of PIK3CA mutation status, interpretation by ESR1- or BRCA1/2-mutation status is limited by the small sample size of patients.
A possible limitation is that this study included only patients of Asian origin; therefore, caution is needed when considering how these results may apply to other racial groups. However, no significant racial and ethnic differences in efficacy parameters have been reported in previous international studies of palbociclib8,9,22,23.
In conclusion, data from the PATHWAY trial showed the clinical benefit of treatment with palbociclib in combination with tamoxifen in patients with HR+/HER2− advanced breast cancer. Although OS results are not yet mature, early OS data showed a trend favoring palbociclib in combination with tamoxifen over placebo with tamoxifen.
Methods
Trial design
PATHWAY (ClinicalTrials.gov number: NCT03423199; study registration date: February 06, 2018) is an international (Japan, Republic of Korea, Taiwan, and Singapore), multicenter, randomized, double-blind, placebo-controlled, phase 3 clinical trial. The protocol was approved by the institutional review board at each trial site, and all patients provided written informed consent.
Patients
Pre-, peri-, or postmenopausal women with locally advanced or metastatic HR+/HER2− breast cancer were eligible if they were candidates to receive tamoxifen as first-line or second-line ET for advanced disease. Patients were excluded if they had received prior treatment with a CDK4/6 inhibitor or tamoxifen. Patients who had disease progression more than 12 months after the completion of adjuvant therapy with tamoxifen were eligible. One previous line of chemotherapy for advanced disease was allowed.
Randomization and treatments
Patients were randomly assigned 1:1 to receive either palbociclib–tamoxifen or placebo–tamoxifen ± goserelin (Supplementary Fig. 1). Patients were stratified by treatment history with ET (first-line or second-line) and by menopausal status (pre/perimenopausal versus postmenopausal) at randomization. Study treatment that was given after recurrence during treatment or within 12 months after completion of adjuvant ET was defined as second-line ET. Patients received either palbociclib (starting dose, 125 mg/day) or placebo orally once daily on day 1 to day 21 followed by 7 days off-treatment for each 28-day cycle, plus tamoxifen 20 mg orally once daily (continuously). Pre/perimenopausal women additionally received goserelin subcutaneously 3.6 mg given every 4 weeks, or a long-acting form 10.8 mg given every 12 weeks.
Dose adjustment was permitted for palbociclib/placebo only. Dose reduction of palbociclib by 1 dose level (to 100 mg/day), and, if needed, by 2 dose levels (to 75 mg/day) was recommended depending on type and severity of the toxicity. Patients were to receive assigned treatment until either disease progression, unacceptable toxicity, death, or withdrawal of consent.
Assessments
Tumor assessments were performed per Response Evaluation Criteria in Solid Tumors version 1.1 at baseline, every 8 weeks for the first 1.5 years, and then every 12 weeks thereafter. Laboratory tests and vital signs were performed on day 1 and day 15 of the first 3 cycles and day 1 of subsequent cycles. AEs were graded with the Common Terminology Criteria for Adverse Events version 4.0. Plasma samples for circulating tumor DNA (ctDNA) analysis were collected on cycle 1 day 1, cycle 2 day 15, and the end of treatment. Mutations in the PIK3CA, ESR1, and BRCA1/2 (somatic or germline mutation) genes were detected using Guardant360 (Guardant Health, Inc., CA, USA).
Outcome measures
The primary endpoint was investigator-assessed PFS, defined as the time from randomization to radiological or clinical disease progression or death due to any cause, whichever occurred first. Secondary endpoints included OS, objective response, duration of response, clinical benefit (defined as a complete response, partial response, or stable disease for 24 weeks or longer), pharmacokinetics, safety, and patient-reported outcomes. Exploratory endpoint included biomarkers obtained through blood sampling. PFS was also assessed by BICR for a randomly selected subgroup of patients (~40%).
Trial oversight
The trial was conducted in accordance with the International Council for Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki. The trial sponsor (National Cancer Center Hospital, Japan; IRB approval number: T4467) and the principal academic investigators designed the trial; Pfizer provided the trial drugs and placebo. Operation of this trial was reported elsewhere26. The trial was supervised by institutional review boards in Japan: National Cancer Center Hospital, Aichi Cancer Center Hospital, National Hospital Organization Osaka National Hospital, National Hospital Organization Hokkaido Cancer Center, National Cancer Center Hospital East, National Hospital Organization Shikoku Cancer Center, Chiba Cancer Center, Kanagawa Cancer Center, Toranomon Hospital, Hyogo Cancer Center, Kindai University Hospital, and Kyusyu Cancer Center; Republic of Korea: Severance Hospital, Seoul National University Hospital, Asan Medical Center, National Cancer Center, Seoul National University Bundang Hospital, and Ajou University Hospital; Taiwan: National Taiwan University Hospital, Koo Foundation Sun Yat-Sen Cancer Center, and Taipei Veterans General Hospital; and Singapore: Singhealth Centralised. Data were collected by the sponsor and analyzed in collaboration with the authors. An independent safety monitoring committee reviewed safety data on an ongoing basis. The authors vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol.
Statistical analysis
A total of 138 PFS events were required based on the 1:1 randomization to have an 80% power to detect a 38% reduction in the risk of disease progression or death for the palbociclib–tamoxifen group, with a 1-sided log-rank test at a significance level of 0.025. Assuming a 10% dropout rate, approximately 180 patients were planned to be randomly assigned to a treatment. The intent-to-treat population was the primary population for evaluating all efficacy endpoints and patient characteristics; the as-treated population was the primary population evaluating treatment administration, compliance, and safety.
A stratified log-rank test was used to compare PFS between the 2 treatment groups. The 95% CI of median PFS was calculated by the Brookmeyer and Crowley method. The stratified Cox Proportional hazards model was used to estimate the treatment HR and the corresponding 95% CI. The stratified analysis was performed with three strata: pre/perimenopausal, postmenopausal, and first-line ET, and postmenopausal and second-line ET. The stratified analysis was originally planned with four strata; however, because of the small sample size of three patients in the pre/perimenopausal and second-line ET strata, the statistical analysis plan was updated before the database lock to combine the pre/perimenopausal and first-line ET and pre/perimenopausal and second-line ET strata. Treatment—Factor interactions were explored for the factors used for subgroup analysis in the PFS. The P values for interactions were calculated by Cox proportional hazard models including the treatment, factor, and their interaction term.
The current OS outcome was evaluated at the time of PFS analysis. The final OS analysis will be performed at least 3 years from randomization of the last patient. OS was evaluated using a stratified log-rank test with HRs and 95% CIs calculated as described for PFS. Reported P values for PFS analyses are 1-sided and P values for interactions are 2-sided. All analyses were performed with SAS, version 9.4 or higher (SAS Institute, Cary, NC).
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request and with the permission of National Cancer Center Hospital.
References
Bardia, A. & Hurvitz, S. Targeted therapy for premenopausal women with HR(+), HER2(−) advanced breast cancer: focus on special considerations and latest advances. Clin. Cancer Res. 24, 5206–5218 (2018).
Yap, Y. S. et al. Insights into breast cancer in the east vs the west: a review. JAMA Oncol. 5, 1489–1496 (2019).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Cardoso, F. et al. ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Ann. Oncol. 25, 1871–1888 (2014).
Thurlimann, B. et al. Anastrozole (‘Arimidex’) versus tamoxifen as first-line therapy in postmenopausal women with advanced breast cancer: results of the double-blind cross-over SAKK trial 21/95-a sub-study of the TARGET (Tamoxifen or ‘Arimidex’ Randomized Group Efficacy and Tolerability) trial. Breast Cancer Res. Treat. 85, 247–254 (2004).
Osborne, C. K. & Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62, 233–247 (2011).
Spring, L. M. et al. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet 395, 817–827 (2020).
Finn, R. S. et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Turner, N. C. et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 373, 209–219 (2015).
Tripathy, D. et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 19, 904–915 (2018).
Yap, Y. S. et al. Ribociclib, a CDK 4/6 inhibitor, plus endocrine therapy in Asian women with advanced breast cancer. Cancer Sci. 111, 3313–3326 (2020).
Hamilton, E. et al. nextMONARCH: abemaciclib monotherapy or combined with tamoxifen for metastatic breast cancer. Clin. Breast Cancer 21, 181–190.e182 (2021).
West, M. T. et al. CDK 4/6 inhibitors are associated with a high incidence of thrombotic events in women with breast cancer in real-world practice. Eur. J. Haematol. 106, 634–642 (2021).
Thein, K. Z. et al. Venous thromboembolism risk in patients with hormone receptor-positive HER2-negative metastatic breast cancer treated with combined CDK 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: a systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res. Treat. 183, 479–487 (2020).
Sonke, G. S. et al. Primary outcome analysis of the phase 3 SONIA trial (BOOG 2017-03) on selecting the optimal position of cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors for patients with hormone receptor-positive (HR+), HER2-negative (HER2-) advanced breast cancer (ABC). J. Clin. Oncol. 41, LBA1000 (2023).
Pagani, O. et al. Adjuvant exemestane with ovarian suppression in premenopausal breast cancer. N. Engl. J. Med. 371, 107–118 (2014).
Pagani, O. et al. Adjuvant exemestane with ovarian suppression in premenopausal breast cancer: long-term follow-up of the combined TEXT and SOFT trials. J. Clin. Oncol. 41, 1376–1382 (2023).
Lu, Y. S., Wong, A. & Kim, H. J. Ovarian function suppression with luteinizing hormone-releasing hormone agonists for the treatment of hormone receptor-positive early breast cancer in premenopausal women. Front. Oncol. 11, 700722 (2021).
Andre, F. et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Andre, F. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann. Oncol. 32, 208–217 (2021).
Turner, N. C. et al. Capivasertib in hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 388, 2058–2070 (2023).
Im, S. A. et al. Palbociclib plus letrozole as first-line therapy in postmenopausal Asian women with metastatic breast cancer: results from the phase III, randomized PALOMA-2 study. J. Glob. Oncol. 5, 1–19 (2019).
Iwata, H. et al. PALOMA-3: phase III trial of fulvestrant with or without palbociclib in premenopausal and postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer that progressed on prior endocrine therapy-safety and efficacy in Asian patients. J. Glob. Oncol. 3, 289–303 (2017).
Fribbens, C. et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast. Cancer. J. Clin. Oncol. 34, 2961–2968 (2016).
Cristofanilli, M. et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 17, 425–439 (2016).
Hata, T. et al. Regulatory and operational challenges in conducting Asian International Academic Trial for expanding the indications of cancer drugs. Clin. Transl. Sci. 14, 1015–1025 (2021).
Acknowledgements
We thank the patients who participated in the trial, their families, the trial investigators and co-investigators, and the study teams at each of the participating sites. The trial was conducted as part of a Clinical Research Collaboration with National Cancer Center Hospital in Japan, Asian academia, and Pfizer. National Cancer Center Hospital is a regulatory sponsor for this study, and Pfizer provided the financial support and study drug. Writing assistance for the first draft of the manuscript was provided by Mahesh Chemudupati, PhD, from Oxford PharmaGenesis Inc. (Newtown, PA, United States), which was in accordance with Good Publication Practice (GPP 2022) and funded by Pfizer Inc. This study was funded by Pfizer Inc. The Clinical Research Support Office was funded/supported by the National Cancer Center Hospital and the Japan Agency for Medical Research and Development [grant numbers 15lk0803003j0001, 16lk0803003j0002, 16lk1203001j0001, 17lk1503003j0001, 18lk1503003j0002, 19lk1503003j0003, 20lk1503003j0004, 20lk0201002j0001, 21lk1503003j0005, 21lk0201005j0001, 22lk1503003j0006, 22lk0201007j0001]. Role of the funding body: Pfizer Inc. had a role in the review of study design, interpretation of data, and the writing of this manuscript and had no role in the data collection or analysis. The Japan Agency for Medical Research and Development had no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
All the authors contributed to drafting the manuscript, provided critical review, and gave final approval to submit the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
Authors declare no competing non-financial interests but the following competing financial interests: Emi Noguchi: speaker honoraria from AstraZeneca, Pfizer, Chugai Pharma, Novartis. Takashi Yamanaka: honoraria from AstraZeneca, Chugai Pharma, Daiichi Sankyo, Eisai, Lilly, Kyowa Kirin, Novartis, Pfizer; consultancy/advisory fees from Daiichi Sankyo. Hirofumi Mukai: honoraria or lecture fee from Takeda and Taiho Pharmaceutical. Naohito Yamamoto: research funding from AstraZeneca and Pfizer. Chi-Feng Chung: advisory role/speaker honoraria: AstraZeneca, Daiichi Sankyo, Pfizer, Roche, and Novartis. Yen-Shen Lu: grant support/research collaborations: Novartis, Pfizer, MSD, Roche, AstraZeneca, ACT Genomics, Advisory board/speaker invitation: Novartis, Pfizer, MSD, Roche, AstraZeneca, Eisai, Eli Lily, Daiichi Sankyo and EuroPharma, Conference support: Novartis, Eisai, and MSD. Dwan-Ying Chang: advisory role/speaker honoraria: Amgen, AstraZeneca, Daiichi Sankyo, Eisai, Eli Lilly, MSD, Novartis, ONO pharma, Pierre-Faber, Pfizer, Roche, Sanofi, TTY Biopharm, and EuroPharma, Conference support: Pfizer, Roche, AstraZeneca, Travel accommodation: Pfizer. Joohyuk Sohn: research funding from MSD, Roche, Novartis, Lilly, Pfizer, Daiichi Sankyo, AstraZeneca, GSK, Sanofi, Boehringer Ingelheim, and Seagen. Kyung-Hun Lee: honoraria or lecture fees from AstraZeneca, Eli Lilly, Novartis, Pfizer, and Everest Medicine. Soo-Chin Lee: honoraria from Pfizer, Novartis, AstraZeneca, ACT Genomics, Lilly, MSD, Roche, Gilead Sciences, Daiichi Sankyo, DKSH; consulting/advisory fees from Pfizer, Novartis, AstraZeneca, MSD, Roche, Gilead Sciences, Daiichi Sankyo; speakers’ fees from Pfizer, Novartis, AstraZeneca, Roche, and MSD; research funding from Pfizer, Eisai, Taiho Pharmaceutical, ACT Genomics, and Karyopharm Therapeutics; travel/accommodation expenses from Amgen, Pfizer, and Roche. Hiroji Iwata: honoraria and research funding from AstraZeneca K.K. and Pfizer, fees for promotional materials from AstraZeneca. Kenichi Watanabe: speaker’s honoraria from Chugai, Eli Lilly, Nippon- Kayaku, Kyowa Kirin, Novartis, Taiho, Eisai, Pfizer, Shionogi, Daiichi Sankyo and AstraZeneca. Kyung Hae Jung: has consulting or advisory role for AstraZeneca, Bixink, Daiichi Sankyo, Eisai, Everest Medicine, MSD, Novartis, Pfizer, Roche, and Takeda Pharmaceuticals. Yuko Tanabe: research funding from MSD. Eriko Tokunaga: personal fees for lectures from Eli Lilly, AstraZeneca, and Daiichi Sankyo. Yoon Sim Yap: Honoraria: Novartis, Pfizer, Lilly/DKSH, AstraZeneca, Eisai, MSD, Specialised Therapeutics, Roche; Research funding: Merck Sharp & Dohme; Travel, accommodations, expenses: DKSH, AstraZeneca. Koji Matsumoto: has received research funding from MSD, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly, and Gilead Sciences; has received honoraria from MSD, Kyowa Kirin, and Chugai. Yoshiko Umeyama: is an employee of Pfizer R&D Japan and owns stock in Pfizer. Kazuki Sudo: honoraria from AstraZeneca, Pfizer, Eisai, Nihon Medi-Physics Co.; research funding from NanoCarrier, Daiichi Sankyo, AstraZeneca, Pfizer, Amgen, PRA Health Sciences, Takeda, and Merck. Aya Kuchiba: honoraria from Chugai Pharma. Kenichi Nakamura: honoraria from Chugai Pharma, Taiho Pharmaceutical, IQVIA, AstraZeneca, Lilly; research funding from Astellas Pharma, Eisai, Otsuka, Ono Pharmaceutical, Daiichi Sankyo, Taiho Pharmaceutical, Takeda, Chugai/Roche, Novartis, Pfizer, Bristol-Myers Squibb Japan, Boehringer Ingelheim Seiyaku, SymBio Pharmaceuticals, Merck, and Servier. Yasuhiro Fujiwara, is now Chief Executive of Pharmaceuticals and Medical Devices Agency, regulatory agency of Japan, and contributed to this trial until March 2019. Kan Yonemori: consulting/advisory fees: Chugai Pharma, Ono Pharmaceutical, Novartis, Eisai, and OncXerna Therapeutics; honoraria from Eisai, Pfizer, AstraZeneca, Novartis, Taiho Pharmaceutical, Lilly Japan, and Daiichi Sankyo/AstraZeneca. Gun Min Kim, Tsutomu Iwasa, Seok Yun Kang, Hiroyuki Yasojima, Kenjiro Aogi, Sung Hoon Sim, Ling-Ming Tseng, Yuki Kojima, Tomomi Hata, Taro Shibata, and Kenji Tamura: no competing financial or non-financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noguchi, E., Yamanaka, T., Mukai, H. et al. A phase 3 study (PATHWAY) of palbociclib plus tamoxifen in patients with HR-positive/HER2-negative advanced breast cancer. npj Breast Cancer 10, 76 (2024). https://doi.org/10.1038/s41523-024-00684-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-024-00684-w
- Springer Nature Limited