
Abstract
Pathogenic variants in INPP5E cause Joubert syndrome (JBTS), a ciliopathy with retinal involvement. However, despite sporadic cases in large cohort sequencing studies, a clear association with non-syndromic inherited retinal degenerations (IRDs) has not been made. We validate this association by reporting 16 non-syndromic IRD patients from ten families with bi-allelic mutations in INPP5E. Additional two patients showed early onset IRD with limited JBTS features. Detailed phenotypic description for all probands is presented. We report 14 rare INPP5E variants, 12 of which have not been reported in previous studies. We present tertiary protein modeling and analyze all INPP5E variants for deleteriousness and phenotypic correlation. We observe that the combined impact of INPP5E variants in JBTS and non-syndromic IRD patients does not reveal a clear genotype–phenotype correlation, suggesting the involvement of genetic modifiers. Our study cements the wide phenotypic spectrum of INPP5E disease, adding proof that sequence defects in this gene can lead to early-onset non-syndromic IRD.
Similar content being viewed by others

Introduction
Inherited retinal degenerations (IRDs) are a group of genetically and clinically heterogeneous disorders characterized by progressive photoreceptor loss due to genetic defects in ~270 genes, inherited in all Mendelian modes1. IRDs manifesting as an isolated phenotype, or non-syndromic IRDs, can be further classified based on their onset and degeneration patterns. Syndromic IRDs manifest as a clinical feature of a syndrome, such as ciliopathies that involve multiple organs and tissues, including the central nervous system, skeletal and reproductive system, kidney, liver, pancreas, lung, and neuroretina2.
Pathogenic variants leading to ciliopathies occur in genes playing either a structural or a functional role in the primary cilium, a specialized organelle protruding from most post-mitotic cells. Cilia act as antennae that “sense” the physical and biochemical stimuli of the cellular environment to promptly initiate the signaling cascades in response to those changes3. Primary cilia play an important role during embryogenesis and organ development and, therefore, ciliary dysfunction often leads to congenital or early-onset disease2. The photoreceptor outer segment is regarded as a specialized primary cilium detecting light stimuli and thus multi-organ ciliopathies often involve retina4. A broad phenotypic spectrum of ciliopathies, ranging from isolated tissues (e.g., retina) to multiple organ involvement have been described for many genes5.
Joubert syndrome (JBTS, OMIM #PS213300) is an example of ciliopathy with retinal involvement. JBTS is a genetically heterogeneous autosomal or X-linked recessive disorder, with currently 36 associated genes (https://www.omim.org/phenotypicSeries/PS213300). The diagnostic hallmark of JBTS is the abnormal development of the mid-hindbrain known as the “molar tooth sign,” a radiological finding detectable on axial magnetic resonance imaging (MRI) of the brain6. JBTS manifests with hypotonia, ataxia, developmental delay, irregular breathing patterns, abnormal eye movements, oculomotor apraxia, and intellectual disability. Extra-neurological findings such as retinal degeneration, coloboma, skeletal abnormalities, cystic kidney disease, liver fibrosis, endocrinological disorders may also be present7.
Pathogenic variants in the Inositol Polyphosphate-5-Phosphatase E gene (INPP5E) on chromosome 9 are a known cause of JBTS (OMIM #213300). INPP5E is a widely expressed ciliary gene8, encoding a 72-kDa (644 amino acid) phosphatase that plays a critical role in controlling ciliary growth and stability via the phosphoinositide 3-kinase signaling pathway9. To date, a total of 34 pathogenic INPP5E variants have been reported, 28 of which in patients with JBTS or MORM (Mental retardation, truncal obesity, retinal dystrophy, and micropenis) syndrome (OMIM #610156)9,10,11,12,13,14,15,16,17,18,19,20,21,22,23. These JBTS cases include eleven patients with no signs of IRD10,15,16,18,20,21,23. In addition, large mutational screening studies identified five early-onset non-syndromic IRD cases, harboring six INPP5E variants11,24,25,26. Since these studies lacked detailed phenotypic descriptions of patients, a clear association between INPP5E and non-syndromic IRD has not been established. Here, we report 16 non-syndromic IRD patients and two cases with an IRD and some JBTS clinical features from 12 unrelated families with pathogenic variants in INPP5E. Our study thus substantiates the involvement of INPP5E variants in non-syndromic retinal disease.
Results
Rare INPP5E variants associated with non-syndromic early-onset IRD
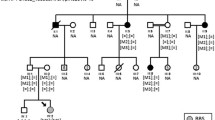
Sequencing analysis of ten recessive non-syndromic IRD families and two unrelated subjects with some JBTS clinical features uncovered ten likely pathogenic variants and five rare variants of unknown significance in INPP5E (see Fig. 1 and Table 1). Thirteen variants were novel, including two present as a complex allele p.[(Ser249Phe);(Arg596Thr)]. All INPP5E variants were rare (AF ≤ 0.0001 in gnomAD), had high CADD Phred scores (>20), and were predicted to be deleterious by several in silico prediction algorithms (see Fig. 2a, Table 1 and Supplementary Table 1)27,28,29,30,31. No other likely pathogenic variants in currently known IRD genes1 (see Supplementary Table 2) segregating with the phenotype were found.

For each family, the specific IRD phenotype diagnosed is mentioned above each pedigree (LCA, Leber Congenital Amaurosis; RCD, Rod Cone Degeneration). Mildly syndromic families C and K are indicated with a hashtag (#). Affected male and female subjects are represented with black squares or circles, respectively. Probands are indicated by a black arrow. The five unaffected sisters in family L are indicated with the superscript 5–10. Novel variants are indicated in bold. When performed, segregation of the INPP5E variants in other family members is shown. First cousin marriage is indicated by a double line. All presented variants refer to the INPP5E transcript NM_019892.5.

a INPP5E secondary structure, tolerance landscape calculated using MetaDome, and distribution of known causal variants. Multiple sequence alignment-derived INPP5E motifs and catalytic domain were highlighted using different colors, while variants were divided into two groups, depending on whether they were found in syndromic or non-syndromic IRD patients. Variants found in our patients are in bold, while novel variants described in this study are additionally highlighted in red. Variants p.(Ser249Phe) and p.(Arg596Thr), found to be part of the same complex allele, are indicated by boxes. Variant Y543*, present in both syndromic and non-syndromic patients, results from two different nucleotide changes: c.1629C>G (JBTS) and c.1629C>A (IRD). b INPP5E tertiary structure. The tridimensional structure was predicted only for C-terminal 349 amino acids (residues 275–623) available on PDB (ID: 2XSW), as the N-terminal half was classified as a disordered region. Two glycerol molecules, acting as a proxy for the larger ligand of this protein (i.e., phosphatidylinositol polyphosphate), are shown in green. Amino acid residues for which missense variants in our patients were found are highlighted in red, except for Serine 249 located in the un-modeled region. c–e Predicted effect of missense variants p.(Arg621Gln) and p.(Arg621Trp) on ligand binding. In the wild-type protein model, Arginine 621 is located in close proximity (3 Å) to one glycerol molecule, which uniquely interacts by establishing one ion bond, indicated by a blue dashed line (c). Missense variants introducing Glutamine (d) or Tryptophan (e) are predicted to increase distance with the glycerol of 4.7 Å and 5.4 Å, respectively, thus disrupting the ion bond.
Two of the novel INPP5E variants were protein-truncating: (p.(Asn159*) and p.(Val587Glyfs*7)), while the remaining were missense. Most of the missense variants clustered in the highly conserved inositol 5-phosphatase catalytic domain (residues 273-621) and seven of them affected arginine residues (see Fig. 2a), which are known to have an important function in the catalysis of the phosphoryl group transfer32.
Affected subjects from five families carried homozygous variants: p.(Arg621Gln) in three unrelated families (families B-D), p.(Val465Ile) in family L, and a complex allele p.[(Ser249Phe); (Arg596Thr)] in family G (see Fig. 1 and Table 1). In three of these families, parents were first cousins (see Fig. 1).
The p.[(Ser249Phe);(Arg596Thr)] complex allele was identified in two unrelated patients, homozygous and compound heterozygous in family G and F, respectively. At present, it is not possible to determine which of the variants or both contribute to disease. The frequency of the p.(Ser249Phe) change is 11-times higher (AF = 0.000067) than of p.(Arg596Thr) (AF = 0.000006) in the gnomAD database (see Table 1), however, both are sufficiently rare to be potentially causal. Ser249 was predicted to be a phosphorylation site for the Protein Kinase C (NetPhos score = 0.84, intervals 0–1, see Supplementary Table 1), whereas Arg596 lies in the catalytic domain, though no specific effect of the p.(Arg596Thr) change was predicted (see Supplementary Table 1).
Apart from p.(Arg621Gln) and the p.[(Ser249Phe);(Arg596Thr)] complex allele, other recurrent missense variants were p.(Arg585His) in families E and K, p.(Arg486Cys) in families H and J (see Fig. 1 and Table 1).
All but one proband carried at least one likely pathologic INPP5E variant (see Fig. 1 and Table 1). The A.II-1 carried two variants of unknown significance (p.(Ser365Leu) and p.(Asp600Glu)). However, given the low mutational tolerance of these two residues and their localization at the catalytic domain, it is likely that they are causal (see Fig. 2a).
Alignment of INPP5E protein sequence in 100 species revealed that most of the missense changes were affecting highly conserved amino acids (identical in ≥98 of species), while variants p.(Ser249Phe), p.(Ser365Leu), p.(Pro526Leu) affected less conserved residues that were identical in 84, 70, and 46 species, respectively (see Supplementary Fig. 1).
Protein modeling and prediction of missense variants at catalytic sites
Modeling of the tertiary structure of INPP5E (Protein Data Bank (PDB) ID: 2XSW; Tresagues et al., unpublished) predicted two sites of potential interaction with a ligand (glycerol molecule used as a proxy of inositol-3-phosphate) (see Fig. 2b). The first interaction site resides in the known catalytic domain where the ligand is predicted to form polar bonds with residues His424, Asn479, Asp477, and His584 (see Supplementary Fig. 2). Three of the likely pathogenic variants identified in this study are located either within the catalytic pocket: p.(Arg557His) and p.(Arg585His) or in its proximity: p.(Thr305Ile) (see Fig. 2b and Supplementary Fig. 2). They have the highest score for deleteriousness according to SuSPect33 (see Supplementary Table 1). The p.(Thr305Ile) change leads to the disruption of the hydrogen bond connecting Gln339 and Thr305 residues, which is thought to result in the alteration of the INPP5E structure (see Supplementary Fig. 3). None of these new or published likely pathogenic INPP5E changes directly affected the residues predicted to bind the IP3 ligand.
The second potential ligand interaction site resides outside of the known catalytic domain and exclusively involves the Arg621 residue. Two of the INPP5E variants detected in our patients (p.(Arg621Gln) and p.(Arg621Trp)) targeted the Arg621 residue. Modeling of the structural changes induced by these two variants showed that both lead to disruption of the polar bond connecting the second glycerol molecules to INPP5E protein (see Fig. 2c–e).
Clinical phenotypes
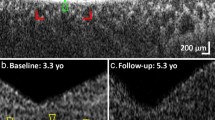
Eight females and four males with INPP5E-associated disease demonstrated features of IRDs that could be separated into two clinical categories. Probands of families A-D had a severe retinal degeneration manifested during early infancy (LCA) whereas the remaining eight had a milder juvenile-onset rod-cone degeneration (RCD) (see Supplementary Table 3). All individuals with LCA had nystagmus as a shared early feature. All four subjects had reduced visual acuity with severely constricted visual fields and undetectable or severely reduced electroretinograms (ERGs). Fundus examination and imaging showed macular and peripheral retinal atrophy. At least two LCA probands (B.II-4 and C.II-1) had structure-function dissociation based on the better foveal structure on optical coherence tomography (OCT) than would be expected from their visual acuities (see Fig. 3 and Supplementary Table 3).

Images show fundus photos (left column), fundus autofluorescence (middle column), and OCTs (right column) for a representative subset of individuals. The specific IRD phenotype of each patient is given in brackets (LCA–Leber congenital amaurosis; RCD–Rod-cone degeneration). Novel INPP5E variants are highlighted in bold.
Individuals with RCD first experienced nyctalopia and impaired dark adaptation beginning typically in childhood and their teens. None of the RCD subjects had nystagmus. Subjects had generally high visual acuities (see Supplementary Table 3). Goldmann perimetry showed mild constriction when available (n = 3). Full-field ERGs were performed in seven of eight subjects with RCD. Scotopic responses were undetectable in all but proband E.II-4 (at age 24) whereas 30 Hz flicker (photopic) responses were present and relatively preserved in five probands (E.II-4, F.II-1, I.II-1, K.II-1, and L.II-1) (see Supplementary Table 3). Fundus examination and wide-field fundus autofluorescence (FAF) imaging showed typical features of RCD in all individuals (see Fig. 3, left and middle panel). Macular OCT imaging showed central ellipsoid zone (EZ) preservation in most patients, and in proband E.II-4 the EZ was robust and identifiable through most of the scanned macula (see Fig. 3). Bilateral cystoid macular edema was present in proband H.III-1. Most individuals in both groups for whom information about refraction was available were myopic.
Two subjects showed extra-ocular features: subject K.II-1 presented with oculomotor apraxia and hypotonia at an early age which resolved, and the individual did not show any neurological or cognitive disability as an adult. Subject C.II-1 was found to have hypoplasia of the inferior cerebellar vermis on brain MRI at the age of 18 during an investigation of headaches (see Supplementary Fig. 4). She had a motor delay in infancy specifically delayed head control and sitting, as well as “lack of strength”, frequent falls, and learning difficulties in childhood. Despite this clinical history, other milestones including speech and walking were met at the appropriate age. This subject completed secondary education at age 18. On a recent neurological examination performed at age 22, mild ataxia and tandem gait disequilibrium was noted. Renal ultrasound performed at age 15 showed no renal anomalies. The remaining ten subjects in this cohort did not have other extra-ocular features.
Meta-analysis of all pathogenic INPP5E variants and their phenotypic correlation
Pathogenic variants in INPP5E can lead to a broad phenotypic spectrum ranging from severe ciliopathies to non-syndromic IRD9,10,11,17,24,25,26. We hypothesized that differences in disease severity are caused by more deleterious variants present in syndromic versus non-syndromic patients. Therefore, we gathered all published pathogenic variants in INPP5E (n = 47) in 34 syndromic (JBTS and MORM)9,10,11,12,13,15,16,17,18,19,20,21,22,23,34 and 17 non-syndromic IRD cases (our families and previously reported cases11,24,25,26) and analyzed their potential effect on protein function (see Figs. 2, 4, and Supplementary Table 4). First, we considered all alleles present in the syndromic cases (68 alleles from 34 patients) and alleles from non-syndromic IRD cases (34 alleles from 17 patients). We noticed that the difference in severity was not the result of a significantly higher frequency of loss-of-function (LoF) alleles in syndromic patients (9/68 alleles) compared to non-syndromic patients (3/34 alleles) (χ2 test, p = 0.5) and only three of the syndromic patients and none of the IRD patients were homozygous for an LoF variant (see Supplementary Table 4 and Fig. 4a). The remaining changes were missense variants, mostly within the inositol polyphosphate catalytic domain, with no apparent clustering based on the disease severity (see Fig. 2a). Only four variants were shared between syndromic and non-syndromic cases: p.(Arg515Trp)10,11, p.(Tyr543*)16,26, p.(Arg585Cys)11,13,16, and p.(Arg621Gln)11,12,15,16,17 (see Supplementary Table 4 and Fig. 4a). Using protein modeling and deleteriousness prediction algorithms we determined the potential impact of each variant on INPP5E function and for each patient added the impact of both variants. Overall, we did not find significant differences in conservation or deleteriousness scores of variants between the syndromic and non-syndromic cases (see Supplementary Tables 1 and 4 and Fig. 4b, Mann-Whitney test p-value > 0.05). One extreme example is the p.(Arg621Gln) variant, which has been observed as a homozygote in two non-syndromic and one mildly syndromic IRD cases in this study and recently reported in one subject with JBTS with no retinal degeneration15. These observations indicate that other genetic factors may play a role in the INPP5E disease manifestation.

a Distribution of the INPP5E variants in all reported INPP5E patients. Four different phenotypes of increasing severity were marked by circles (non-syndromic IRD), diamonds (IRD-mild JBTS, reported in this study), squares (JBTS with IRD), and triangles (JTBS without IRD). b Violin plot of the cumulative CADD Phred scores for the INPP5E variants in syndromic (JBTS) and non-syndromic IRD cases.
Discussion
We report non-syndromic IRD patients from ten families and two mildly syndromic JBTS cases with rare variants in INPP5E. Pathogenic variants in INPP5E are mainly known to cause systemic disorders, JBTS10 and MORM9, characterized by severe neurological manifestations and extra-neurological symptoms which may differ both quantitatively and qualitatively in each affected individual, even within the same family10,12,16,21,34. Therefore, the mostly non-syndromic patients described in this study are on a milder spectrum of the INPP5E disease. Although non-syndromic IRD due to INPP5E variants has been reported before in large mutational screening studies11,24,25,26, our report provides further evidence of the involvement of INPP5E variants in isolated retinal disease. We describe in detail retinal phenotypes of all the probands and report extraocular symptoms present in two patients. In addition, we present a thorough analysis of the functional impact that the identified variants may have on the INPP5E protein, using in-silico protein modeling tools. Our meta-analysis of all the published INPP5E variants and their potential phenotypic correlation illustrates the broad spectrum of phenotypes caused by the INPP5E-associated disease.
All probands described here were initially referred to as IRD patients at the time of their first visit. Ten of 12 presented with retinal degeneration with no other extra-ocular symptoms thus there was no indication for further clinical investigation other than ophthalmological. In probands, C.II-1 and K.II-1 mild ciliopathy features were identified during childhood. These symptoms, however, resolved during development and no cognitive or major neurological disability was present in adulthood. Hypoplasia of the inferior cerebellar vermis in C.II-1 was a secondary finding discovered by MRI performed to investigate the source of persistent headaches. Although this anatomical finding is less pronounced than the molar tooth sign usually detected in JBTS (see Supplementary Fig. 4), it is likely due to the homozygous p.(Arg621Gln) change in INPP5E and possibly additional genomic variability carried by this subject. A review of the exome sequencing (ES) data for this proband revealed a known pathogenic splicing variant (c.3290-2A>G) in CPLANE1, a gene associated with JBTS (OMIM # 614615) and Orofaciodigital syndrome VI (OFD, OMIM # 277170) that share some clinical features such as the molar tooth sign, vermis hypoplasia, and developmental delay35. The presence of this single allele in a recessive ciliopathy gene is however not sufficient to consider it as a modifier of the phenotype in patient C.II-1. A review of the genome sequencing data of proband K.II-1, with childhood oculomotor apraxia and hypotonia, did not return any rare coding variants (MAF < 0.001) nor CNVs in any of the Joubert or ataxia with oculomotor apraxia (OMIM# 208920, APTX)36,37 genes. Since brain MRI was not performed on the remaining cases we cannot rule out subclinical anatomical changes in these patients.
Of the 14 variants described in this study, 12 were novel and mainly resulting in missense changes of conserved amino acid residues in the phosphatase catalytic domain. Eight patients carried two likely pathogenic variants, two patients carried one likely pathogenic and one variant of unknown significance, and two cases carried two rare variants of unknown significance. Two variants in our study, p.(Arg621Gln) and p.(Arg621Trp), affected the same residue. Both variants were predicted to disrupt a unique polar bond between Arginine 621 and a potential ligand. Homozygous p.(Arg621Gln) and p.(Arg621Trp) changes were found in five patients, three non-syndromic LCA patients (this study and24), one mildly syndromic LCA case (C.II-1, this study), and one JBTS case without apparent retinal involvement15. The p.(Arg621Gln) change has also been associated with non-syndromic IRD cases and with JBTS without retinal involvement in a compound heterozygous scenario11,16. Unfortunately, at present, the paucity of genotyped INPP5E patients makes it impossible to explain the phenotypic discrepancies in patients carrying the p.(Arg621Gln) variant. Nevertheless, the frequency at which Arginine 621 is mutated suggests that this amino acid constitutes a critical residue for the INPP5E function and together with the putative ligand binding by Arg621, warrants expansion of the catalytic domain of INPP5E to this position. Of the 19 known pathogenic variants present in a homozygous state only three (p.[(Ser249Phe); (Arg596Thr)], p.(Val465Ile), p.(Arg621Trp)) resulted in a non-syndromic retinal degeneration, which may imply a hypomorphic or photoreceptor-specific impact of these variants on INPP5E function. These residues may also be important for photoreceptor-specific interactions with other ciliary proteins. Further functional studies will be needed to understand the impact of the identified INPP5E variants on the phosphatase activity or interactions with other proteins.
In order to understand the broad phenotypic spectrum of INPP5E-associated disease, we used several deleteriousness prediction algorithms and protein modeling to uncover the impact of each variant on the protein function. We have not found significant differences between the syndromic and non-syndromic cases or between the LCA and RCD cases, analyzing the combined impact of both INPP5E variants in each patient. The lack of a clear correlation of predicted variant impact on phenotype indicates that other genetic factors may play a role. Previous studies have shown that the INPP5E function in the cilium is dependent on other ciliary proteins, such as ARL3 and TULP3, and defects in those proteins lead to reduced or absent INPP5E localization to primary cilia38,39. Moreover, genetic modifiers in cis or trans to the primary disease variant(s) have been reported in many IRD studies where they influence disease penetrance, severity, and progression40. For example, the AHI1 variant p.(Arg830Trp) modifies the relative risk of retinal degeneration greater than seven-fold within a nephronophthisis cohort41. Similarly, resequencing of the TTC21B gene in a large group of clinically diverse ciliopathies showed that variants in this gene account as severity modifiers in ~5% of ciliopathy patients42. Although the number of genotyped samples with specific disease phenotypes is not large enough to support an unquestionable genotype-phenotype association, the rapid increase of high-throughput exome and genome sequencing in standard diagnostic protocols will help to validate some of these associations in the near future.
In conclusion, we provided further evidence for the involvement of INPP5E variants in non-syndromic IRD and demonstrated that these variants also account for previously underdiagnosed retinal degeneration patients.
Methods
Ethics statement
The study was approved by the institutional review board of all participating institutions (Partners HealthCare System for families E-G, the Boston Children’s Hospital Committee on Clinical Investigation for family A, Instituto de Oftalmologia Dr. Gama Pinto for families C, D, and H, the Institutional Review Boards and ethics committees of Moorfields Eye Hospital for families I-K, the institutional review board at Hadassah-Hebrew University Medical Center for family B, the Ethics Committee at Rambam Health Care Campus for family L) and adhered to the Declaration of Helsinki. Informed consent was obtained from all individuals on whom genetic testing and further molecular evaluations were performed.
Clinical evaluation
Twelve probands with autosomal recessive retinal degeneration, who were part of larger IRD cohorts from five medical centers, were enrolled in this study. Four probands were ascertained from two different medical centers in Boston, USA (Massachusetts Eye and Ear and Boston Children’s Hospital), three in the United Kingdom (Moorfields Eye Hospital), three in Portugal (Instituto de Oftalmologia Dr. Gama Pinto), and two in Israel (Hadassah-Hebrew University Medical Center, Rambam Health Care Campus).
Clinical evaluation was performed by experienced ophthalmologists according to previously published protocols and included functional and structural assessments43,44,45,46.
For proband C.II-1, brain MRI was performed using a GE Signa HDxt 1.5T scanner (GE Medical Systems, Milwaukee, WI). The JBTS and control cases were scanned on a 3T scanner (Verio and Vida, Siemens Healthcare, Erlangen, Germany). Scanning protocols included unenhanced 3D T1 weighted Imaging and T2 spin-echo weighted imaging, which were sufficient to make the first diagnosis.
Genetic analysis
Blood samples were obtained from probands, and when possible their parents, affected, and unaffected siblings. DNA was isolated from peripheral blood lymphocytes by standard procedures. Four probands (E.II-4, F.II-1, G.II-1, and A.II-1) were sequenced using the Genetic Eye Disease (GEDi) panel, described previously47. The GEDi version used in this study (v6) targeted exons of 278 known IRD genes (see Supplementary Table 2)1. The NGS data from the GEDi panel was analyzed using Genome Analysis Toolkit (GATK) version 348 and annotated using the Variant Effect Predictor (VEP) tool49 with additional annotations taken from the Genome Aggregation Database (gnomAD)27, the Genomic Evolutionary Rate Profiling (GERP)28, SIFT29, PolyPhen230, CADD Phred31, and retinal expression50. To detect possible copy number variations gCNV software was used as before51. The relatedness of the families sequenced with GEDi panel was excluded using Peddy52. Exome sequencing (ES) for five probands was performed at different facilities (B.II-4, Pronto Diagnostics Ltd; C.II-1, D.II-4 and H.III-1, Novogene (HK); L.II-1, Otogenetics Corporation), as previously described45,53,54. Finally, three patients (I.II-1, J.II-1, K.II-1) underwent genome sequencing (GS, Genomics England) according to previously published protocols27,55. In these patients, CNVs were interrogated by MANTA56 and CANVAS57 algorithms and direct inspection of the read data using IGV.
Variant validation and phasing
All presented variants refer to the INPP5E transcript NM_019892.5. Variant segregation was performed by Sanger sequencing (primers in Supplementary Table 5) or analysis of NGS reads. For F.II-1, the three INPP5E variants detected were phased by cloning and Sanger sequencing (see Supplementary Fig. 5). Briefly, genomic DNA from the proband was amplified using Takara-LA (Takara Bio USA, Inc.) and primers spanning the region containing all variants. The amplified fragment was then cloned into the pCR2.1 plasmid, TA cloning kit (Invitrogen) and Sanger sequenced. Sanger sequencing was performed on ABI 3730xl (Applied Biosystems) using BigDye Terminator v3.1 kits (Life Technologies). Sequence analysis was done using SeqManPro (Lasergene 11, DNAStar Madison, WI, USA), in which variants were considered to be in trans when they were never present on the same clone.
Multiple sequence alignment, protein modeling, and prediction of missense variants
Multiple sequence alignment of the human INPP5E protein and 99 orthologues was generated using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/)58 and sequences were retrieved from the UniProt Knowledgebase (UniProtKB, https://www.uniprot.org/help/uniprotkb)59. Tridimensional structure of the INPP5E protein, its putative catalytic sites, and mutated residues was generated with a protein modeling software (PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) using the crystal structure of human INPP5E as an input (Protein Data Bank (PDB) ID: 2XSW). The mutation tolerance at INPP5E protein residues was analyzed using MetaDome (https://stuart.radboudumc.nl/metadome/)60, while the impact of specific missense variants on INPP5E structure and function, was predicted by using four prediction algorithms: SIFT (https://sift.bii.a-star.edu.sg/)29, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)30, Missense3D (http://www.sbg.bio.ic.ac.uk/~missense3d/)61 and SuSPect (http://www.sbg.bio.ic.ac.uk/suspect)33. NetPhos 3.1 Server (http://www.cbs.dtu.dk/services/NetPhos/)62 was used to predict phosphorylation sites.
Preprints
An earlier version of this manuscript has been published on MedRxiv.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Variants are available through ClinVar (Accession codes SCV001573305.1; SCV001573306.1; SCV001573392.1; SCV001573393.1; SCV001573394.1; SCV001573395.1; SCV001573396.1; SCV001573397.1; SCV001573398.1; SCV001573399.1; SCV001573400.1; SCV001573477.1; SCV001573478.1; SCV001573513.1; SCV001573569.1; SCV001573570.1; SCV001573637.1; SCV001573638.1; SCV001573767.1) and in Supplemental Materials.
Code availability
Software used in this study: PyMOL Molecular Graphics System, Version 1.2r3pre; GraphPad Prism, version 9.0.2; SeqManPro (Lasergene v11).
References
Daiger S. P., Sullivan L. S., Bowne S. J., R. B. RetNet. Retinal Information Network. https://sph.uth.edu/retnet/.
Hildebrandt, F., Benzing, T. & Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533–1543 (2011).
Wheway, G., Nazlamova, L. & Hancock, J. T. Signaling through the primary cilium. Front. Cell Dev. Biol. 6, 8 (2018).
Bujakowska, K. M., Liu, Q. & Pierce, E. A. Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb. Perspect. Biol. 9, a028274 (2017).
Chen, H. Y., Kelley, R. A., Li, T. & Swaroop, A. Primary cilia biogenesis and associated retinal ciliopathies. Semin. Cell Dev. Biol. 110, 70–88 (2021).
Maria, B. L. et al. ‘Joubert syndrome’ revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 12, 423–430 (1997).
Brancati, F., Dallapiccola, B. & Valente, E. M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 5, 1–10 (2010).
The Genotype-Tissue Expression (GTEx) Project. www.gtexportal.org.
Jacoby, M. et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027–1031 (2009).
Bielas, S. L. et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036 (2009).
Stone, E. M. et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331 (2017).
Brooks, B. P. et al. Joubert syndrome: ophthalmological findings in correlation with genotype and hepatorenal disease in 99 patients prospectively evaluated at a single center. Ophthalmology 125, 1937–1952 (2018).
Toma, C. et al. Macular staphyloma in patients affected by Joubert syndrome with retinal dystrophy: a new finding detected by SD-OCT. Doc. Ophthalmol. 137, 25–36 (2018).
Fadaie, Z. et al. Identification of splice defects due to non-canonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 40, 2365–2376 (2019).
Radha Rama Devi, A., Naushad, S. M. & Lingappa, L. Clinical and molecular diagnosis of Joubert syndrome and related disorders. Pediatr. Neurol. 106, 43–49 (2020).
Travaglini, L. et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert Syndrome and related disorders. Eur. J. Hum. Genet. 21, 1074–1078 (2013).
Tsurusaki, Y. et al. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J. Hum. Genet. 58, 113–115 (2013).
Sönmez, F. et al. Development of end-stage renal disease at a young age in two cases with Joubert syndrome. Turk. J. Pediatr. 56, 458–461 (2014).
Kroes, H. Y. et al. Joubert syndrome: genotyping a Northern European patient cohort. Eur. J. Hum. Genet. 24, 214–220 (2016).
Suzuki, T. et al. Molecular genetic analysis of 30 families with Joubert syndrome. Clin. Genet. 90, 526–535 (2016).
De Goede, C. et al. Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders. Eur. J. Paediatr. Neurol. 20, 286–295 (2016).
Hardee, I. et al. Defective ciliogenesis in INPP5E-related Joubert syndrome. Am. J. Med. Genet. Part A 173, 3231–3237 (2017).
Shetty, M., Ramdas, N., Sahni, S., Mullapudi, N. & Hegde, S. A homozygous missense variant in INPP5E associated with Joubert syndrome and related disorders. Mol. Syndromol. 8, 313–317 (2017).
Wang, X. et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J. Med. Genet. 50, 674–688 (2013).
Xu, Y. et al. Mutation analysis in 129 genes associated with other forms of retinal dystrophy in 157 families with retinitis pigmentosa based on exome sequencing. Mol. Vis. 21, 477–486 (2015).
Birtel, J. et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 8, 4824 (2018).
Genome Aggregation Database (GnomAD). https://gnomad.broadinstitute.org.
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
O’Brien, P. J., Lassila, J. K., Fenn, T. D., Zalatan, J. G. & Herschlag, D. Arginine coordination in enzymatic phosphoryl transfer: Evaluation of the effect of Arg166 mutations in Escherichia coli alkaline phosphatase. Biochemistry 47, 7663–7672 (2008).
Yates, C. M., Filippis, I., Kelley, L. A. & Sternberg, M. J. E. SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J. Mol. Biol. 426, 2692–2701 (2014).
Khan, S. et al. BBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan. Ann. Hum. Genet. 83, 477–482 (2019).
Lopez, E. et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum. Genet. 133, 367–377 (2014).
Date, H. et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 29, 184–188 (2001).
Moreira, M. C. et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 29, 189–193 (2001).
Alkanderi, S. et al. ARL3 mutations cause Joubert syndrome by disrupting ciliary protein composition. Am. J. Hum. Genet. 103, 612–620 (2018).
Han, S. et al. TULP3 is required for localization of membrane-associated proteins ARL13B and INPP5E to primary cilia. Biochem. Biophys. Res. Commun. 509, 227–234 (2019).
Kousi, M. & Katsanis, N. Genetic modifiers and oligogenic inheritance. Cold Spring Harb. Perspect. Med. 5, a017145 (2015).
Louie, C. M. et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 42, 175–180 (2010).
Davis, E. E. et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196 (2011).
Scott, H. A. et al. Expanding the phenotypic spectrum in RDH12-associated retinal disease. Cold Spring Harb. Mol. case Stud. 6, a004754 (2020).
Moye, A. R. et al. Mutations in ARL2BP, a protein required for ciliary microtubule structure, cause syndromic male infertility in humans and mice. PLoS Genet. 15, e1008315 (2019).
Haer-Wigman, L. et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 24, 3742–3751 (2015).
Men, C. J. et al. The importance of genetic testing as demonstrated by two cases of CACNA1F-associated retinal generation misdiagnosed as LCA. Mol. Vis. 23, 695–706 (2017).
Consugar, M. B. et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 17, 253–261 (2015).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Farkas, M. H. et al. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics 14, 486 (2013).
Zampaglione, E. et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 22, 1079–1087 (2020).
Pedersen, B. S. & Quinlan, A. R. Who’s who? Detecting and resolving sample anomalies in human DNA sequencing studies with Peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Sharon, D. et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 41, 140–149 (2020).
Peter, V. G. et al. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene. Genet. Med. 21, 2734–2743 (2019).
Taylor, R. L. et al. Association of steroid 5α-reductase type 3 congenital disorder of glycosylation with early-onset retinal dystrophy. JAMA Ophthalmol. 135, 339–347 (2017).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Roller, E., Ivakhno, S., Lee, S., Royce, T. & Tanner, S. Canvas: versatile and scalable detection of copy number variants. Bioinformatics 32, 2375–2377 (2016).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Bateman, A. et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Wiel, L. et al. MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 40, 1030–1038 (2019).
Ittisoponpisan, S. et al. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 431, 2197–2212 (2019).
Blom, N., Gammeltoft, S. & Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1361 (1999).
Acknowledgements
This work was supported by grants from the National Eye Institute [R01EY012910 (E.A.P.), R01EY026904 (K.M.B./E.A.P.), and P30EY014104 (MEEI core support)], the Jürg Tschopp MD–Ph.D. Scholarship (V.G.P.), the Swiss National Science Foundation [31003A_176097 (C.R.)], the Foundation Fighting Blindness (EGI-GE-1218-0753-UCSD, K.M.B./E.A.P. and BR-GE-0214-0639-TECH to T.B., D.S., and E.B.), and the Curing Kids Foundation (K.M.B.). Supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (R.B., M.M., A.R.W., G.A.), Moorfields Eye Charity (M.M.), and Retina UK (M.M.). The authors would like to thank the patients and their family members for their participation in this study and the Ocular Genomics Institute Genomics Core members for their experimental assistance. We specifically would like to thank Cassandra Amarello and Hilary Scott for their technical assistance. The authors would like to thank the Exome Aggregation Consortium, the Genome Aggregation Database (gnomAD), and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about and http://gnomad.broadinstitute.org/about.
Author information
Authors and Affiliations
Contributions
R.S. performed most of the experiments, data analysis, and led the manuscript writing. I.D analyzed clinical data and wrote the manuscript. V.G.P., R.B.A., E.M.P., N.E.W., A.B.F., L.C.S., B.R., E.B., A.B.S., M.N., M.M., T.B.Y., E.A.P., C.R., A.R.W., G.A., D.S., R.M.H. analyzed clinical data and reviewed the manuscript. E.Z., A.A.T., A.L., M.Q. performed part of the genetic analysis. V.D. performed a brain MRI. K.M.B. guided the experimental design, aided in variant analysis, and contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sangermano, R., Deitch, I., Peter, V.G. et al. Broadening INPP5E phenotypic spectrum: detection of rare variants in syndromic and non-syndromic IRD. npj Genom. Med. 6, 53 (2021). https://doi.org/10.1038/s41525-021-00214-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-021-00214-8
- Springer Nature Limited
This article is cited by
-
Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa
European Journal of Human Genetics (2024)
-
Inpp5e Regulated the Cilium-Related Genes Contributing to the Neural Tube Defects Under 5-Fluorouracil Exposure
Molecular Neurobiology (2024)
-
Novel variants identified in five Chinese families with Joubert Syndrome: a case report
BMC Medical Genomics (2023)