

Abstract
The sodium (Na) superionic conductor is a key component that could revolutionize the energy density and safety of conventional Na-ion batteries. However, existing Na superionic conductors are primarily based on a single-anion framework, each presenting inherent advantages and disadvantages. Here we introduce a family of amorphous Na-ion conductors (Na2O2–MCly, M = Hf, Zr and Ta) based on the dual-anion framework of oxychloride. Benefiting from a dual-anion chemistry and with the resulting distinctive structures, Na2O2–MCly electrolytes exhibit room-temperature ionic conductivities up to 2.0 mS cm−1, wide electrochemical stability windows and desirable mechanical properties. All-solid-state Na-ion batteries incorporating amorphous Na2O2–HfCl4 electrolyte and a Na0.85Mn0.5Ni0.4Fe0.1O2 cathode exhibit a superior rate capability and long-term cycle stability, with 78% capacity retention after 700 cycles under 0.2 C (1C = 120 mA g−1) at room temperature. The discoveries in this work could trigger a new wave of enthusiasm for exploring new superionic conductors beyond those based on a single-anion framework.
Similar content being viewed by others

Main
Rechargeable Na-ion batteries (NIBs) are emerging as a viable substitute for lithium-ion batteries, especially for large-scale, economical energy storage, due to the Earth’s abundant sodium resources1,2,3. Despite this potential, the advancement of NIB technology is hindered by two primary challenges: (1) their relatively low energy density (~160 Wh kg−1), which undermines their market competitiveness, and (2) the use of flammable organic liquid electrolytes, which raises substantial safety concerns in practical applications4. To address these issues, the development of all-solid-state NIBs (ASSNIBs) is proposed as the optimal solution, offering several key advantages. First, replacing flammable liquid electrolytes with solid-state electrolytes (SSEs) substantially enhances the safety of NIBs5,6. Second, bipolar stacking markedly increases the volumetric energy density of ASSNIBs7. Third, if SSEs can resolve the long-lasting issue of Na dendrite growth, ASSNIBs using metallic Na anodes have the potential to achieve a maximum energy density.
The pursuit of advanced ASSNIBs has spurred rapid advancements in sodium-ion SSEs, a crucial component of these batteries8,9,10. Recent developments include various forms of SSEs: organic (polymer) SSEs, inorganic SSEs and hybrid composites11. Among these, inorganic SSEs can be further categorized into ceramic oxides, sulfides, halides and borohydrides according to their distinct anionic frameworks12. However, each type of SSE offers unique benefits and drawbacks. For instance, oxides provide a broad electrochemical stability window and excellent thermal stability, yet their mechanical rigidity necessitates high-temperature sintering to reduce interfacial and grain boundary impedance during device assembly13,14. Sulfides are noted for their high room-temperature ionic conductivities (~10−3 S cm−1), as well as their malleability and ease of forming intimate solid–solid interfaces via cold pressing15,16. However, they are challenged by narrow electrochemical stability windows and poor environmental resistance17. Halide SSEs, by contrast, offer impressive high-voltage and dry-air stability, but their low room-temperature ionic conductivities (~10−5–10−6 S cm−1) fall short of industrial requirements18,19. Borohydride SSEs exhibit superior ionic conductivity and good compatibility with metallic Na anodes, but their oxidation and thermal stability remain substantial concerns20,21. In essence, the limitations of these SSEs primarily stem from their reliance on single-anion frameworks, inherently limiting their performance capabilities. Thus, exploring innovative structural designs for sodium-ion SSEs based on mixed-anion frameworks could overcome the limitations of single-anion frameworks, potentially enabling the development of versatile, high-performance SSEs that could revolutionize ASSNIB technology.
In this work, we introduce a class of sodium-ion conductors featuring a dual-anion sublattice of oxychlorides (Na2O2–MCly, NMOC; M = Hf, Zr, Ta; y = 4, 5). These NMOC electrolytes achieve high ionic conductivities, up to 2.0 mS cm−1 at 25 °C, facilitated by the structure of oxychlorides, which enable rapid sodium-ion transport through the synergistic effects of bridging and non-bridging oxygen with distinct functionalities. Additionally, these SSEs exhibit several advantageous properties: a broad electrochemical stability window, excellent formability and a moderate Young’s modulus. When the Na2O2–HfCl4 (NHOC) electrolyte is directly paired with a layered oxide Na0.85Mn0.5Ni0.4Fe0.1O2 cathode without additional surface coating, the resulting ASSNIBs exhibit a remarkable rate performance and cycling stability over 700 cycles at 0.2 C, outperforming previous reports. This study paves the way for the development of new-generation Na-ion SSEs, advancing ASSNIB performance.
Synthesis and characterization of amorphous NMOC SSEs
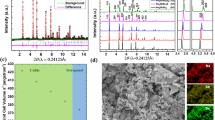
The oxychloride structure is exploited in developing sodium-ion superionic conductors because of the extended ionic bond lengths and substantial polarizabilities of oxygen and chlorine ions22. As a proof of concept, sodium peroxide (Na2O2) and hafnium tetrachloride (HfCl4) were initially selected to synthesize various stoichiometric ratios of oxychloride sodium-ion conductors (xNa2O2–HfCl4; 0.8 ≤ x ≤ 1.2) through a one-step mechanochemical process under an inert atmosphere. X-ray diffraction (XRD) analysis of these samples (Fig. 1a) revealed no distinct diffraction peaks at the stoichiometric ratio of x = 1.0 (that is, Na2O2–HfCl4 or NHOC), indicating an amorphous phase. This amorphous structure was further verified by additional synchrotron XRD, transmission electron microscopy imaging and diffraction patterns (Supplementary Fig. 1). Notably, an impurity phase of NaCl was prevalent in compositions with x > 1.0, while another impurity phase, Na2HfCl6, was more pronounced in lower Na2O2 molar ratios. The increase in Na2HfCl6 content was marked by stronger diffraction peaks as the Na2O2 content decreased.

a, Lab-based XRD patterns of xNa2O2–HfCl4 (0.8 ≤ x ≤ 1.2) and Na2HfCl6 SSEs synthesized employing an identical methodology. The crystalline impurity phase of NaCl is marked with asterisks. 2θ, the angle between the incident beam and diffracted beam. b, The temperature (T )-dependent ionic conductivities (σ) of the as-prepared xNa2O2–HfCl4 (0.8 ≤ x ≤ 1.2) SSEs. c–f, Lab-based XRD patterns (c and d) and Nyquist plots of the EIS measurement results (e and f) of NTOC and NZOC SSEs at 25 °C. Z′ represents the real part of the impedance, and Z″ represents the imaginary part of the impedance. g, Comparison of the ionic conductivity at 25 °C and activation energy (Ea) for the amorphous NMOC SSEs. h, Electrochemical stability windows of NMOC SSEs in comparison with that of the Na3PS4 electrolyte17.
Temperature-dependent ionic conductivities of the xNa2O2–HfCl4 (0.8 ≤ x ≤ 1.2) samples were evaluated through electrochemical impedance spectroscopy (EIS; Supplementary Fig. 2), revealing that room-temperature ionic conductivity gradually increased with an increasing Na2O2-to-HfCl4 ratio, reaching a peak of 0.52 mS cm−1 at x = 1.0 in amorphous NHOC (Fig. 1b and Supplementary Fig. 3). However, conductivity decreased with further increases in sodium peroxide content. The high ionic conductivity of the NHOC electrolyte benefits partly from its amorphous nature, wherein the intrinsic ionic conduction is unaffected by grain boundaries (Supplementary Fig. 2f)23. On the other hand, reductions in ionic conductivity in samples other than NHOC were linked to the presence of crystalline impurity phases of NaCl or Na2HfCl6. The ionic conductivity of amorphous NHOC was substantialy higher than that of its single-anion counterpart, Na2HfCl6 (0.02 mS cm−1), which was synthesized employing an identical methodology yet crystallized into a monoclinic structure (Supplementary Fig. 4 and Supplementary Tables 1 and 2). Correlating with the ionic conductivity results, Na+-ion transport within NHOC exhibits a lower migration barrier of 0.36 eV, compared to 0.42 eV across the well-defined channels in crystalline Na2HfCl6 (Supplementary Fig. 5). These enhancements can be attributed to the intrinsic structural characteristics of the oxychloride NHOC electrolyte, which will be further discussed in subsequent sections. Additionally, NHOC was identified as an electronic insulator relative to its fast Na+-ion migration (Supplementary Fig. 6), beneficial for inhibiting electrical leakage and ensuring prolonged operational life in ASSNIBs24.
To validate the versatility of the oxychloride framework for sodium-ion superionic conductors, Na2O2–TaCl5 (NTOC) and Na2O2–ZrCl4 (NZOC) were synthesized using binary compound precursors in equivalent molar ratios. Both materials formed amorphous phases with high bulk ionic conductivities of 2.0 and 0.42 mS cm−1 at room temperature, respectively (Fig. 1c–f), surpassing their crystalline halide counterparts, NaTaCl6 and Na2ZrCl6, as detailed in Supplementary Figs. 7 and 8 and Supplementary Tables 3 and 4. Furthermore, the notably lower activation energies of 0.30 eV for NTOC and 0.35 eV for NZOC suggest more efficient sodium-ion transport.
In addition to their appealing ion transport behaviour, these oxychloride electrolytes also exhibit beneficial mechanical properties and oxidation stability. Supplementary Figs. 9, 10 and 11a demonstrate that amorphous NMOC electrolytes can form dense morphology through a simple cold-pressing process, suggesting good deformability, which aids in resolving interfacial contact issues with cathode materials5. Importantly, their moderate Young’s modulus promises the maintenance of such intimate interfacial contact during long-term battery cycling (Supplementary Fig. 12)5,25. This property contrasts sharply with that of their crystalline halide counterparts, where obvious interparticle voids and grain boundaries are apparent in cold-pressed electrolyte pellets (Supplementary Figs. 11b, 13 and 14). On the other hand, the electrochemical stability windows of NHOC, NZOC and NTOC SSEs were determined to be in the ranges of 1.83–4.03 V, 1.97–3.94 V and 2.24–4.01 V (versus Na+/Na15Sn4), respectively (Fig. 1h and Supplementary Fig. 15). Their relatively low density (Supplementary Table 5) and high electrochemical oxidative stability make these NMOC SSEs promising in advancing high-energy-density ASSNIBs, although some compatibility issues with Na15Sn4 anodes were noted (Supplementary Figs. 16–18).
Structural analysis of amorphous NMOC SSEs
To gain insights into the relationship between local structures and Na+ diffusion within NMOC SSEs, comprehensive structural analyses were conducted. Raman spectra of the NHOC electrolyte exhibit the characteristic signatures at ~167 and 333 cm−1 (Fig. 2a), indicating the presence of discrete [HfCl6]2− octahedral units derived from the original HfCl4 structure26,27. Wavelet-transformed extended X-ray absorption fine structure (EXAFS) and Fourier-transform EXAFS analyses at the Hf L3-edge spectrum identified the presence of oxygen and provided additional structural details of NHOC. These analyses revealed two main peaks corresponding to Hf–O and Hf–Cl interactions, suggesting complex local bonding environments (Fig. 2b and Supplementary Fig. 19). The EXAFS fitting results (Fig. 2c) estimated the average coordination numbers of O and Cl around Hf in the first coordination sphere to be 2.3 and 3.5, respectively, with detailed coordination information summarized in Supplementary Table 6. Pair distribution function (PDF) analysis, G(r), helped clarify both the short- and medium-range structural configurations in NMOC SSEs (Fig. 2e). Consistent with the EXAFS results, the Hf atoms are closest to being surrounded by oxygen and chlorine in NHOC. Additionally, the PDF profile revealed specific distances that correspond to different types of connections between Hf-centred polyhedra. The Hf–Hf peak at about 3.4 Å mainly corresponds to the O edge-sharing connection of Hf-centred polyhedra, while distances of around 3.9 Å are due to both Cl edge-sharing connections and O corner-sharing connections28,29. Notably, the Raman characteristic vibrational frequencies and PDF features of the NHOC closely resemble those of NZOC but differ from those of NTOC (Fig. 2a,e), likely due to the chemical and structural similarities between Hf4+ and Zr4+ ions. The coordination information around Zr atoms in NZOC was also examined via wavelet-transformed EXAFS and Fourier-transform EXAFS fitting at the Zr K-edge, with detailed results presented in Supplementary Fig. 20 and Supplementary Table 7.

a, Raman spectra of amorphous NMOC SSEs and crystalline HfCl4, ZrCl4 and TaCl5 references. Insets are the representative local configurations of crystalline HfCl4, ZrCl4 and TaCl5, respectively. b, Wavelet-transformed EXAFS contour plots of NHOC SSE at Hf L3-edge. The original EXAFS signal χ(k) is weighted by k2, and k represents wavenumber. R + ΔR represents the radial distance, and ΔR indicates the distance correction due to phase shifts. c, Fourier-transform (FT) EXAFS fitting for the Hf L3-edge spectrum of NHOC SSE. Black and red circles represent the magnitude (Mag.) and real (Re.) part of the FT experimental (Exp.) EXAFS, respectively. d, Computed structure of amorphous NHOC at 500 K generated from melt-and-quench AIMD simulations, and relative populations of the Hf-centred clusters in the computed atomic configuration. e, PDF analysis of synchrotron total scattering data for amorphous NMOC electrolytes and the metal oxide and metal chloride references. The r represents the interatomic distance. f, Wavelet-transformed EXAFS contour plots of NTOC SSE at Ta L3-edge. The original EXAFS signal χ(k) is weighted by k3, and k represents wavenumber. g, FT EXAFS fitting for the Ta L3-edge spectrum of NTOC SSE. h,i, The main possible building blocks of NHOC or NZOC (h) and NTOC (i) SSEs. The ellipsis indicates the presence of other types of M-centred polyhedra as the basic building blocks of NMOC SSEs.
In addition to the experimental results, the theoretical atomic arrangement of amorphous NHOC was investigated by ab initio molecular dynamics (AIMD) simulations at different melt-quenching temperatures. The pair correlation functions (g(r)) of the generated model structure at 500 K most closely match the experimentally measured PDF profile in terms of the shapes and positions of the prominent Hf–O and Hf–Cl peaks (Supplementary Fig. 21). The minor discrepancies likely stem from challenges in characterizing the exact structures of experimentally synthesized amorphous materials30. Figure 2d shows a typical atomic structure of simulated NHOC from AIMD calculations. The existence of HfCl6 and different types of Hf–O–Cl polyhedral building blocks, including HfOCl5, HfO2Cl3, HfO3Cl2, HfO2Cl5 and so on, can be confirmed in the short-range order, and the relative populations of Hf-centred clusters are calculated and presented.
For the amorphous NTOC electrolyte, the presence of octahedral [TaCl6]− clusters was initially confirmed by Raman signals at 349, 369 and 405 cm−1 (Fig. 2a)31. Furthermore, Ta L3-edge wavelet-transformed EXAFS and the EXAFS fitting results confirmed the presence of Ta–O–Cl polyhedrons, with a centred Ta surrounded on average by two oxygens at 1.84 Å and 3.6 chlorines at 2.35 Å (Fig. 2f,g and Supplementary Table 8). The local geometries of primary Ta–O–Cl units were roughly determined via interpreting the PDF profiles (Fig. 2e). To be specific, the existence of both Ta–O–Cl tetrahedron and octahedron configurations could be proposed based on the Ta–O distance at around 1.93 Å (refs. 32,33). In the second-nearest region, the most distinct feature in the PDF profile is an intense peak at ~3.8 Å, which mainly corresponds to Ta–Ta pairs between Ta-centred polyhedra connecting via corner-shared oxygen (Supplementary Fig. 22)34.
Structural analyses of NMOC SSEs reveal that NHOC and NZOC feature complex Hf- or Zr-centred units with various interconnections, whereas NTOC mainly consists of [TaCl6]− clusters and O corner-sharing connectivity within Ta–O–Cl polyhedra (Fig. 2h,i). These structures, derived from mixed-valent anion chemistry, make NMOC species outstanding Na+-ion conductors. First, oxygen’s involvement causes local structural rearrangements that give NMOC an amorphous nature, which stands in contrast to crystalline halide counterparts that lack oxygen. This amorphous state in NMOC SSEs eliminates resistive grain boundaries, benefiting the high ionic conductivity5,35. Second, in NMOC, oxygen is predominantly coordinated with multivalent M4+/5+ cations instead of Na+ ions, existing as bridging and non-bridging oxygen atoms (Supplementary Fig. 23). Non-bridging oxygen aids in forming a relatively open framework that is conducive to Na+-ion conduction36. Bridging oxygen, as per the Anderson–Stuart model, primarily serves to connect M-centred polyhedra, enlarging the ‘doorway’ radius for easier access of Na+ ions within the structural network37,38. Furthermore, bridging oxygen also acts as a weak trap for Na+, easing the release of Na+ from Coulomb traps and enabling effective Na+ mobility between its initial and final sites39. These factors contribute to the dramatically enhanced Na+ conductivities and reduced activation energies in NMOC compared to the halide counterparts. It is important to note that an excess of oxygen beyond the optimal Na2O2/HfCl4 (1:1) ratio creates an O-rich environment around Hf atoms and leads to NaCl precipitation, which hampers Na+-ion conductivity (Fig. 1a,b and Supplementary Fig. 24). Furthermore, differences in local structures among NHOC, NZOC and NTOC influence their activation energies and thermal stability. Despite the comparable sodium atom ratios among the three SSEs (Supplementary Fig. 10), NTOC’s local structure possesses more oxygen corner-sharing units that lead to a higher degree of distortion around Na sites40,41, creating efficient pathways for fast Na+ transport with a low energy barrier (Supplementary Figs. 25 and 26), allowing NTOC to achieve the highest ionic conductivity and lowest activation energy among the NMOC electrolytes (Fig. 1g). By contrast, the robust local structure connectivity and bonds in NHOC and NZOC result in better thermal stability (Supplementary Fig. 27)42.
ASSNIBs based on amorphous NMOC SSEs
The electrochemical performance of the NMOC SSEs was evaluated in ASSNIBs, with the cell configuration depicted in Fig. 3a. The layered oxide Na0.85Mn0.5Ni0.4Fe0.1O2 (NMNFO) served as the cathode material (Supplementary Fig. 28), chosen for its appropriate voltage range and established electrochemical performance in liquid-based NIBs43. Among the three oxychloride electrolytes, the NHOC-based ASSNIBs demonstrated superior electrochemical performance. As shown in Supplementary Fig. 29, an initial discharge capacity of 125.5 mAh g−1 with a coulombic efficiency exceeding 99.9% was achieved by the NHOC-based ASSNIB at 0.1 C, comparable to that of the liquid-based cell using the NMNFO cathode43. The overlapping charge and discharge profiles during the initial three cycles indicated a highly reversible phase transition of the NMNFO cathode within NHOC-based ASSNIBs.

a, Schematic diagram of NMOC-based ASSNIBs. CNTs, carbon nanotubes. b, Initial galvanostatic intermittent titration technique charge and discharge curves of NHOC-based ASSNIB at 0.1 C. The blue and green lines represent charge and discharge profiles, respectively. The red balls represent the charge and discharge open circuit voltage. c, Na+ diffusion coefficients (DNa+) at different electrochemical states of NHOC-based ASSNIB. d,e, The second charge and discharge curves (d) and rate capability (e) of the NHOC-based ASSNIB under various current densities at room temperature. f,g, Galvanostatic voltage profiles (f) and specific capacities (g) of ASSNIBs as a function of cycle number, running at 0.2 C and room temperature (r.t.). All the NHOC-based ASSNIBs were tested in the voltage range of 2.0–4.0 V versus Na+/Na15Sn4.
The favourable electrochemical process in the ASSNIBs was further confirmed by galvanostatic intermittent titration technique results, which show a low polarization deviation and effective Na+-ion transport dynamics during the initial cycle (Fig. 3b,c and Supplementary Fig. 30). The rate capability of the designed ASSNIBs was evaluated by increasing current densities from 0.1 to 1 C at room temperature. The NHOC-based ASSNIB delivered reversible discharge capacities of 119.7, 111, 100, 81.9 and 31.4 mAh g−1 at 0.1, 0.2, 0.3, 0.5 and 1.0 C, respectively (Fig. 3d,e). While the limited ionic conductivity of the Na3PS4 interlayer (Supplementary Fig. 31) notably affected the achievable capacities at higher rates, the cell retained 95.3% of its initial capacity when the C rate was returned to 0.1 C. Impressively, the NHOC-based ASSNIB maintained 78% capacity retention over 700 cycles at 0.2 C, with an average coulombic efficiency around 99.9% (Fig. 3f,g). This performance ranks among the best for all-solid-state sodium batteries operating at room temperature, in terms of capacity retention and cycle numbers (Supplementary Fig. 32 and Supplementary Table 9).
The electrochemical performance of NZOC- and NTOC-based ASSNIBs was also studied. ASSNIBs employing NZOC electrolyte exhibited a slightly inferior rate capability and cycling stability compared to those with NHOC electrolyte. Within the operational voltage window of 2.1–3.8 V, the ASSNIB equipped with NZOC electrolyte delivered specific capacities of 116.6, 103.1, 91.1, 73.5 and 17.3 mAh g−1 at 0.1, 0.2, 0.3, 0.5 and 1 C, respectively (Supplementary Fig. 33a–d). Furthermore, the NZOC electrolyte enables the ASSNIB to sustain 700 cycles at a 0.2 C rate, which yields a capacity retention of 72%. By sharp contrast, despite NTOC having the highest ionic conductivity among the three electrolytes, NTOC-based ASSNIBs manifested a suboptimal electrochemical performance (Supplementary Fig. 33e–h). At 0.1 C within a voltage range of 2.3–3.8 V, the NTOC-based ASSNIB exhibited a reversible capacity of 106.6 mAh g−1, but with an increase in current density, capacity dramatically dropped to only 7.1 mAh g−1 at 1 C. Additionally, a notable decline in reversible capacity was evident during cycling at 0.2 C, with only 43% capacity retention after 400 cycles. Consistent with the cycling stability, performance decay in NTOC-based cells was evident during the initial cycles, contrasting with the essentially overlapped cyclic voltammetry curves in the NHOC-based system (Supplementary Fig. 34).
Interfacial compatibility between NMNFO cathode and NMOC SSEs
Interfacial compatibility, encompassing mechanical, chemical and electrochemical aspects, is essential for the long-term performance of ASSNIBs25. Scanning electron microscopy images and elemental mapping of the NHOC-based cathode reveal that NMNFO particles are well embedded within the NHOC SSE, with the particle edges fully encapsulated (Fig. 4a and Supplementary Fig. 35). This indicates effective interfacial contact, achieved through cold pressing, attributed to NHOC’s excellent deformability and adhesion to the NMNFO cathode. The advantageous mechanical properties of the NHOC SSE are further demonstrated by the close interfacial adhesion between the NHOC electrolyte and the cathode composite layer (Fig. 4b), as well as the seamless NHOC–Na3PS4 interface (Supplementary Fig. 36). Analogous favourable physical interfacial contact was observed in NZOC- and NTOC-based ASSNIBs, facilitating effective ionic paths between different cell components. Despite the challenge of maintaining such integrity in battery components, with stress-induced cracks and delamination often degrading performance44, these issues were negligible in cycled NHOC-based ASSNIBs (Supplementary Figs. 37–39). The structural durability benefits partially from NHOC’s suitable Young’s modulus, which compensates for volume expansion during the sodiation of the cathode. Additionally, an appropriate stack pressure plays a vital role in maintaining the cell performance and should not be overlooked (Supplementary Fig. 40)45.

a,b, Scanning electron microscopy images of the NHOC-based cathode composite (a) and the interphase between the cathode composite and NHOC electrolyte layer (b). c,d, Ex situ Hf 4f (c) and Cl 2p (d) X-ray photoelectron spectra of cathode composites at different charge–discharge states, that is, the initial state, fully charged to 4 V (C-4V), fully discharged to 2 V (D-2V), after three cycles and after five cycles at 0.2 C. The spin doublet components are shaded with the same colour for each spectrum. e, Intermittent galvanostatic voltage profile of NHOC-based ASSNIB in the initial three cycles. The ASSNIB was cycled at 20 mA g−1, and each impedance spectrum was recorded after a rest period of 1 h to allow for reaching equilibrium. The time (h) here represent only the charging and discharging time, excluding any resting periods. f, Distribution of relaxation times spectra calculated from EIS measurements. The τ represents relaxation time, and γ (τ) stands for distribution function of relaxation times. g, Two-dimensional intensity colour map of the charge- and discharge-dependent distribution of relaxation times curves abstracted from f.
Chemical and electrochemical compatibility of the NHOC SSE with the NMNFO cathode was subsequently investigated by X-ray photoelectron spectroscopy. The Hf 4f and Cl 2p X-ray photoelectron spectra of the cathode composite at various electrochemical states showed no noticeable peak shifts (Fig. 4c,d), suggesting the excellent chemical and electrochemical stability of the NHOC SSE when paired with a bare NMNFO cathode. This stability was corroborated by consistent Hf L3-edge X-ray absorption fine structure (XAFS) spectra of the NHOC-based cathode after cycling (Supplementary Fig. 41). Similarly, no notable changes in the chemistry of the NZOC-based cathode were observed in Zr K-edge XAFS spectra after 50 cycles. Accordingly, the prolonged, stable cycling of NHOC- and NZOC-based ASSNIBs stems not only from their superior structural integrity but also from the excellent chemical and electrochemical compatibility between the electrolyte and the NMNFO cathode. By stark contrast, the Ta L3-edge XAFS spectra of the NTOC cathode showed a slight shift towards higher energy after cycling (Supplementary Fig. 41c), indicating its electrochemical instability with the NMNFO cathode. Comparative morphological analyses on pristine and post-cycled NTOC-based ASSNIBs further suggested that the formation of an unfavourable electrode–electrolyte interphase, rather than structural destruction, was responsible for the low coulombic efficiency and substantial capacity decay, particularly in the initial 75 cycles (Supplementary Fig. 42). Addressing these compatibility issues between the highly ionic conductive NTOC electrolyte and cathode materials is expected to substantially enhance the electrochemical performance of ASSNIBs.
In situ impedance spectra collected during the first three cycles showed how resistance evolved in the NHOC-based ASSNIB (Fig. 4e). Analysis of the distribution of relaxation times linked dominant time constants in impedance measurements to specific electrochemical behaviours within the ASSNIBs46. The distribution of relaxation times spectra, derived from the impedance data (Fig. 4f and Supplementary Fig. 43), showed six distinct peaks. Peaks above 105 Hz (P1) likely originate from the contact resistance at current collectors, electrode interfaces and electrode particles47,48. The modestly intense P2 and P3 peaks, showing minimal variation, are attributed to ion transport across the Na3PS4 interlayer and the NHOC electrolyte layer, respectively. Peaks P4 and P5 were associated with ion transport at the anode and cathode interfaces49,50. Relaxation peaks related to solid-state diffusion have the largest time constants (frequency below 0.1 Hz) and are labelled as P6 in the distribution of relaxation times spectrum49,50. Changes observed in peaks P4 and P6, which were greatly affected by electrochemical states (Fig. 4g), indicated relatively sluggish reaction kinetics at the end of the sodiation process, consistent with the observed voltage hysteresis changes in Supplementary Fig. 30. The change of the P4 peak could be related to the interfacial kinetics between the Na–Sn and SSE interface, which strongly depend on the Na content in the Na–Sn alloy anode. This phenomenon is similar to the changes in In–SSE interfacial resistance in solid-state Li-ion batteries51. On the cathode side, solid-state diffusion becomes sluggish at the end of discharging due to increased energy requirements for further sodiation as Na+ sites in the cathode structure are approaching saturation. Crucially, the consistency in relaxation peak changes across the initial cycles confirmed the excellent reversibility of the electrochemical process when integrating NHOC SSE with a bare NMNFO cathode.
In conclusion, this study presents a breakthrough in the development of ASSNIBs through the introduction of a family of dual-anion-based sodium superionic conductors, NMOCs. These NMOC electrolytes, leveraging a dual-anion sublattice of oxychlorides, have demonstrated exceptional ionic conductivities up to 2.0 mS cm−1, favourable mechanical ductility and high oxidative stability. Our findings reveal that the enhanced Na+-ion mobility is primarily facilitated by the innovative structure of the oxychloride framework, which optimizes ion transport pathways and minimizes migration barriers through synergistic effects of bridging and non-bridging oxygens. Notably, NHOC-based ASSNIBs have shown outstanding cycling stability, maintaining 78% capacity after 700 cycles at 0.2 C at room temperature, highlighting the critical role of interfacial compatibility in extending battery life and efficiency. These findings open pathways for the development of next-generation SSEs and highlight the profound implications of adopting mixed-anion systems in enhancing energy storage technologies, potentially transforming future approaches to battery design and application.
Methods
Material synthesis
The oxychloride xNa2O2–MCly (M = Hf, Zr and Ta; 0.8 ≤ x ≤ 1.2), Na3PS4 and sodium metal halide (Na2ZrCl6, Na2HfCl6 and NaTaCl6) solid electrolytes used in this work were synthesized by a mechanochemical method using high-energy ball milling. All preparations and sample treatments were carried out under an Ar atmosphere (O2 < 1 ppm, H2O < 1 ppm). HfCl4 (Sigma-Aldrich, 98%), ZrCl4 (Sigma-Aldrich, ≥99.5%), TaCl5 (Sigma-Aldrich, ≥99.99%), Na2O2 (Sigma-Aldrich, 97%) and NaCl (Sigma-Aldrich, >99%) were used as the starting materials without further purification. For the preparation of oxychloride NMOC SSEs, an ~1 g stoichiometric mixture of metal chloride (ZrCl4, HfCl4 or TaCl5) and Na2O2 was placed in an 80 ml ZrO2 ball mill jar with 40 g ZrO2 balls (four 10-mm-diameter (D10) balls, eight D8 balls and the rest being D5 balls), and then milled at 500 rpm for 10 h (Retsch Emax). The synthesis procedures of the sodium metal halides (Na2ZrCl6, Na2HfCl6 and NaTaCl6) were like that of the oxychloride NMOC electrolytes, with metal chlorides (ZrCl4, HfCl4 or TaCl5) and NaCl used as the starting precursors.
For the synthesis of Na3PS4, the starting materials of Na2S (Sigma-Aldrich, 99%) and P2S5 (Sigma-Aldrich, 99%) were milled at 170 rpm for 2 h and then 500 rpm for 10 h in an 80 ml ZrO2 ball mill jar. The ball-milled product was extracted from the jars in the glove box, pelletized and then loaded into vacuumed quartz tubes. The Na3PS4 was obtained after being annealed at 270 °C for 2 h. The obtained Na3PS4 was extracted from the quartz tubes and manually ground into powder for future use. The Na15Sn4 anode was prepared by mixing stoichiometric amounts of Na (Sigma-Aldrich, 99.95% trace metals basis) and Sn (Sigma-Aldrich, 99%) in an Ar-filled glove box until a dark grey powder was obtained.
The NMNFO cathode material was synthesized by mixing a stoichiometric amount of Na2CO3·H2O (Alfa Aesar, 99%), Mn2O3 (Sigma-Aldrich, 99%), NiO (Alfa Aesar, 99%) and Fe2O3 (Alfa Aesar, 99%) thoroughly using planetary ball milling at 550 rpm for 12 h. Afterward, the mixture was pressed into pellets and annealed in a tube furnace under oxygen. The solid-state synthesis followed a two-step process. First, the furnace was heated to 500 °C with a ramping rate of 5 °C min–1 and kept isothermal for 5 h. Then, the furnace temperature was increased to 900 °C at the same ramping rate of 5 °C min–1 and kept there for 14 h. Afterward, the furnace was cooled to room temperature with a rate of 3 °C min–1.
Characterizations
The lab-based XRD patterns were collected using a Bruker AXS D8 Advance with Cu Kα radiation (wavelength, λ = 1.5418 Å). Regular tests were conducted at a scan rate of 10° min–1 from 10 to 80°. Scanning electron microscopy images and elemental mapping were obtained using a Hitachi S-4800 field-emission scanning electron microscope equipped with energy dispersive spectroscopy. High-resolution transmission electron microscopy imaging and selected area electron diffraction patterns of the NHOC electrolyte were performed by a Cs-corrected Titan Themis 80-300 transmission electron microscope operated at 300 kV. Raman spectra were collected on a HORIBA Scientific LabRAM Raman spectrometer equipped with a 532.03 nm laser. During XRD and Raman characterizations, all samples were placed into airtight holders to avoid possible air exposure. The X-ray photoelectron spectroscopy data were collected with a monochromatic Al Kα source (1,486.6 eV) using a Thermo Scientific K-Alpha spectrometer. The thermal stability of the NMOC electrolytes was examined using a Mettler Toledo TGA2 thermogravimetric analyser, and the measurements were carried out at a heating rate of 10 °C min−1 from room temperature to 800 °C. The densities of the NMOC SSEs were determined by a True Density Meter (Anton Paar), and the measurements were conducted using approximately 3 g of NMOC electrolyte powder to minimize experimental errors. In situ nanoindentation tests were conducted using an Alemnis Nanoindenter system inside an Apreo-2S field-emission gun scanning electron microscope. Tests were conducted under vacuum at room temperature to maintain environmental consistency.
The synchrotron-based powder XRD and PDF measurements were collected using the Brockhouse High Energy Wiggler beamline at the Canadian Light Source (CLS) with a wavelength of 0.3497 Å. The samples were loaded into 0.8-mm-inner-diameter polyimide capillaries and sealed with epoxy in an Ar-filled glove box. The XRD Rietveld refinement and PDF fittings were conducted by GSAS-2 and PDFgui software52,53. The synchrotron X-ray computed tomography was carried out at the Biomedical Imaging and Therapy Facility 05B1-1 beamline at the CLS. X-ray absorption spectroscopy at the Na K-edge and O K-edge was collected from beamlines 7.3.1 and 8.0.1.4 at the Advanced Light Source, Lawrence Berkeley National Laboratory. X-ray absorption spectroscopy at the Cl K-edge was carried out on the soft X-ray microcharacterization beamline at CLS. The Hf L3-edge, Ta L3-edge and Zr K-edge X-ray absorption fine structure data were measured at the Hard X-ray MicroAnalysis beamline at the CLS. All X-ray absorption spectroscopy data were analysed with the Athena software, and the EXAFS data were processed with the Artemis program54.
Atomic structure characterization of NHOC and NTOC
The NHOC structure model is composed of HfCl4 and Na2O2, while the NTOC structure model is composed of TaCl5 and Na2O2. The amorphous structures of NHOC and NTOC configurations were prepared by a heat-and-quench method based on AIMD simulations. All AIMD simulations were performed with a time step of 2 fs in an NVE ensemble (constant number of atoms (N), volume (V) and energy (E)). We heated the NHOC and NTOC to 1,200 K for about 5 ps, and then quenched them to different temperatures of 500 K, 600 K or 1,000 K at a speed of 0.1 K fs–1. Afterward, the amorphous structures were equilibrated at three different temperatures of 500 K, 600 K and 1,000 K for 10 ps. The pair correlation functions g(r), coordination numbers and neighbouring clusters within 3 Å were calculated by averaging over 100 different configuration samples in the equilibrate state.
Given the complexity of identifying the amorphous structure compared to the crystal structure, AIMD simulations were conducted to directly observe the Na+ migration pathways and the migration barriers in the amorphous NHOC and NTOC electrolytes. These pathways were derived from the observed Na-ion hopping pathways using identical supercell models, confirming that the NTOC electrolyte with more oxygen corner-sharing frameworks is beneficial for fast Na+-ion transport. Furthermore, we calculate the Na migration barrier along the oxygen and chlorine corner-sharing pathways within 4.0 Å for NHOC and NTOC SSEs. The ion diffusion pathways and their associated energy profiles were assessed using the nudged-elastic-band method. The initial and final structures were fully relaxed, with the intermediate images interpolated between them. In the nudged-elastic-band calculations, energy and force convergence criteria were set at 1 × 10−6 eV per atom and 1 × 10−4 eV Å–1, respectively. The energy barriers (Ea) were determined by computing the difference between the maximum and initial energies along the diffusion pathways, serving to represent the energy barrier surmounted by mobile ions. To ensure a consistent comparison across various pathways, the energy profiles were referenced to the lowest energy site within each migration pathway.
Conductivity measurement
The ionic conductivity of the as-prepared samples was measured by a.c. impedance spectroscopy. For the temperature-dependent EIS measurement, ~120–130 mg of the samples was cold pressed into pellets with a diameter of 10 mm at ~330 MPa, and two stainless steel rods served as blocking electrodes. Impedance spectroscopy was performed using a VMP3 potentiostat/galvanostat (BioLogic) in the frequency range between 1,000 mHz and 7 MHz at different temperatures, and the amplitude was 20 mV. The electronic conductivity of the sample was evaluated according to the direct current polarization measurement on a cold-pressed pellet with applied voltages from 0.2 V to 1.0 V at room temperature.
The activation energies of the as-prepared solid electrolytes were calculated using the following equation:
where σ is the ionic conductivity, σ0 is the Arrhenius prefactor, T is absolute temperature, Ea is the activation energy and kB is the Boltzmann constant.
Electrochemical characterizations
The electrochemical stability of NMOC solid electrolytes was determined by cyclic voltammetry and linear sweep voltammetry measurements using a versatile multichannel potentiostat 3/Z (VMP3) with a Na15Sn4 | Na3PS4 | NMOC | NMOC + carbon black (weight ratio, NMOC/carbon black = 8:2) cell configuration. For battery assembly, 100 mg NMOC powder was first cold pressed into a SSE pellet. After that, a 10 mg working electrode (NMOC + carbon black) and 30 mg Na3PS4 were dispersed evenly, with one on each side of the solid electrolyte pellet, and then pressed at ~300 MPa. Finally, 50 mg Na15Sn4 alloy was uniformly pressed on the Na3PS4 side at ~400 MPa to act as the anode material. The cathodic and anodic stabilities of NMOC were measured with a positive scan range from the open-circuit voltage to 6 V, and a negative scan range from the open-circuit voltage to 0 V, respectively. The scan rate for cyclic voltammetry and linear sweep voltammetry characterizations was 0.1 mV S−1. The stability of NMOC electrolytes towards Na15Sn4 was studied by conducting time-resolved EIS and cycling stability tests on Na15Sn4 | NMOC | Na15Sn4 symmetric cells, which were assembled by pressing 50 mg Na15Sn4 powder on both sides of the cold-pressed NMOC electrolyte pellet (140 mg).
The cathode composite for ASSNIBs was prepared by mixing NMNFO active material powder with NMOC SSE using an agate mortar and pestle, with a weight ratio of 6:4, and then 2 wt% carbon nanotubes were added as the conductive material. For battery assembly, 100 mg as-synthesized NMOC was compressed at ~330 MPa to form a SSE layer. Then, 8 mg of the cathode composite and 30 mg Na3PS4 were dispersed evenly, with one on each side of the solid electrolyte pellet, and further pressed at ~330 MPa. Finally, 50 mg Na15Sn4 alloy was pressed on the Na3PS4 side at ~440 MPa. The cells were cycled under an external pressure of ~110 MPa. All the batteries were assembled in the Ar-filled glove box and then given a rest period of 2 h to reach equilibrium before the electrochemical measurement. In situ EIS measurements (2 MHz to 100 mHz) were recorded using the VMP3 potentiostat/galvanostat (BioLogic) during the initial three cycles of the ASSNIB. The cell was cycled at 20 mA g−1 for 1 h, and impedance spectra were recorded after a 1 h rest during both the charge and discharge processes.
Data availability
Data supporting findings from this work are available within this Article and the Supplementary Information. All other relevant data supporting findings are available from the corresponding author on request. Source data are provided with this paper.
Code availability
The code used to generate the atomic configurations of NHOC in the present work is available from the corresponding author upon request.
References
Hwang, J.-Y., Myung, S.-T. & Sun, Y.-K. Sodium-ion batteries: present and future. Chem. Soc. Rev. 46, 3529–3614 (2017).
Vaalma, C., Buchholz, D., Weil, M. & Passerini, S. A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 3, 18013 (2018).
Peng, J. et al. Ice-assisted synthesis of highly crystallized Prussian blue analogues for all-climate and long-calendar-life sodium ion batteries. Nano Lett. 22, 1302–1310 (2022).
Yang, H.-L. et al. Progress and challenges for all-solid-state sodium batteries. Adv. Energy Sustain. Res. 2, 2000057 (2021).
Chi, X. et al. An electrochemically stable homogeneous glassy electrolyte formed at room temperature for all-solid-state sodium batteries. Nat. Commun. 13, 2854 (2022).
Yang, C., Xin, S., Mai, L. & You, Y. Materials design for high-safety sodium-ion battery. Adv. Energy Mater. 11, 2000974 (2021).
Heo, J. W., Banerjee, A., Park, K. H., Jung, Y. S. & Hong, S.-T. New Na-ion solid electrolytes Na4−xSn1−xSbxS4 (0.02 ≤ x ≤ 0.33) for all-solid-state Na-ion batteries. Adv. Energy Mater. 8, 1702716 (2018).
Kim, J.-J., Yoon, K., Park, I. & Kang, K. Progress in the development of sodium-ion solid electrolytes. Small Methods 1, 1700219 (2017).
Hou, W. et al. Solid electrolytes and interfaces in all-solid-state sodium batteries: progress and perspective. Nano Energy 52, 279–291 (2018).
Lin, X. et al. A dual anion chemistry-based superionic glass enabling long-cycling all-solid-state sodium-ion batteries. Angew. Chem. Int. Ed. 63, e202314181 (2024).
Lu, Y., Li, L., Zhang, Q., Niu, Z. & Chen, J. Electrolyte and interface engineering for solid-state sodium batteries. Joule 2, 1747–1770 (2018).
Zhao, Q., Stalin, S., Zhao, C.-Z. & Archer, L. A. Designing solid-state electrolytes for safe, energy-dense batteries. Nat. Rev. Mater. 5, 229–252 (2020).
Lin, X. et al. Reviving anode protection layer in Na-O2 batteries: failure mechanism and resolving strategy. Adv. Energy Mater. 11, 2003789 (2021).
Singh, K., Chakraborty, A., Thirupathi, R. & Omar, S. Recent advances in NASICON-type oxide electrolytes for solid-state sodium-ion rechargeable batteries. Ionics 28, 5289–5319 (2022).
Zhang, Z. et al. Na11Sn2PS12: a new solid state sodium superionic conductor. Energy Environ. Sci. 11, 87–93 (2018).
Hayashi, A., Noi, K., Sakuda, A. & Tatsumisago, M. Superionic glass-ceramic electrolytes for room-temperature rechargeable sodium batteries. Nat. Commun. 3, 856 (2012).
Tang, H. et al. Probing solid–solid interfacial reactions in all-solid-state sodium-ion batteries with first-principles calculations. Chem. Mater. 30, 163–173 (2018).
Kwak, H. et al. Na2ZrCl6 enabling highly stable 3 V all-solid-state Na-ion batteries. Energy Storage Mater. 37, 47–54 (2021).
Wu, E. A. et al. A stable cathode-solid electrolyte composite for high-voltage, long-cycle-life solid-state sodium-ion batteries. Nat. Commun. 12, 1256 (2021).
Sadikin, Y., Brighi, M., Schouwink, P. & Černý, R. Superionic conduction of sodium and lithium in anion-mixed hydroborates Na3BH4B12H12 and (Li0.7Na0.3)3BH4B12H12. Adv. Energy Mater. 5, 1501016 (2015).
Udovic, T. J. et al. Exceptional superionic conductivity in disordered sodium decahydro-closo-decaborate. Adv. Mater. 26, 7622–7626 (2014).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Li, F. et al. Amorphous chloride solid electrolytes with high Li-ion conductivity for stable cycling of all-solid-state high-nickel cathodes. J. Am. Chem. Soc. 145, 27774–27787 (2023).
Han, F. et al. High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes. Nat. Energy 4, 187–196 (2019).
Hao, F. et al. Taming active material-solid electrolyte interfaces with organic cathode for all-solid-state batteries. Joule 3, 1349–1359 (2019).
Salyulev, A. B. & Vovkotrub, E. G. Raman spectroscopy study of products of reactions of zirconium and hafnium tetrachlorides with indium and thallium monochlorides. Russ. J. Appl. Chem. 86, 687–690 (2013).
Rak, J., Gutowski, M., Dokurno, P., Thanh, H. V. & Bl/ażejowski, J. Theoretical studies on structure, thermochemistry, vibrational spectroscopy, and other features of ZrX2−6 (X=F,Cl,Br,I): coulombic energy in inorganic and organic hexahalogenozirconates. J. Chem. Phys. 100, 5810–5820 (1994).
Firth, F. C. N. et al. Exploring the role of cluster formation in UiO family Hf metal–organic frameworks with in situ X-ray pair distribution function analysis. J. Am. Chem. Soc. 143, 19668–19683 (2021).
Christensen, R. S., Kløve, M., Roelsgaard, M., Sommer, S. & Iversen, B. B. Unravelling the complex formation mechanism of HfO2 nanocrystals using in situ pair distribution function analysis. Nanoscale 13, 12711–12719 (2021).
Lacivita, V. et al. Resolving the amorphous structure of lithium phosphorus oxynitride (Lipon). J. Am. Chem. Soc. 140, 11029–11038 (2018).
Xie, T., Brockner, W. & Gjikaj, M. New ionic liquid compounds based on tantalum pentachloride TaCl5: synthesis, structural, and spectroscopic elucidation of the (μ-oxido)-chloridotantalates(V) [BMPy][TaCl6], [BMPy]4[(TaCl6)2(Ta2OCl10)], and [EMIm]2[Ta2OCl10]. Z. Anorg. Allg. Chem. 636, 2633–2640 (2010).
Benjamin, S. L., Hyslop, A., Levason, W. & Webster, M. [Cl5Ta(μ-O)TaCl3{iPrS(CH2)2SiPr}] and [(TaCl4)2(μ-O)(μ-Me2Se2)]: two chalcogenoether complexes of Ta2OCl8 with very different geometries. Acta Crystallogr. C 67, m221–m223 (2011).
Tanaka, Y. et al. New oxyhalide solid electrolytes with high lithium ionic conductivity >10 mS cm−1 for all-solid-state batteries. Angew. Chem. Int. Ed. 62, e202217581 (2023).
Shyam, B. et al. Measurement and modeling of short and medium range order in amorphous Ta2O5 thin films. Sci. Rep. 6, 32170 (2016).
Sun, G. et al. Electrochemically induced crystalline-to-amorphization transformation in sodium samarium silicate solid electrolyte for long-lasting sodium metal batteries. Nat. Commun. 14, 6501 (2023).
Caurant, D. et al. Structural investigations of borosilicate glasses containing MoO3 by MAS NMR and Raman spectroscopies. J. Nucl. Mater. 396, 94–101 (2010).
Kim, Y., Saienga, J. & Martin, S. W. Anomalous ionic conductivity increase in Li2S + GeS2 + GeO2 glasses. J. Phys. Chem. B 110, 16318–16325 (2006).
Anderson, O. L. & Stuart, D. A. Calculation of activation energy of ionic conductivity in silica glasses by classical methods. J. Am. Ceram. Soc. 37, 573–580 (1954).
Minami, T., Hayashi, A. & Tatsumisago, M. Recent progress of glass and glass-ceramics as solid electrolytes for lithium secondary batteries. Solid State Ion. 177, 2715–2720 (2006).
Jun, K. et al. Lithium superionic conductors with corner-sharing frameworks. Nat. Mater. 21, 924–931 (2022).
Zhang, S. et al. A family of oxychloride amorphous solid electrolytes for long-cycling all-solid-state lithium batteries. Nat. Commun. 14, 3780 (2023).
Lu, K. & Mahapatra, M. K. Network structure and thermal stability study of high temperature seal glass. J. Appl. Phys. 104, 074910 (2008).
Xiao, B. et al. A general strategy for batch development of high-performance and cost-effective sodium layered cathodes. Nano Energy 89, 106371 (2021).
He, Y., Lu, C., Liu, S., Zheng, W. & Luo, J. Interfacial incompatibility and internal stresses in all-solid-state lithium ion batteries. Adv. Energy Mater. 9, 1901810 (2019).
Doux, J.-M. et al. Pressure effects on sulfide electrolytes for all solid-state batteries. J. Mater. Chem. A 8, 5049–5055 (2020).
Wan, T. H., Saccoccio, M., Chen, C. & Ciucci, F. Influence of the discretization methods on the distribution of relaxation times deconvolution: implementing radial basis functions with DRTtools. Electrochim. Acta 184, 483–499 (2015).
Li, X. et al. Highly stable halide-electrolyte-based all-solid-state Li–Se batteries. Adv. Mater. 34, 2200856 (2022).
Chen, X., Li, L., Liu, M., Huang, T. & Yu, A. Detection of lithium plating in lithium-ion batteries by distribution of relaxation times. J. Power Sources 496, 229867 (2021).
Gargh, P. et al. Correlating capacity fade with film resistance loss in fast charging of lithium-ion battery. J. Power Sources 485, 229360 (2021).
Illig, J., Schmidt, J. P., Weiss, M., Weber, A. & Ivers-Tiffée, E. Understanding the impedance spectrum of 18650 LiFePO4-cells. J. Power Sources 239, 670–679 (2013).
Zhang, W. et al. Interfacial processes and influence of composite cathode microstructure controlling the performance of all-solid-state lithium batteries. ACS Appl. Mater. Interfaces 9, 17835–17845 (2017).
Toby, B. H. & Von Dreele, R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Farrow, C. L. et al. PDFfit2 and PDFgui: computer programs for studying nanostructure in crystals. J. Phys. Condens. Matter 19, 335219 (2007).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Research Chair Program (CRC), the Canada Foundation for Innovation (CFI), the Ontario Research Fund, the Canadian Light Source (CLS) at the University of Saskatchewan and the University of Western Ontario. Part of this work was conducted at the Advanced Light Source, which is a US Department of Energy Office of Science User Facility, under contract no. DE-AC02-05CH11231. M.Y. acknowledges support from the National Natural Science Foundation of China (52302302). X. Lin acknowledges support from the CLSI Graduate and Post-Doctoral Student Travel Support Program. We appreciate the help of the Hard X-ray MicroAnalysis (HXMA) beamline scientists at CLS, N. Chen and W. Chen. We also thank Q. Sun for helpful discussions.
Author information
Authors and Affiliations
Contributions
X. Lin designed and conducted the experiments, performed most of the characterizations and draughted the manuscript. S.Z. helped with the synchrotron-related data analysis and writing. M.Y. helped with the computational simulations, structural analyses and manuscript writing. B.X. helped with the cathode material synthesis and data analysis. Y.Z., C.W., J. Luo, X. Li and J. Liang assisted in electrochemical/physical property testing and interpreting and organizing the data. J.F., W.L., F.Y. and J.G. helped with the synchrotron-related measurements and experimental results analysis. H.D. conducted the energy dispersive spectroscopy characterization. B.F. and H.A. performed nanoindentation measurements and data interpretation. G.K. helped with the PDF characterization and data analysis. X.S. supervised the whole project. All authors contributed to the discussions and revisions of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks Ainara Aguadero and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–43 and Tables 1–9.
Supplementary Data 1
Computational data of amorphous NHOC electrolyte.
Source data
Source Data Fig. 1
XRD data plotted in Fig. 1a,c,d, ionic conductivity data plotted in Fig. 1b,g, Nyquist data plotted in Fig. 1e,f and electrochemical stability data plotted in Fig. 1h.
Source Data Fig. 2
Raman spectra data plotted in Fig. 2a, EXAFS data plotted in Fig. 2c,g, AIMD data plotted in Fig. 2d and PDF data plotted in Fig. 2e.
Source Data Fig. 3
Electrochemical data plotted in Fig. 3b–g.
Source Data Fig. 4
X-ray photoelectron spectroscopy data plotted in Fig. 4c,d, galvanostatic data plotted in Fig. 4e and relaxation time data plotted in Fig. 4f.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, X., Zhang, S., Yang, M. et al. A family of dual-anion-based sodium superionic conductors for all-solid-state sodium-ion batteries. Nat. Mater. (2024). https://doi.org/10.1038/s41563-024-02011-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41563-024-02011-x
- Springer Nature Limited