
Abstract
Intellectual disability (ID) is a highly heterogeneous disorder with hundreds of associated genes. Despite progress in the identification of the genetic causes of ID following the introduction of high-throughput sequencing, about half of affected individuals still remain without a molecular diagnosis. Consanguineous families with affected individuals provide a unique opportunity to identify novel recessive causative genes. In this report, we describe a novel autosomal recessive neurodevelopmental disorder. We identified two consanguineous families with homozygous variants predicted to alter the splicing of ATP9A which encodes a transmembrane lipid flippase of the class II P4-ATPases. The three individuals homozygous for these putatively truncating variants presented with severe ID, motor and speech impairment, and behavioral anomalies. Consistent with a causative role of ATP9A in these patients, a previously described Atp9a−/− mouse model showed behavioral changes.
Similar content being viewed by others
Introduction
Intellectual disability (ID) or delayed psychomotor development are common and highly heterogeneous phenotypes of genetic origin, affecting 1–3% of the general population1,2 which pose a significant socio-economic burden on the affected families, the health care system, and society in general3. Despite considerable progress in genetic diagnosis after the introduction of high throughput sequencing technologies, the genetic cause of more than half of ID cases remains undetermined4. The leading genetic cause of ID in individuals from outbred populations is de novo variants5,6; in contrast a substantial fraction of autosomal recessive (AR) disorders cause ID in families with multiple affected individuals that practice consanguinity7. It is estimated that worldwide 10.4% of marriages occur among close relatives8. Consanguinity increases the extent of homozygous genomic regions and brings to homozygosity deleterious alleles resulting in birth defects and infant mortality9,10. Large consanguineous families with (multiple) affected individuals thus provide a unique opportunity to identify novel recessive causative genes.
P4-ATPases are transmembrane lipid flippases11, that function in vesicles formation and trafficking. They regulate the asymmetric distribution of phospholipids in membranes of eukaryotic cells11,12. There are 14 different P4-ATPases in humans that can be phylogenetically grouped in five classes13. ATP9A and its 75% similar paralog ATP9B are the unique members of class II. They are the only P4-ATPase that do not require the CDC50 β-subunit for normal function and cellular localization14. They show different intracellular and tissue distribution: ATP9A is found in early and recycling endosomes and at a lower level at the plasma membrane, while ATP9B is only found in the trans-Golgi network12,14,15,16. Similarly, the genes encoding ATP9A and ATP9B present with overlapping but different expression patterns with ATP9A mainly expressed in the brain (Human Protein Atlas, GTEx). Suggestive of an important role of ATP9A in intercellular communication, this P4-ATPase inhibits extracellular vesicles release15,16.
Here we report two consanguineous families with homozygous pathogenic variants predicted to alter ATP9A splicing and we propose ATP9A as a novel cause of a recessive neurodevelopmental disorder.
Results
Clinical report
We identified three affected individuals from two unrelated consanguineous families of Pakistani and Iranian origin. The main clinical features of the affected individuals are reported in Table 1 and in Fig. 1.

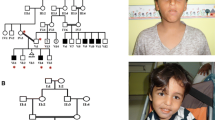
The pedigrees and the available genotypes of the Pakistani (family 1, top) and Iranian families (family 2, bottom) are depicted on the left. Sanger sequencing chromatograms confirming the segregation of the ATP9A NM_006045.3:c.799 + 1 G > T (six top traces) and NM_006045.3:c.327 + 1 G > T variants (bottom five traces) are shown on the right. Inserts showing the facial features of the two affected sisters IV:1 and IV:7 of family 1 are presented below the Pakistani pedigree.
Family 1 is from the Khyber Pakhtunkhwa region of Pakistan. As indicated in the pedigree, the unaffected parents (III:3 and III:4), who are first cousins, have six children. The oldest and youngest siblings (IV:1 and IV:7) exhibited similar clinical features that include delayed childhood milestones, severe ID, mild hypotonia, attention deficit hyperactivity disorder (ADHD), aggressive behavior, bilateral eye squints, and impaired vision. The oldest affected daughter (IV:1) presented with microcephaly (<1st percentile, −3.12 SD), however the head circumference of the second affected sibling, the youngest daughter, (IV:2) is in the normal range (39th percentile). We could not perform brain magnetic resonance imaging (MRI) because the family lives in a very remote area and did not agree to travel due to COVID19 outbreak and the high rate of infections in the region. While the other siblings (IV:2, IV:3, IV:4, and IV:5) were unaffected, we note that pregnancy IV:6 was not carried to term (Fig. 1).
The proband (IV:1) of the Iranian family 2 is the only child born from a couple of first cousins (Fig. 1). Childbirth was unremarkable. The parents noticed a delay in the development of both language and walking (18 months). The proband started epileptic episodes at 3 years of age and seizures were controlled with sodium valproate. An abnormal EEG with epileptiform activity was reported. Brain MRI was normal. At the time of the last visit, the child did not present motor paralysis or coordination deficit, but he had an abnormal gait. At 11 years of age, height, weight, and head circumference were in the normal range with 140 cm, 45 kg, and 53 cm, respectively. Eye contact was impaired and there was complete language dysfunction. He is presenting with severe ID, prominent stereotypic movement disorder, and autistic features. The proband has arched eyebrows with round, downturned eyes, thin lips, bulbous nose, and a short philtrum. The proband’s cousin was also reported to be affected by a neurodevelopmental disorder. He is presenting with moderate ID, autistic features, and epilepsy. However, he does not have any motor or coordination problem. The different severity of ID, growth parameters, and the absence of motor impairment are possibly indicative of a different genetic etiology.
Exome analysis
In family 1, whole-exome sequencing (WES) was performed in the proband (IV:1) to exclude variants in genes previously reported to cause ID or developmental delay. Subsequently, SNP-array was performed in both affected individuals (IV:1 and IV:7), parents (III:3 and III:4) and an unaffected sibling (IV:3). Homozygosity mapping revealed a 2.5 Mb region of homozygosity (chr20[GRCh37]:49010965-51638043) common in both patients (IV:1 and IV:7) but not in the parents (III:3 and III:4) and an unaffected sibling (IV:3). In total, six homozygous variants from the WES data of the proband (IV:1) were present in the segregating ROH (chr20:49010965-51638043) (as mentioned in the Supplementary Table 1), but the splicing variant (NM_006045.3:c.799 + 1 G > T) in ATP9A was the only mutation with the MAF < 1% (in any of the population in the gnomAD database). (Fig. 1). The variant was not present in gnomAD17, Bravo (https://bravo.sph.umich.edu/freeze5/hg38/) or our local database of >500 Pakistani controls. Its segregation in the family was confirmed by Sanger sequencing, in particular, the youngest sister and second affected sibling is homozygous for this variant (Fig. 1). The change at the conserved first nucleotide of the donor splice site was predicted to cause abnormal splicing by SpliceAI17 (score DS_DL = 0.99), MaxEntScan18 (MaxEntScan_diff = 8.504), and NNsplice19. RNA samples from affected individuals were not available to assess RNA splicing.
Our search for more cases led to the identification of a second family. The WES of proband IV:1 from family 2 also revealed the presence of a homozygous splicing variant in ATP9A, a base pair substitution in intron 3 of ATP9A (NM_006045.3:c.327 + 1 G > T). This variant is absent from the gnomAD17 and Bravo databases, the Iranome (i.e. 800 healthy individuals from eight different Iranian ethnic groups, http://www.iranome.ir/) and our local database of >250 Iranian controls. Multiple predictions tools indicated a likely loss of the canonical donor splice site (NNsplice, SpliceAI score DS_DL = 0.95, MaxEntScan_diff = 8.504). The abnormal splicing could either result in the skipping of the inframe exon 3, leading to the deletion of 38 amino acid residues, or utilization of an alternative donor site resulting in partial intronic retention and the appearance of a premature stop codon. Testing of the aberrant RNA splicing was not possible due to the unavailability of the patient’s RNA or cells. Sanger sequencing confirmed the segregation of the potentially causative variant (Fig. 1), i.e., the variant is heterozygous in the proband’s parents (III:2 and III:3), his aunt (III:4) and absent in his uncle (III:1). Homozygosity mapping of proband 1 revealed that the ATP9A variant is embedded in a putative 6.83 Mb region of homozygosity (ROH) (chr20[GRCh37]: 45358223-52192534). While we did not find any likely pathogenic variants in known ID genes in proband IV:1 of family 2 (based on the Panelapp gene list for ID20; Supplementary Table 2), we cannot exclude those variants besides the ATP9A one might play a role in the patient’s phenotype. In particular, we identified homozygous variants in CCDC88C (NM_001080414.4: c.1126 C > T, p.Arg376Trp) and ZNF407 (NM_017757.3: c.5497 > T, p.Pro1833Ser), two genes previously implicated in neurodevelopmental disorders but associated with phenotypes different than the one found in our proband. Bi-allelic variants in CCDC88C were associated with a form of congenital hydrocephalus21,22,23, while variants in ZNF407 have been recently implicated in an AR form of ID with microcephaly, short stature, hypotonia, and ocular anomalies24,25.
Discussion
Autosomal recessive ID is characterized by extensive genetic heterogeneity. Still, many patients do not receive a molecular diagnosis, suggesting that a considerable number of causative genes have not yet been identified4,26. We described three individuals from two consanguineous families with different homozygous splicing variants in canonical splice sites of the ATP9A gene. All three patients present with severe ID, motor delay, speech and fine motor impairment, and behavioral anomalies. Both affected sisters (IV:1 and IV:7) of family 1 had an attention deficit hyperactivity disorder-like phenotype combined with aggressiveness, whereas proband IV:1 from family 2 presented with autistic features, including prominent stereotypic movements, and lack of eye contact.
ATP9A is under constraint (intolerance to missense variants z-score = 4.15; pLI = 1; LOEUF = 0.2) according to gnomAD27. Its yeast homolog, NEO1, was shown to be an essential gene28, while the absence of the C. elegans orthologous TAT-5 resulted in disrupted cell adhesion and morphogenesis in worms’ embryos29. Whereas ablation of the mouse orthologous Atp9a did not diminish survival, the Atp9a−/− mice engineered and phenotyped by the International Mouse Phenotyping Consortium were hyperactive and showed a significant increased exploration in new environment reminiscent of the behavioral symptoms of our patients30,31. Depletion of ATP9A were lethal in human hepatoma HepG2 cells but not in other cell lines including HeLa, HEK293T, MCF-7, and THP-1, suggesting that the absence of ATP9A could be tolerated in certain tissues but not in others12,15. ATP8A2, another P4-ATPase highly expressed in the brain, has been implicated in a recessive disorder characterized by cerebellar ataxia, ID, and disequilibrium syndrome (CAMRQ, MIM 615268), or severe hypotonia, ID, and optic atrophy with or without encephalopathy32,33,34,35,36. A de novo balanced translocation leading to haploinsufficiency of this gene has been also proposed as the cause of moderate ID and hypotonia37.
Downregulation of ATP9A has been associated with a significant increase of extracellular vesicles release, in particular the exosome15,16. Extracellular vesicles release is an important form of intercellular communication that enables the transport of several different signaling molecules—including proteins and RNA—without the need of direct cell-to-cell contacts. It is involved in a wide range of biological processes, such as blood coagulation and immune response38,39. Different physiological roles in the central nervous system have been proposed for extracellular vesicles, including neurite outgrowth and neuronal survival38,40. Depletion of ATP9A reduces the plasma membrane expression of the glucose transporter GLUT1 and increases its level in the endosome, altering its recycling12. Deficiency of GLUT1 has been associated with a neurological disorder with a variable phenotype including epilepsy, movement disorders, mild to severe ID, and acquired microcephaly in some cases41,42. Similarly, alteration in the recycling endosomal processes by mutations in the SLC9A6 sodium exchanger have been associated with Christianson syndrome (MIM 300243), a neurodevelopmental disorder characterized by ID, speech impairment, epilepsy, postnatal microcephaly, truncal ataxia, and hyperactivity43,44.
Since the original submission of this paper and the deposit of our data in medrxiv, a study describing additional ATP9A cases was published45. This latter study reports three affected individuals from two consanguineous families with homozygous loss of function variants, p.(Arg290*) and c.642 + 1 G > A; p.(Ser184Profs*16) in ATP9A, and phenotypic manifestations similar to our study. Patients are all presenting with mild or severe ID, motor and speech delay. Behavioral anomalies, including attention deficit, were also reported in all affected individuals. All patients were noted to have microcephaly, a feature observed only in individual IV:1 of family 1 but not in her sister, IV:7. They were all reported to have short stature and failure to thrive, which are not observed in our patients. In the other cohort, strabismus was reported for only another affected individual but not in his brother, while here it is observed in both affected Pakistani sisters. Combined with ours, these results strengthen the hypothesis of the causative role of ATP9A biallelic truncation variants in a novel neurodevelopmental syndrome.
In conclusion, we describe a novel AR neurodevelopmental disorder. In two unrelated consanguineous families, we identified variants predicted to affect the splicing of ATP9A. The three individuals homozygous for these putatively truncating variants presented with severe ID, motor and speech impairment, and behavioral anomalies. Consistent with a causative role of ATP9A in the patients’ phenotypes, Atp9a−/− mouse model showed behavioral changes.
Methods
Recruitment
The current study was approved by the IRBs of the Khyber Medical University, Peshawar, Pakistan, and the University Hospitals of Geneva, Switzerland (Protocol number: CER 11-036). Informed consent forms were obtained from guardians of all affected individuals who participated in this study. Informed consent was obtained for the publication of photos from the guardians of the affected individuals of family 1.
Exome sequencing
The proband IV:1 of family 1 was subjected to exome sequencing (ES). DNA was enriched using SureSelect Human All Exon v6 capture kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq 4000 platform, with an average coverage of 120x at each nucleotide position. ES data were analyzed with an in-house customized pipeline8 that is based on published algorithms including BWA, SAMtools46, PICARD (http://broadinstitute.github.io/picard/) and (GATK)47. Initial screening for known or novel pathogenic mutations in the reported ID genes was performed. The 720 K SNP array was performed in parents (III:3 and III:4), affected (IV:1 and IV:7) and unaffected individuals (IV:3 and IV:5) of family 1to identify Runs of Homozygosity (ROH) using PLINK as described previously48,49,50. ROH and exome sequencing data were analyzed with CATCH51 to determine variants that were present in ROHs of patients (IV:1 and IV:7) but not in normal individuals of family 1. Subsequently, the variants were filtered manually by using the criteria described in published studies49,50.
The exome of IV:1 from family 2 was captured using the xGen Exome Research Panel v2 (Integrated DNA Technologies) and sequenced using the Illumina HiSeq 4000 platform according to the manufacturer’s protocols. The overall mean-depth base coverage was 153-fold and 97% of the targeted region was covered at least 20-fold. Read mapping and variant calling were performed as described52 using the Varapp software53. Homozygous and hemizygous variants with a MAF < 1% in the general population (1000genome, EVS, gnomAD) were retained and screened for variants in reported ID genes (Supplementary Table 1). Homozygosity mapping was performed with AutoMap, which uses Variant Call Format (VCF) files from WES54.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The identified variants have been submitted to ClinVar under accession numbers SCV001911505-506.
References
Iqbal, Z. & van Bokhoven, H. Identifying genes responsible for intellectual disability in consanguineous families. Hum. Hered. 77, 150–160 (2014).
Vissers, L. E. L. M., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016).
Maulik, P. K., Mascarenhas, M. N., Mathers, C. D., Dua, T. & Saxena, S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 32, 419–436 (2011).
Bruel, A.-L. et al. Next-generation sequencing approaches and challenges in the diagnosis of developmental anomalies and intellectual disability. Clin. Genet. 98, 433–444 (2020).
Vissers, L. E. L. M. et al. A de novo paradigm for mental retardation. Nat. Genet. 42, 1109–1112 (2010).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762 (2020).
Antonarakis, S. E. Carrier screening for recessive disorders. Nat. Rev. Genet. 20, 549–561 (2019).
Marchi, N. et al. Close inbreeding and low genetic diversity in Inner Asian human populations despite geographical exogamy. Sci. Rep. 8, 9397 (2018).
Tadmouri, G. O. et al. Consanguinity and reproductive health among Arabs. Reprod. Health 6, 17 (2009).
El-Attar, L. M., Bahashwan, A. A., Bakhsh, A. D. & Moshrif, Y. M. The prevalence and patterns of chromosome abnormalities in newborns with major congenital anomalies: a retrospective study from Saudi Arabia. Intractable Rare Dis. Res. advpub, (2021).
Andersen, J. P. et al. P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front. Physiol 7, 275 (2016).
Tanaka, Y. et al. The phospholipid flippase ATP9A is required for the recycling pathway from the endosomes to the plasma membrane. Mol. Biol. Cell 27, 3883–3893 (2016).
Van der Mark, V. A., Elferink, R. P. J. O. & Paulusma, C. C. P4 ATPases: flippases in health and disease. Int. J. Mol. Sci. 14, 7897–7922 (2013).
Takatsu, H. et al. ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J. Biol. Chem. 286, 38159–38167 (2011).
Naik, J. et al. The P4-ATPase ATP9A is a novel determinant of exosome release. PLoS ONE 14, e0213069 (2019).
Xu, X. et al. Effects of ATP9A on extracellular vesicle release and exosomal lipid composition. Oxid. Med. Cell. Longev. 2020, e8865499 (2020).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e24 (2019).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in genie. J. Comput. Biol. 4, 311–323 (1997).
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565 (2019).
Drielsma, A. et al. Two novel CCDC88C mutations confirm the role of DAPLE in autosomal recessive congenital hydrocephalus. J. Med. Genet. 49, 708–712 (2012).
Ekici, A. B. et al. Disturbed Wnt signalling due to a mutation in CCDC88C causes an autosomal recessive non-syndromic hydrocephalus with medial diverticulum. Mol. Syndromol. 1, 99–112 (2010).
Ruggeri, G. et al. Bi-allelic mutations of CCDC88C are a rare cause of severe congenital hydrocephalus. Am. J. Med. Genet. A 176, 676–681 (2018).
Kambouris, M. et al. Mutations in zinc finger 407 [ZNF407] cause a unique autosomal recessive cognitive impairment syndrome. Orphanet. J. Rare Dis. 9, 80 (2014).
Zahra, Q. et al. Biallelic ZNF407 mutations in a neurodevelopmental disorder with ID, short stature and variable microcephaly, hypotonia, ocular anomalies and facial dysmorphism. J. Hum. Genet. 65, 1115–1123 (2020).
Bamshad, M. J., Nickerson, D. A. & Chong, J. X. Mendelian gene discovery: fast and furious with no end in sight. Am. J. Hum. Genet. 105, 448–455 (2019).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Prezant, T. R., Chaltraw, W. E. & Fischel-Ghodsian, N. Identification of an overexpressed yeast gene which prevents aminoglycoside toxicity. Microbiology 142, 3407–3414 (1996).
Wehman, A. M., Poggioli, C., Schweinsberg, P., Grant, B. D. & Nance, J. The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr. Biol. 21, 1951–1959 (2011).
Brown, S. D. M. & Moore, M. W. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm. Genome 23, 632–640 (2012).
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011).
Emre Onat, O. et al. Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur. J. Hum. Genet. 21, 281–285 (2013).
Alsahli, S., Alrifai, M. T., Al Tala, S., Mutairi, F. A. & Alfadhel, M. Further delineation of the clinical phenotype of cerebellar ataxia, mental retardation, and disequilibrium syndrome type 4. J. Cent. Nerv. Syst. Dis. 10, 1179573518759682 (2018).
Guissart, C. et al. ATP8A2-related disorders as recessive cerebellar ataxia. J. Neurol. 267, 203–213 (2020).
Martín-Hernández, E. et al. New ATP8A2 gene mutations associated with a novel syndrome: encephalopathy, intellectual disability, severe hypotonia, chorea and optic atrophy. Neurogenetics 17, 259–263 (2016).
McMillan, H. J. et al. Recessive mutations in ATP8A2 cause severe hypotonia, cognitive impairment, hyperkinetic movement disorders and progressive optic atrophy. Orphanet J. Rare Dis. 13, 86 (2018).
Cacciagli, P. et al. Disruption of the ATP8A2 gene in a patient with a t(10;13) de novo balanced translocation and a severe neurological phenotype. Eur. J. Hum. Genet. 18, 1360–1363 (2010).
Raposo, G. & Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383 (2013).
Yáñez-Mó, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066 (2015).
Wang, S. et al. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J. Neurosci. 31, 7275–7290 (2011).
Klepper, J. et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 5, 354–365 (2020).
Wang, D., Pascual, J. M. & De Vivo, D. In GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, Seattle, 1993).
Kerner-Rossi, M., Gulinello, M., Walkley, S. & Dobrenis, K. Pathobiology of Christianson syndrome: linking disrupted endosomal-lysosomal function with intellectual disability and sensory impairments. Neurobiol. Learn. Mem. 165, 106867 (2019).
Morrow, E. M. & Pescosolido, M. F. In GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, Seattle, 1993).
Vogt, G. et al. Biallelic truncating variants in ATP9A cause a novel neurodevelopmental disorder involving postnatal microcephaly and failure to thrive. J. Med. Genet. https://doi.org/10.1136/jmedgenet-2021-107843 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Ansar, M. et al. Biallelic variants in LINGO1 are associated with autosomal recessive intellectual disability, microcephaly, speech and motor delay. Genet. Med. 20, 778–784 (2018).
Ansar, M. et al. Bi-allelic variants in DYNC1I2 cause syndromic microcephaly with intellectual disability, cerebral malformations, and dysmorphic facial features. Am. J. Hum. Genet. 104, 1073–1087 (2019).
Santoni, F. A. et al. Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res. 24, 349–355 (2014).
Alfaiz, A. A. et al. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum. Mutat. 35, 447–451 (2014).
Delafontaine, J. et al. Varapp: a reactive web-application for variants filtering. Preprint at bioRxiv https://doi.org/10.1101/060806 (2016).
Quinodoz, M. et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 12, 518 (2021).
Acknowledgements
We thank the probands and their families for their participation in this study. This work was supported by grants from the Swiss National Science Foundation (31003A_182632) and the Lejeune Foundation (JLF #1838) to A.R. and the Childcare Foundation to S.E.A. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
F.M. and A.R. wrote the manuscript with the help of S.E.A. and M.A. F.M. and M.A. performed WES, in silico analysis of the variant and Sanger sequencing. S.A.P. and H.M.A.B. provided clinical information for family 1. H.D., A.T., S.G.F., and M.C. collected genomic DNAs and clinical information for family 2. S.E.A., M.A., and A.R. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mattioli, F., Darvish, H., Paracha, S.A. et al. Biallelic truncation variants in ATP9A are associated with a novel autosomal recessive neurodevelopmental disorder. npj Genom. Med. 6, 94 (2021). https://doi.org/10.1038/s41525-021-00255-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-021-00255-z
- Springer Nature Limited
This article is cited by
-
Variant-specific pathophysiological mechanisms of AFF3 differently influence transcriptome profiles
Genome Medicine (2024)
-
Regulation of phospholipid distribution in the lipid bilayer by flippases and scramblases
Nature Reviews Molecular Cell Biology (2023)
-
ATP9A deficiency causes ADHD and aberrant endosomal recycling via modulating RAB5 and RAB11 activity
Molecular Psychiatry (2023)