

Abstract
Faithful DNA replication is essential for genome integrity1,2,3,4. Under-replicated DNA leads to defects in chromosome segregation, which are common during embryogenesis5,6,7,8. However, the regulation of DNA replication remains poorly understood in early mammalian embryos. Here we constructed a single-cell genome-wide DNA replication atlas of pre-implantation mouse embryos and identified an abrupt replication program switch accompanied by a transient period of genomic instability. In 1- and 2-cell embryos, we observed the complete absence of a replication timing program, and the entire genome replicated gradually and uniformly using extremely slow-moving replication forks. In 4-cell embryos, a somatic-cell-like replication timing program commenced abruptly. However, the fork speed was still slow, S phase was extended, and markers of replication stress, DNA damage and repair increased. This was followed by an increase in break-type chromosome segregation errors specifically during the 4-to-8-cell division with breakpoints enriched in late-replicating regions. These errors were rescued by nucleoside supplementation, which accelerated fork speed and reduced the replication stress. By the 8-cell stage, forks gained speed, S phase was no longer extended and chromosome aberrations decreased. Thus, a transient period of genomic instability exists during normal mouse development, preceded by an S phase lacking coordination between replisome-level regulation and megabase-scale replication timing regulation, implicating a link between their coordination and genome stability.
Similar content being viewed by others


Main
Aneuploidy is relatively common during early mammalian embryogenesis, although the exact frequency is unclear5,6,7,8. Factors contributing to this include the lack of stringent cell-cycle checkpoints and problems during chromosome segregation5. Moreover, unreplicated DNA due to replication stress could add another layer to this complexity1,2,3,4, as reported in human cleavage-stage embryos9. However, understanding of the regulation of DNA replication during early mammalian embryogenesis is lacking owing to the lack of reliable methodology.
Recently, we and others developed a single-cell DNA replication sequencing technology—scRepli-seq10,11. This technology, which enables high-resolution genome-wide profiling of the replication state in single S-phase cells, confirmed that replication is regulated at the level of megabase (Mb)-sized domains even in single cells, replicated in a defined temporal order in a cell-type-specific manner12,13. Early and late replication timing (RT) domains correspond well with the euchromatic (A compartment) and heterochromatic (B compartment) compartments defined by Hi-C, respectively14,15. scRepli-seq can also detect aneuploidy at a high resolution13.
Thus, scRepli-seq is an excellent method for studying replication regulation, inferring the three-dimensional (3D) genome organization dynamics and assessing the aneuploidy rate during early mouse embryogenesis. With this in mind, we constructed a single-cell genome-wide DNA replication atlas of pre-implantation mouse embryos.
RT emerges abruptly at the 4-cell stage
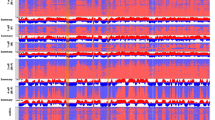
We generated binarized scRepli-seq data16 of BDF1 (F1 hybrid between C57BL/6 and DBA2 mice) embryos at the 4-, 8- and 16-cell and blastocyst stages (Fig. 1a), during which the 3D genome gradually becomes somatic-cell like17,18,19. We generated whole-S RT profiles10,16 with single cells covering the entire S phase (Fig. 1b). We used 80 kb bins—our current resolution limit for reliable scRepli-seq binarization (Supplementary Note 1 and Extended Data Fig. 1a,b). The 4-cell RT profile was already similar to that of mouse embryonic stem cells (mESCs) and was largely maintained thereafter with only small local differences (Fig. 1b and Extended Data Fig. 1c). t-Distributed stochastic neighbour embedding (t-SNE) analysis of mid-S-phase cells confirmed gradual RT changes after the 4- and 8-cell stages (Fig. 1c). RT profiles of 8-cell embryos and inner cell mass (ICM) were similar, with only around 4% RT changes throughout the genome, which coincided with regions subject to developmental RT regulation20 (Extended Data Fig. 1d,e). Notably, RT profiles of ICM and trophectoderm (TE) cells were highly similar (Fig. 1b,c and Extended Data Fig. 1c,f), suggesting that the A/B compartments do not change significantly at the onset of embryonic/extraembryonic lineage segregation.

a, Schematic of scRepli-seq profiling. WGA, whole-genome amplification. b, Binarized whole-S scRepli-seq profiles of 4-, 8- and 16-cell and blastocyst (ICM and TE) BDF1 embryos, along with BrdU immunoprecipitation (IP) population RT profiles of mESCs and averaged scRepli-seq profiles calculated from mid-S-phase cells with 30–70% replication scores. Each row represents a single-cell replication profile (80 kb bins). Cells throughout the S phase were analysed and ordered by their percentage replication score. Four-cell embryos were sampled at hourly intervals (1–11 h after 2-to-4-cell division) on embryonic day 2. Eight- and 16-cell embryos were sampled every 2 h on embryonic days 2.5 and 3, respectively. The durations that cover the entire S phase of each cell cycle were predetermined based on EdU-incorporation competency. c, t-SNE analysis of mid-S-phase cells (30–70% replication score, excluding chromosome X) in 4-, 8- and 16-cell embryos, ICM and TE, using the log2[median] RT scores. d, Binarized scRepli-seq profiles of 1-, 2- and 4-cell B6MSM embryos, along with the BrdU-IP population RT profiles of mESCs. One-cell embryos were sampled at hourly intervals (4–10 h after fertilization); 2- and 4-cell embryos were sampled hourly during 3–8 h and 5–13 h after their latest cell division, respectively. Most bins were called ‘replicated’ in 1- and 2-cell embryos (asterisks), reflecting the lack of regional copy-number differences due to gradual and uniform replication (Extended Data Fig. 2c). e, The MAD score distribution16 of 1-, 2- and 4-cell blastomeres and mESCs was analysed using scRepli-seq. Cell-cycle phases of each cell (or sampling timepoint) were estimated by EdU staining as described in Fig. 2a–c. Each dot represents a single cell. f, Overview of the EdU staining experiment in g. g, Representative EdU staining patterns of metaphase chromosomes derived from MC12 cells and 1-, 2- and 4-cell embryos. The line plots show the EdU (green) and DAPI (magenta) signal intensities along the white lines. Identical magnification was used for all images (chromosome size variations are probably a batch effect of colcemid treatment). T and C, the telomeric and centromeric ends, respectively. Scale bar, 5 μm (g).
In prezygotic genome activation (pre-ZGA) embryos, the paternal and maternal genomes undergo epigenetic reprogramming with distinct kinetics21,22. We analysed pre-ZGA embryos of B6MSM mice (F1 hybrid between C57BL/6 and MSM mice) to distinguish the haplotypes. Notably, 1- and 2-cell embryos had no RT domains that were detectable throughout S phase by scRepli-seq, whereas 4-cell embryos clearly exhibited RT domains (Extended Data Fig. 1g and Supplementary Note 1, haplotype-resolved at 400 kb resolution; Fig. 1d, non-haplotype-resolved at 80 kb). In 1- and 2-cell embryos, almost all genomic bins were categorized as replicated (blue), giving the impression that these cells have completed genome replication abruptly in early S phase (Fig. 1d and Extended Data Fig. 1g). However, this cannot be true, as our sampling covered the entire S phase at hourly intervals and the 5-ethynyl-2′-deoxyuridine (EdU)-positive S phase lasted 4–5 h. Rather, it most likely reflected the lack of regional differences in copy number during S phase; scRepli-seq seems unable to detect replicated and unreplicated segments.
Consistently, tag or read density profiles of scRepli-seq data from individual mid-S-phase cells of 1- and 2-cell embryos did not exhibit bimodal distribution but showed a single peak using 200, 80 or 40 kb bins (Extended Data Fig. 2a,b (left) and Supplementary Note 1; although 40 kb is reaching or slightly exceeding the scRepli-seq resolution limit), and binary calling could hardly separate unreplicated and replicated bins (Extended Data Fig. 2a,b (right)). This resulted in a seemingly completely replicated or completely unreplicated output depending on the binarization mode chosen (Extended Data Fig. 2c). Thus, scRepli-seq is unable to resolve replicated and unreplicated segments in 1- and 2-cell S phase, suggesting uniform replication.
This view was supported by the scRepli-seq tag density profile transition during S-phase progression. In 1- and 2-cell embryos, cells showed a single peak throughout S phase, indicating the lack of copy-number differences among genomic bins (Extended Data Fig. 2d,e). However, the 4-cell S-phase profiles were bimodal with the larger peak height gradually increasing, reflecting the gradual increase of replicated bins as S phase progressed (Extended Data Fig. 2d,e). Furthermore, log2[(mappability-corrected S phase reads)/median] (log2[median]) value profiles (close to raw data) lacked the high/low contrast along the length of chromosomes in 1- and 2-cell embryos, in contrast to in 4-cell embryos (Extended Data Fig. 1g). These data are consistent with gradual and uniform replication in 1- and 2-cell embryos at 40–80 kb resolution.
Furthermore, the median absolute deviation (MAD) score, a measure of bin-to-bin data variability, was constant during S-phase progression in 1- and 2-cell embryos (Fig. 1e), regardless of the bin-size settings or haplotype (Extended Data Fig. 2f,g). This was in sharp contrast to mESCs and 4-cell embryos, which exhibited inverted V-shape patterns with the MAD score peaking at mid-S phase (Fig. 1e), reflecting the presence of equal numbers of fully replicated and fully unreplicated bins at mid-S phase and minimum heterogeneity at the earliest and latest S phase.
Lastly, we prepared metaphase chromosome spreads after 1 h EdU labelling at the earliest S-phase period (Fig. 1f). While cultured (somatic) cells and 4-cell embryos exhibited stripe patterns of early-RT domains, 1- and 2-cell embryos showed uniform staining, indicating a lack of RT domains (except for the centromeres; discussed later) (Fig. 1g). Taken together, we conclude that, in 1- and 2-cell mouse embryos, an RT program is completely absent and the chromosomes are replicated in a gradual and uniform manner. Then, a somatic-cell-like RT program abruptly commences at the 4-cell stage.
The sole exception on the otherwise uniformly replicated chromosomes of 1- and 2-cell embryos was the heterogeneously late-replicating domains on the paternal chromosomes in 1-cell embryos (Extended Data Fig. 1g). While this could be a feature of the MSM chromosomes, we speculate that this represents the remnants of strong heterochromatin structures (late-replicating B-compartment domains) derived from the sperm19 that become reprogrammed by the 2-cell stage (Extended Data Fig. 2h–k).
Nuclear compartments strengthen in 4-cell embryos
As RT correlates with A/B compartments, the somatic-cell-like RT emergence might reflect nuclear compartment emergence. Thus, we analysed the spatiotemporal patterns of replication, that is, replication foci, which change in somatic cells from early to late S as follows: (1) throughout the nuclear interior; (2) in the nuclear periphery and nucleoli; and (3) in internal heterochromatic foci (Extended Data Fig. 3a; mESCs). We labelled 1- and 2-cell embryos with EdU at 1 h intervals and 4-cell embryos at 2 h intervals and observed replication foci patterns (Fig. 2a). As replication foci patterns in early embryos were slightly different from in somatic cells, we classified them into a total of eight patterns (Fig. 2b,c). In 1-cell embryos, S phase was short (around 5 h) and the entire maternal pronuclei were uniformly EdU stained throughout S phase, followed by a brief period of nucleolar precursor body (NPB) staining (Fig. 2d). In the paternal pronuclei, the nuclear periphery and NPB were EdU stained during mid-S phase (Fig. 2d), which may correspond to the heterogeneously late-RT domains (Extended Data Fig. 1g). As centromeres accumulate around the NPBs23 and were not replicated in early-S phase (Fig. 1g), they are presumably replicated during late-S phase in both pronuclei (Fig. 2d). In 2-cell embryos, S phase was still short (approximately 4 h), but the EdU pattern changed slightly, starting from the entire nuclei in early-S phase to the nuclear membrane and NPB, and to the internal foci (Fig. 2d). However, the typical mid-S phase pattern (nuclear periphery/nucleoli) was still absent (Fig. 2d). These results are consistent with the nuclear compartments being absent or still incomplete in 1- and 2-cell embryos17,18,19.

a, The EdU-labelling scheme. b, Representative nuclear EdU staining (replication foci) patterns (Methods). c, Exemplary replication foci images. The colour code represents the EdU staining patterns in b. Green, EdU; magenta, histone H3. Scale bar, 10 μm. d, Replication foci pattern dynamics. The colour code is as in b. The numbers (n) of nuclei and embryos are shown. e, scRepli-seq analysis of SCNT embryos, collected at 1 h intervals covering the S phase. f, Binarized whole-S scRepli-seq profiles of SCNT embryos (80 kb bins, 2-somy mode59). Each row represents a single cell, ordered by their sampling order or percentage replication score. Averaged scRepli-seq RT (avg scRT) was calculated by averaging S-phase cells. Heterogeneously late-RT regions (hetero late) were regions with an average scRepli-seq RT of <0.50. RT class definitions have been described previously20; CE, constitutively early; CL, constitutively late; D, developmentally regulated. PC1 is the A/B compartment profile based on cumulus cell Hi-C60. Embryos were sampled hourly (1 cell, 3–10 h after strontium activation; 2 cell, 1–8 h after cell division). SCNT S-phase lengths were predetermined by EdU. Asterisks indicate data reflecting gradual and uniform replication (Fig. 1d). g, DNA fibre spreading assay. Embryos (early S) or mESCs (control, asynchronous) were labelled with IdU/CldU in vivo followed by fibre extension and immunostaining. h, Representative images of three fork categories. IdU and CldU (green), CldU (magenta) and ssDNA (blue) signals on DNA fibres. Scale bar, 4.55 μm (20 kb). i, DNA fibre classification based on fork categories. j, The IOD between the immobile single-dot forks and IOD on mobile 8-cell fibres (Extended Data Fig. 5b). k, The mobile fork speed. l, G1-to-S-phase lengths revealed by PCNA live-cell imaging. The numbers (n) of cells from a total of 26 embryos are shown. Error bars represent the mean ± s.d. (j–l).
In 4-cell embryos, S phase was clearly longer (around 11 h; Fig. 2d), and the nuclear periphery/internal foci pattern emerged during mid/late-S phase as in mESCs (Fig. 2d), suggesting nuclear compartment strengthening. To test whether the nuclear periphery/internal foci staining pattern indeed emerged at 4-cell mid-S phase, such cells were analysed using scRepli-seq after imaging (Extended Data Fig. 3a), which validated their mid-S phase identity (Extended Data Fig. 3b). Likewise, cells with uniform or internal foci patterns were at early-S or late-S phase, respectively (Extended Data Fig. 3b). These somatic-cell-like spatiotemporal replication foci patterns were also observed in 8-cell embryos (Extended Data Fig. 3c). Consistent with the emergence of the heterochromatic replication foci pattern, histone H3 lysine 9 dimethylation (H3K9me2) showed strong discrete nuclear foci in 4-cell but not 1- and 2-cell embryos (Extended Data Fig. 3d–f). These data suggest that the emergence of the somatic-cell-like RT program in 4-cell embryos is accompanied by a considerable strengthening of nuclear compartments, which may be accompanied by chromatin tethering to the nuclear lamina.
The near coincidence of nuclear compartment strengthening and the emergence of the somatic-cell-like RT program led us to examine whether the former could trigger the latter. Although we have no means to manipulate the nuclear compartment strength, we reasoned that we could use somatic cell nuclear transfer (SCNT) into enucleated oocytes using cumulus cells as donors, followed by scRepli-seq (Fig. 2e). As the cumulus cell nuclei maintain compartment strength to some extent even after SCNT and show stronger A/B compartmentalization compared with control fertilized embryos24, we could instead examine whether precocious emergence of somatic-cell-like RT could be observed in SCNT embryos.
Somatic-cell-like RT was non-existent in SCNT 1-cell embryos as in control embryos (Fig. 2f and Extended Data Fig. 4a), indicating that partial presence of A/B compartments is insufficient for somatic-cell-like RT emergence (although rigorous testing will ultimately require us to perform simultaneous Hi-C). However, in SCNT 2-cell embryos, large late-replicating domains appeared in some but not all cells (Fig. 2f (hetero late)), which corresponded well with constitutively late-replicating, B-compartment regions in somatic cells (Fig. 2f and Extended Data Fig. 4a–c). They also corresponded well with the heterogeneously late-RT domains on the paternal chromosomes in 1-cell embryos (Extended Data Figs. 2a,b and 4d,e), suggesting that they are the most preferential late-replicating domains on the mouse genome. Accordingly, the SCNT 2-cell embryos showed some ‘somatic’ signatures such as small deviations from the flat MAD score plots of the control embryos (Extended Data Fig. 4f). However, overall, SCNT 2-cell embryos were also similar to 2-cell control embryos without a somatic-cell-like RT program (Fig. 2f and Extended Data Figs. 2e and 4a), placing the SCNT 2-cell profile somewhere in between the control 2-cell and 4-cell profiles.
Discordance between 4-cell RT and fork speed
While the abrupt RT emergence was surprising, the coordinated strengthening of nuclear compartments is consistent with the known close relationship between them2,12,13. However, there is one major paradox with the ‘embryonic’ DNA replication program regarding how one can achieve gradual and uniform replication of all 80 kb bins across the genome in an approximately 5 h S phase if the replication forks travel at 1–2 kb per min as stated in the literature12,25,26. The logical conclusion is that replication forks must be extremely slow to achieve an approximately 5 h S phase with high and uniform origin density. To test this, we analysed the fork speed in early embryos using a standard DNA fibre spreading assay, in which early-S-phase cells were labelled sequentially with nucleoside analogues iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU) for 30 min each (Fig. 2g). As a control, we used mESCs and confirmed a fork speed of 1–2 kb per min as expected26 (Fig. 2k).
Notably, we found that 63–82% of all DNA fibres with IdU/CldU signals at the 1-, 2- and 4-cell stages were categorized as immobile forks (Fig. 2h,i and Extended Data Fig. 5a). That is, IdU + CldU or CldU-only labels formed single dots that were around 12–22-kb apart from each other (Fig. 2j). It was challenging to measure the fork speed of these tiny dots. However, we used an anti-single-stranded DNA (ssDNA) antibody that generated ‘dotty’ signal patterns, and these dots were also preferential sites of detection by our anti-IdU/CldU antibodies when IdU and CldU were incorporated. This enabled us to estimate the immobile dot size in base pairs and, in turn, their fork speed, which was <22–147 bp per min assuming bidirectional forks (or <43–147 bp per min assuming unidirectional forks) (Supplementary Note 2). Among all of the fibres, 11–24% contained mobile forks in 1-, 2- and 4-cell embryos with an average speed of 0.3–0.4 kb per min (median mobile fork speeds at the 1-, 2- and 4-cell stages of 0.38, 0.29 and 0.35 kb per min, respectively; Fig. 2i,k). The remaining 7–15% of fibres contained intermediate-speed forks (Fig. 2i).
Upon transition to the 8-cell stage, fork types changed substantially; the immobile forks almost disappeared, and the majority became mobile with an average speed of approximately 0.76 kb per min, which is less than, albeit comparable to, that of mESCs at around 1.2 kb per min (Fig. 2i,k). Consistently, the inter-origin distance (IOD) was also greater at 56.1 kb (median) in 8-cell embryos (Fig. 2j and Extended Data Fig. 5b).
We also used orthogonal measurements. First, the early-S-phase median IODs were around 21.9, 13.7 and 12.3 kb at the 1-, 2- and 4-cell stages, respectively (Fig. 2j and Extended Data Fig. 5b). Next, the S-phase length (EdU-incorporation period) was approximately 5, 4 and 11 h at the 1-, 2- and 4-cell stages, respectively (Fig. 2d). Moreover, the binarized scRepli-seq data showed almost no bin-to-bin difference throughout S phase in 1- and 2-cell embryos at 80 kb resolution (Fig. 1d). Taken together, the 1-, 2- and 4-cell immobile fork speed estimate was <33 bp per min assuming bidirectional forks (<66 bp per min assuming unidirectional forks) (Supplementary Note 2).
Combining the two independent estimates, the immobile forks (63–82% of all forks) should be <33 bp per min assuming bidirectional forks (<66 bp per min assuming unidirectional forks) in 1-, 2- and 4-cell embryos. The fastest forks (mobile; 11–24% of all forks) have a speed of around 300–400 bp per min, and the remaining intermediate forks (7–15% of all forks) must be in between at 33–300 bp per min assuming bidirectional forks (66–300 bp per min assuming unidirectional forks). Thus, the majority of forks in 1-, 2- and 4-cell embryos are approximately 4–10 times slower than previously reported27, with an average IOD of 12–22 kb (Supplementary Note 2). Upon transition to the 8-cell stage, forks abruptly accelerated. As somatic-cell-like RT emerged one cell cycle earlier (Fig. 1b,d), we identified a transient discordance between RT and fork regulation at the 4-cell stage.
scRepli-seq data support slow 4-cell fork speed
In the 4-cell S phase, the immobile bidirectional forks travelled at <33 bp per min and the median IOD was around 12 kb. Without additional origin firing, a given immobile bidirectional fork must travel around 6.0 kb at <33 bp per min, which takes approximately 3 h. This is much longer than the current consensus of about 1 h to replicate Mb-sized replication domains using forks travelling at 1–2 kb per min with an IOD of 100–200 kb (refs. 2,12,28). This raises the question of why the 4-cell RT profile is somatic-cell like (Fig. 1b).
Revisiting the whole-S scRepli-seq data, we found that tag-density bimodality was less prominent in 4-cell than in 8-cell embryos or mESCs at 30–40% S phase (that is, cells with 30–40% of all genomic bins replicated based on binarized scRepli-seq data) (Extended Data Fig. 5c). Such a feature disappeared at 50–60% S phase. Thus, while the forced binarization of 4-cell scRepli-seq data generated a somatic-cell-like whole-S-phase RT profile (Fig. 1d), it reflected a state containing ‘relatively replicated’ and ‘completely unreplicated’ bins during the first half of S phase. Thus, 4-cell S phase is compartmentalized into early and late halves, and replication completion of individual Mb-sized replication domains takes longer (>4 h) than in 8-cell embryos and mESCs (Supplementary Note 2).
Slower forks could also explain the longer 4-cell S phase. As S-phase length estimation is challenging after the 8-cell stage owing to a loss of synchrony among blastomeres, we measured the G1-to-S-phase length using proliferating cell nuclear antigen (PCNA) live imaging, which confirmed a longer G1-to-S-phase length in 4-cell compared with in 8-cell and 2-cell embryos (Fig. 2l).
In summary, while the 4-cell RT domain organization appeared somatic-cell like, the fork speed and IOD inside individual replication domains were almost identical to in 1- and 2-cell embryos. This suggests that RT regulation and replisome regulation are temporally uncoordinated during the 4-cell stage, giving rise to a transitional S-phase in between embryonic (1 and 2 cell) and somatic (after the 8-cell stage) S-phase types. Even during this transitional 4-cell S phase, the intra-S-phase checkpoint was functional, as in somatic cells29 (Extended Data Fig. 5d–i).
Transitional 4-cell S phase is error prone
The transitional 4-cell S phase exhibits both embryonic (slow forks) and somatic (somatic-cell-like RT) properties. We wondered whether this uncoordinated state is linked to cellular stress. As scRepli-seq is capable of high-resolution karyotyping, we examined the chromosome aberration frequency in G1- and S-phase cells.
Chromosome aberrations can be either whole-chromosome loss or gain (whole), or chromosome breaks (break). We detected both using scRepli-seq analysis of 4-, 8- and 16-cell embryos derived from in vitro development (Fig. 3a). We collected all cells of an embryo, which were annotated with the embryo-of-origin information, enabling us to identify pairs of sister cells with complementary errors (Fig. 3a). We determined at which cell division the segregation error had occurred, in a manner similar to lineage barcoding30, except that the barcoding is done endogenously by the spontaneous errors. Notably, more than 13% of cells exhibited de novo abnormalities during 4-to-8-cell division in vitro, the majority of which were of the break type (Fig. 3b, Extended Data Fig. 6a,b and Supplementary Table 1). Abnormalities were not observed during 2-to-4-cell and 8-to-16-cell divisions in vitro. Similar error rates were observed in embryos collected in vivo from the oviduct, with high error rates observed only during the 4-to-8-cell division (Fig. 3b). Live-cell imaging confirmed that chromosome bridges formed and eventually broke into unbalanced chromosome mass or fragments more frequently during 4-to-8-cell division (Fig. 3c, Extended Data Fig. 6c and Supplementary Video 1). Consistent with the low frequency of whole-type errors, we never observed anaphase with merotelic centromeres remaining in the middle of the spindle in our live-cell imaging data (Fig. 3c and Supplementary Note 3).

a, Detection of de novo chromosome aberrations using scRepli-seq. Sister cells with reciprocal chromosome gain/loss are shown. b, Increased de novo errors during the 4-to-8-cell division as revealed by scRepli-seq. Errors were gain or loss of whole chromosomes (whole) or chromosome fragments (break). The number (n) of cell divisions is shown. c, Early-anaphase chromosome bridges were detected using live-cell imaging (major-satellite–mClover, centromeres, green; H2B–mCherry, chromosomes, magenta) and categorized by their late-anaphase fates: breakage into unbalanced chromosome mass (unbalanced) or breakage leaving an acentric fragment. Representative images are shown (min:s, time after anaphase onset). Merotelic (chromosome with centromere left in the middle) was not observed. The number (n) of cell divisions observed live are shown. The arrowheads indicate chromosome bridges; asterisks indicate unbalanced chromosome mass and acentric fragments. d, Representative early/mid/late-S breakpoints revealed by AneuFinder59 (Extended Data Fig. 6d). e, Breakpoints were predominantly late replicating in vitro and in vivo (4-to-8-cell division). The number (n) of breaks is shown. f, Live-cell imaging of mEGFP–SLX4 (DNA repair foci, green) and H2B–mCherry (chromosomes, magenta) until the 16-to-32-cell division. The arrowheads indicate SLX4 foci; the asterisk indicates chromosome fragments. g, The number and duration of mEGFP–SLX4 foci. The number (n) of nuclei observed live is shown. h, Replication stress is increased during 4-cell late-S phase. The numbers (n) of nuclei from 8 (2-cell), 15 (4-cell) and 10 (8-cell) embryos are shown. p-CHK1, replication stress marker, green; histone H3, magenta. i, DNA damage is increased during 4-cell late-S phase. The numbers (n) of nuclei from 26 (2-cell), 20 (4-cell) and 19 (8-cell) embryos are shown. The 4-to-8-cell M-phase image (bottom) shows γH2AX (DNA damage marker, green) enrichment on a chromosome arm (arrowhead). Histone H3, magenta. Scale bars, 5 μm (c, f, h and i). Error bars represent the mean ± s.d. (e, g–i).
Clues about the underlying molecular mechanism came from the breakpoints (Fig. 3d and Extended Data Fig. 6d). During the 4-to-8-cell division in vitro, around 90% of the breakpoints were located in mid/late-RT regions, while 100% were in late-RT regions in vivo (Fig. 3e and Extended Data Fig. 6b), suggesting under-replication of late-RT regions as a potential cause of breaks. We therefore analysed the localization of MUS81–EME1–SLX4 complex foci, which are involved in unreplicated DNA repair in G2/M phase31. The monomeric enhanced green fluorescent protein (mEGFP)-tagged SLX4 foci and SLX4-positive duration clearly increased at the 4-to-8-cell division but not at the other cell divisions during G2/M (Fig. 3f,g).
Consistently, phosphorylated CHK1 and γH2AX, markers of replication stress and DNA damage, respectively, were significantly increased in 4-cell late-S phase (Fig. 3h,i). Furthermore, low-dose aphidicolin treatment, which induces replication stress, increased severe chromosome aberrations specifically during the 4-to-8-cell division (Extended Data Fig. 7a; with 30 ng ml−1 aphidicolin, 2-to-4-cell and 8-to-16-cell divisions were accompanied by mild (orange) but not severe (dark grey) chromosome aberrations, suggesting a higher basal replication stress level in 4-cell embryos). Furthermore, severe breakpoints were observed predominantly in late-RT domains (Extended Data Fig. 7b), consistent with transitional 4-cell S phase being under replication stress and error prone.
To obtain more direct evidence suggesting a link between replication stress and chromosome aberrations, we recorded cell-cycle progression and chromosome segregation of developing embryos using live imaging of mEGFP–PCNA and histone H2B–mCherry (Extended Data Fig. 7c and Supplementary Video 2). PCNA foci not only mark S phase, but their spatial patterns also reflect S-phase time, with internal replication foci patterns marking late-S phase32. These PCNA features enabled us to measure the durations of G1-to-late-S, late-S, G2 and M phases (Extended Data Fig. 7d–f). We identified error cells exhibiting chromosome bridges during the 4-to-8-cell division (Fig. 4a) and tracked these cells back in time to assess their cell-cycle lengths (Fig. 4b), which demonstrated that the 4-cell G1-to-late-S period was significantly extended (Fig. 4b–d and Extended Data Fig. 7g). Thus, chromosome aberrations are more likely to occur in cells that exhibit S-phase extension, suggesting replication stress and under-replication as potential sources of the errors.

a, Live imaging of mEGFP–PCNA (S-phase marker, green) and H2B–mCherry (chromosomes, magenta) in mouse embryos. Magnified z-projection snapshots of 4-cell sister blastomeres are shown, along with 3D-reconstructed images of 2-to-4-cell and 4-to-8-cell divisions (h:min, time after 2-cell anaphase onset). The image frame colours reflect the cell-cycle phases in b. The asterisks show PCNA foci (late-S phase marker). b–d, Extended S phase often precedes chromosome bridge formation during 4-to-8-cell division (error). In b, each line represents a single cell, and the average cell-cycle durations are shown in c, based on durations plotted in d. The number (n) of cells from 24 embryos is shown. e, Nucleoside-supplementation experiment. Embryos cultured for 6 h in +nucleoside medium from 4-cell G1 were sequentially labelled with IdU and CldU for 30 min each, then analysed using the DNA fibre spreading assay (f and g). Control embryos were sampled simultaneously at mid/late-S phase. f, DNA fibre classification of +nucleoside 4-cell embryos based on fork categories (Fig. 2h). Control 4-cell embryos differ slightly from Fig. 2i, possibly reflecting differences between mid/late-S phase (f) and early-S phase (Fig. 2i). g, Mobile fork speed. h, Nucleosides suppressed p-CHK1 at the 4-cell late-S phase. The numbers of nuclei from 30 (control) and 12 (+nucleosides) embryos are shown. i, Nucleosides suppressed γH2AX at the 4-cell late-S phase. Histone H3, magenta. The numbers (n) of nuclei from 23 (control) and 15 (+nucleosides) embryos are shown. j, Nucleosides suppressed chromosome segregation errors. The chromosome-aberration frequency at anaphase during 4-to-8-cell division was monitored using live-cell imaging. The numbers (n) of cell divisions analysed live are shown. Scale bars, 10 μm (a (blue and grey boxes)) and 5 μm (a (magenta boxes), h and i). Error bars represent the mean ± s.d. (d, g–i).
Lastly, to test the causal relationship between replication stress and chromosome segregation errors, we used nucleoside supplementation (Fig. 4e), which accelerates forks and reduces replication stress in human embryonic stem cells (hESCs)33,34. Nucleoside supplementation from 4-cell G1 led to a slight decrease and increase in the rate of immobile and intermediate forks, respectively (Fig. 4f; although not statistically significant on the basis of a χ2 test). Moreover, nucleoside supplementation modestly but significantly accelerated 4-cell mobile forks (Fig. 4g; the average speed changed from 0.39 to 0.53 kb per min; n = 26–32; P = 0.0156), led to significant downregulation of phosphorylated CHK1 and γH2AX (Fig. 4h,i) and significantly reduced errors during 4-to-8-cell division (Fig. 4j and Supplementary Note 3; from 21.1% to 4.26%, n = 33–42; P = 0.0377). Our results are consistent with a model in which replication stress involving slow forks contributes to chromosome segregation errors in 4-cell mitosis.
Notably, the proportion of immobile forks was slightly decreased in mid/late-S phase (Fig. 4f) compared with in early-S phase (Fig. 2i). Future studies are warranted to investigate whether there is a slight fork acceleration towards the later half of S phase.
Discussion
Here we analysed the regulation of DNA replication during early mouse embryogenesis genome-wide in substantial detail. First, we found the complete absence of RT during the 1- and 2-cell stages, when embryonic DNA replication proceeds gradually and uniformly throughout the genome. Second, we demonstrated that the 2-to-4-cell transition is accompanied by an abrupt emergence of somatic-cell-like RT, coincident with nuclear compartment strengthening (Fig. 5). However, the abrupt RT emergence was uncoordinated with fork-speed regulation. We propose that 4-cell embryos undergo transient replication stress and show genomic instability, exhibiting frequent chromosome segregation errors due to under-replication during the transitional S phase (Fig. 5). These observations suggest a link between genome stability and coordination of replisome-level and RT-level regulation, and have implications for embryonic genome regulation from the standpoint of molecular, developmental and evolutionary biology. Moreover, they provide insights for future clinical applications in in vitro fertilization clinics regarding strategies to minimize chromosome aberrations common in early embryos5,6,7.

During the embryonic S phase in 1- and 2-cell embryos, DNA replication proceeds gradually and uniformly across the entire genome in the absence of Mb-sized RT domain structure. This is achieved by numerous replication forks with a median IOD of 12–22 kb that travel extremely slowly, with the majority estimated to move at <33 bp per min bidirectionally (or <66 bp per min assuming unidirectional forks). The only exceptions are the centromeres in 1- and 2-cell embryos and the sperm-derived heterochromatin in 1-cell embryos, which replicate later in S phase. At the 4-cell stage, a somatic-cell-like RT program commences abruptly, which is accompanied by a marked strengthening of nuclear compartments. Despite this abrupt switch, the forks were still extremely slow and the median IOD was still around 12 kb, giving rise to a transitional 4-cell S phase, in which the RT regulation mode is somatic, while the replisome-level regulation mode is still embryonic. This uncoordinated regulatory state led to S phase extension in 4-cell embryos, and the cells with the most extended S phase frequently showed chromosome breaks during the 4-to-8-cell division that probably arose due to under-replication of late-replicating domains after an S phase with increased replication stress, DNA damage and repair. After the 8-cell stage, forks accelerate (approximately 0.76 kb per min), IODs become larger (median, 56.1 kb) and DNA replication proceeds in a coordinated manner again, resulting in a reduction in genomic instability (somatic S phase).
In non-mammalian species, the cell cycle and replication in early embryos are unique5,35,36,37,38. In Drosophila and Xenopus embryos, multiple rapid S–M phase cycles are observed after fertilization, with an S phase of around 10 and 20 min, respectively37,39. In these organisms, it is believed that replication before ZGA is uniformly initiated from origins distributed throughout the genome3,39,40,41,42 and, after ZGA, G1/G2 phases and a somatic cell-like RT program emerge coincident with the acquisition of somatic-cell-like 3D genome organization43,44,45,46,47.
By contrast, in mouse embryos, replication foci patterns have suggested the presence of some degree of 3D genome arrangement before ZGA23,48,49. Moreover, in contrast to in Drosophila, Xenopus and zebrafish pre-ZGA embryos, G1/G2 phases exist in mouse zygotes and 1- and 2-cell S phase is 4–5 h long5. Given these reports, uniform replication in pre-ZGA mouse embryos was completely unexpected. However, all our data are consistent with each other and Hi-C data showing unstructured genome before ZGA17,18,19. Thus, uniform replication and a lack of strong euchromatic/heterochromatic compartments before ZGA is evolutionarily conserved in metazoans, whereas the extremely slow fork movement is unique to mice and perhaps humans9,27.
Notably, while the relatively unstructured pre-ZGA 3D genome was conserved in zebrafish and medaka50,51, somatic-cell-like RT was reported in pre-ZGA zebrafish embryos, despite the extremely short S phase52. As with the controversy regarding the 3D genome state before ZGA50,53, further studies are needed to resolve this discrepancy.
Our data raise more questions than answers. First, the question of why forks are extremely slow only in mouse (and human) pre-ZGA embryos. While the mechanism is unclear, chromosome segregation errors were much fewer in 2-cell embryos than in 4-cell mouse embryos. The slower forks and longer S phase might have contributed to higher replication fidelity in mice and were selected during evolution. Notably, slow forks were conserved in 2-cell-like cells, while uniform RT was not27. In fact, the 2-cell-like cells exhibited somatic-cell-like RT27, resembling the 4-cell embryos from a replication regulation standpoint. Notably, a recent study revealed a lower frequency of chromosomal abnormalities in mouse versus human early-cleavage embryos, despite the similarly slow fork speed9. The difference between mice and humans is an important area of future investigation54,55,56.
Second, regarding what could trigger the abrupt emergence of somatic-cell-like RT, replication foci patterns suggest nuclear compartment strengthening at the 4-cell stage, when certain genomic regions start to interact with the nuclear lamina57. However, while the A/B compartment changes are candidate upstream events that might cause RT changes15, nuclear compartment strengthening may not be the decisive factor based on our SCNT experiments. Given its timing, 2-cell major ZGA may be involved, although suppressing transcription without inhibiting replication is technically challenging17,18.
Third, regarding why replisome and RT regulations are temporally uncoordinated, the presence of such a period suggests that they are controlled independently. Nevertheless, forks accelerate abruptly in 8-cell embryos, which becomes relatively error-free. Thus, there seems to be a strong constraint to coordinate replisome regulation with RT in developing mouse embryos, which may be important for genome integrity. Moreover, fork acceleration by nucleosides reduced chromosome segregation errors during 4-to-8-cell transition (Fig. 4j). Given these observations, it is all the more surprising that the erroneous transitional 4-cell S phase was not eliminated during evolution.
Fourth, as chromosome abnormalities are detrimental to development, error cells must be excluded from the embryos. While error cells could simply be eliminated, they could, for example, go to the extraembryonic lineage. Nevertheless, the physiological importance of maintaining the peculiar 4-cell S phase remains a mystery. As cell divisions are synchronous within an embryo until the 4-to-8-cell division, there could be a trade-off between cell-cycle synchrony and replication fidelity.
Finally, while our manuscript was under revision, a separate study reported that the somatic-cell-like RT program is established gradually from the 1-cell stage in mice58. By contrast, our study shows an abrupt RT emergence at the 4-cell stage. Future studies are needed to clarify this difference.
Methods
Animals
All animal experiments conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Committee of Laboratory Animal Experimentation of the RIKEN Center for Biosystems Dynamics Research. B6D2F1 (C57BL/6 × DBA/2) and C57BL/6 mice, aged 8–10 weeks, were used to produce oocytes and sperm. For allele-specific analysis, C57BL/6 female mice and MSM/Ms male mice were used to produce oocytes and sperm, respectively. To eliminate the effect of individual differences between mice, multiple mice were used in each experiment as follows. Figure 1b, 2 (4-cell), 4 (8-cell), 2 (16-cell) and 4 (ICM and TE) mice; Fig. 1d, 10 mice each (1-cell, 2-cell and 4-cell); Fig. 2d, 4 (1-cell), 3 (2-cell) and 3 (4-cell) mice; Fig. 2f, 3 (1-cell), 3 (2-cell) and 4 (4-cell and 8-cell) mice; Fig. 3b, 4 in vitro and 2 in vivo (2-to-4-cell), 8 in vitro and 7 in vivo (4-to-8-cell), and 4 in vitro and 2 in vivo (8-to-16-cell) mice; Fig. 3c, 15 mice each (2-to-4-cell, 4-to-8-cell and 8-to-16-cell); Fig. 3g, 3 mice each (2-to-4-cell, 4-to-8-cell, 8-to-16-cell and 16-to-32-cell); Fig. 3h, 4 (2-cell) and 3 (4-cell and 8-cell) mice; Fig. 3i, 5 (2-cell, 4-cell, and 8-cell) mice; Fig. 4d, 4 mice; Fig. 4h–i, 6 mice; Fig. 4j, 16 mice; Extended Data Fig. 7a, 7 (2-to-4-cell DMSO), 6 (2-to-4-cell aphidicolin 15 ng ml−1, 30 ng ml−1, and 60 ng ml−1), 8 (2-to-4-cell aphidicolin 75 ng ml−1), 16 (4-to-8-cell DMSO), 6 (4-to-8-cell aphidicolin 15 ng ml−1, 30 ng ml−1 and 60 ng ml−1), 6 (4-to-8-cell aphidicolin 75 ng ml−1), 12 (8-to-16-cell DMSO), 8 (8-to-16-cell aphidicolin 15 ng ml−1, 30 ng ml−1 and 60 ng ml−1) and 6 (8-to-16-cell aphidicolin 75 ng ml−1) mice.
Oocyte and embryo collection
The temperature, humidity and light cycle of mouse cages were maintained at 20–24 °C, 45–65% and 12 h–12 h dark–light, respectively. Mature oocytes were collected from the oviducts of eight- to ten-week-old female mice that had been induced to superovulate with 5 IU of equine chorionic gonadotropin (eCG, ASKA Pharmaceutical) followed by 5 IU of human chorionic gonadotropin (hCG; ASKA Pharmaceutical) 48 h later. Cumulus-oocyte complexes were collected from the oviducts approximately 16 h after hCG injection. Cumulus-oocyte complexes were placed in M2 medium and treated with 0.1% (w/v) bovine testicular hyaluronidase. After several minutes, the cumulus-free oocytes were washed twice and then moved to Chatot, Ziomek and Bavister medium (CZB). Mature metaphase II (MII) oocytes were subjected to in vitro fertilization (IVF), intracytoplasmic sperm injection (ICSI) or SCNT. Developmentally arrested or delayed embryos at each embryonic day were excluded from further analysis. In vivo developed zygotes and embryos were collected from the oviducts of pregnant mice that had been induced to superovulate and mated with male mice. For in vitro development, in vivo developed zygotes were cultured in CZB. As we pooled embryos from multiple females before single-embryo sampling, our dataset is not suitable for female-to-female variability assessment. When indicated, embryos were cultured in the presence of nucleosides (Sigma-Aldrich, EmbryoMax Nucleosides, 100×; at a final concentration of 2×) or aphidicolin (FUJIFILM Wako). In Extended Data Fig. 7a,b, embryos were monitored under the microscope every 30 min, and embryos that had just completed cell division were judged to be in G1 and transferred to the medium containing aphidicolin. The subsequent M phase was monitored by live imaging as in Extended Data Fig. 7a. Embryos that had just completed the 4-to-8-cell division were collected for scRepli-seq as in Extended Data Fig. 7b.
Fertilization
IVF was performed according to the manufacturer’s instructions using CARD MEDIUM (Cosmo Bio). To limit fertilization, we set the insemination time to 1 h. Intracytoplasmic sperm injection with sperm heads was performed as described previously61. In brief, the sperm head was separated from the tail by applying several piezo pulses to the neck region, and the head was then injected into an oocyte. After 20 min of recovery at room temperature, injected oocytes were cultured in CZB.
Single-cell (blastomere) collection
Collection of single blastomeres was performed using micromanipulation. Embryos were transferred into M2 medium supplemented with 5 μg ml−1 cytochalasin B (Sigma-Aldrich) for 10 min. The zona pellucida was then cut using the LYKOS laser system (Hamilton Thorne) in a micromanipulation chamber, which was placed onto a warmed stage (37 °C) in an inverted microscope (Olympus). After cutting the zona pellucida, a fire-polished injection pipette (inner diameter, 30 µm) was inserted through the hole of the zona pellucida, and single blastomeres were isolated. For late-8-cell- and 16-cell-stage embryos, embryos were treated with TrypLE Express (Gibco) for 5 min before single-cell isolation. TE and ICM cells were isolated as previously described62. Microsurgical isolation of single cells was performed by micromanipulation. After single-cell isolation, the cell was washed twice with PBS. The cell in 0.5 µl PBS was transferred to a tube with 6 µl sampling buffer.
SCNT
SCNT was performed as previously described61. In brief, groups of MII oocytes were transferred into droplets of M2 medium containing 500 µg ml−1 cytochalasin B on the microscope stage to collect the MII spindle. Oocytes undergoing microsurgery were held with a holding pipette. A hole was made in the zona pellucida through the application of several piezo-pulses using an enucleation pipette. The MII spindle was aspirated into the pipette with a minimal volume of ooplasm, and the resulting enucleated oocytes were transferred into CZB. For nuclear injection, donor cumulus cells were gently aspirated in and out of the injection pipette to broken plasma membranes. Each nucleus was injected into an enucleated oocyte, and these reconstructed oocytes were kept in the incubator until activation. Reconstructed oocytes were parthenogenetically stimulated by incubation in CZB supplemented with 10 mM SrCl2, 2 mM ethylene glycol tetraacetic acid (EGTA), and 5 µM latrunculin A for 10 h, and then cultured in CZB.
Single-cell DNA replication profiling (scRepli-seq) using mouse embryos
scRepli-seq experiments using mouse embryos were performed as previously reported with slight modifications. In brief, unfixed single blastomeres (derived from BDF1 or B6MSM strain embryos) were collected into 8-well PCR tubes with 6 μl cell lysis buffer (288 μl of H2O, 2 μl of 10 mg ml−1 proteinase K (Sigma-Aldrich, P4850), 32 μl of 10× single-cell lysis and fragmentation buffer (Sigma-Aldrich, L1043)), incubated at 55 °C for 1 h and then at 99 °C for 4 min for gDNA isolation and fragmentation. For scRepli-seq analysis, we analysed all blastomeres of an embryo unless there was accidental damage to the cell/sample. For scRepli-seq experiments after EdU staining (Extended Data Fig. 3b,c), after taking the photographs of EdU-stained cells (according to the protocol in Extended Data Fig. 3a), the cells were manually collected by a mouth pipette under the microscope into 12 μl of cell lysis buffer and incubated at 55 °C for 16 h (not 1 h). The remaining whole-genome amplification process (SeqPlex enhanced DNA amplification kit, Sigma-Aldrich, SEQXE) and next-generation sequencing (NGS) library construction (NGS LTP library preparation kit, KAPA, KK8232) were basically performed according to the manufacturer’s instructions. The samples were processed for NGS on the Illumina Hiseq 1500 or Hiseq X Ten system (80-bp-length single-read or 150-bp-length paired-end read sequencing).
Immunostaining
Embryos were fixed with 2% paraformaldehyde in PBS-polyvinyl alcohol (PVA) (pH 7.4) for 30 min. After blocking and permeabilization in PBS-PVA containing 1 mg ml−1 BSA (PBS-PVA-BSA) and 0.1% Triton X-100, the embryos were incubated with appropriate primary antibodies overnight at 4 °C, washed several times in PBS-PVA-BSA and incubated with secondary antibodies for 90 min at room temperature. DNA was counterstained with 40 µg ml−1 of Hoechst 33342. Finally, the embryos were washed and transferred to BSA-PVA for imaging with a Zeiss LSM780 confocal microscope. The following primary antibodies were used: mouse anti-γH2A.X (phosphorylated Ser139) (1:200, Abcam, ab22551); rabbit anti-histone H3 (1:200, Abcam, ab62706); mouse anti-histone H3 (1:200, Abcam, ab195277); mouse anti-histone H3K9me2 (1:200, Monoclonal Antibody Institute, Japan (MABI), MABI0317); rabbit anti-phosphorylated-CHK1 (Ser345) (133D3) (1:200, Cell Signaling Technology, 2348S) antibodies. The secondary antibodies were Alexa Fluor 488 goat anti-mouse IgG (H+L) (A11029); goat anti-rabbit IgG (H+L) (A11034); Alexa Fluor 555 goat anti-mouse IgG (H+L) (A21424) (1:400, Invitrogen).
Quantification of fluorescence signals
To quantify the levels of H3K9me2, p-Chk1 or γH2AX relative to the levels of histone H3, we obtained the mean signal intensity for H3K9me2, p-CHK1 or γH2AX within the nuclei (Ime_nuc). We then subtracted the mean cytoplasmic signal intensity (Ime_cyto), which was obtained from a region near the nuclei, from the Ime_nuc value (Ime_nuc − Ime_cyto). Similarly, we determined the histone H3 level within the same nuclei (IH3_nuc − IH3_cyto). Finally, we calculated the ratio between the two values (Ime_nuc − Ime_cyto)/(IH3_nuc − IH3_cyto).
EdU staining
For each embryo, the timing of its fertilization or cleavage was recorded by observation using stereomicroscopy every 30 min. At each hour after fertilization or cleavage, embryos were collected, treated with 20 µM EdU for 30 min, and then fixed in 3.7% paraformaldehyde in PBS-PVA (pH 7.4) for 30 min. EdU staining was performed using Click-iT Plus EdU Alexa Fluor 555 or 594 Imaging Kit (Invitrogen). After EdU staining, samples were incubated with mouse anti-histone H3 (1:200, Abcam, ab195277) primary antibody at 4 °C overnight, washed several times in PBS-PVA-BSA and incubated with Alexa Fluor 488 goat anti-mouse IgG (H+L) (A11029) secondary antibody for 120 min at room temperature. The embryos were finally washed and transferred to BSA–PVA for imaging on the Zeiss LSM780 confocal microscope. The images were reconstructed into 3D with Imaris software. In Fig. 2b–d and Extended Data Fig. 3a–c, we categorized EdU staining patterns based on spatial distribution and intensity. Images of nuclei were subjected to auto-thresholding with the ‘default’ algorism in Fiji software, and the thresholded patterns were manually categorized into ‘uniform’, ‘nuclear periphery + NPBs’, ‘NPBs’, ‘nPBs + internal foci’, ‘nuclear periphery + internal foci’, and ‘internal foci’. The ‘uniform’ category was further divided into two groups: those with nuclear EdU intensity 1.5× higher relative to cytoplasmic intensity (strong) and the others (weak). If the nuclear EdU intensity was less than 1.1 times the cytoplasmic intensity, it was categorized as ‘no signal’. For the experiment shown in Extended Data Fig. 3a, EdU-stained cells after Hoechst 33342 treatment (20 μM, for 30 min at 37 °C) were analysed using fluorescence-activated cell sorting using the Sony SH800 cell sorter using the single-cell mode (SH800 v.2.1).
EdU staining of metaphase chromosome spreads
The MC12 cells (cultured in 10% FBS/DMEM medium) and embryos (1-cell, 2-cell and 4-cell; C57BL/6 strain) were labelled with 20 μM EdU for 60 min in the presumptive early-S phase. After several hours of cultivation without EdU, the cells and embryos were treated with 0.01 μg ml−1 colcemid for 120 min to synchronize to M phase. The cells and embryos, from which the zona pellucida was removed by acidic Tyrode’s solution, were exposed to hypotonic 1% FBS/PBS supplemented with 0.075 M KCl for 10–20 min. The cells and embryos were then fixed with 3:1 methanol:acetic acid solution at −20 °C for 30 min (cells) or room temperature for a few minutes (embryos), and the chromosome spreads were prepared on glass slides. EdU staining of metaphase chromosomes was performed using the Click-iT EdU Cell Proliferation Kit (Invitrogen), and EdU fluorescence signal intensity was analysed with ImageJ.
DNA fibre spreading assay
More than 30 embryos (C57BL/6 strain) at each developmental stage were collected. 1-, 2- and 4-cell embryos were collected at early S phase. Likewise, most 8-cell embryos were also collected at early-S phase (although some cells may not be at early S, as cells lose cell cycle synchrony after the 8-cell stage). Embryos were labelled with 100 μM 5-iodo-2′-deoxyuridine (IdU) for 30 min just before collection. After three quick washes with KSOM medium, the embryos were labelled with 100 μM 5-chloro-2′-deoxyuridine (CldU) for 30 min. The labelling reaction was stopped by washing the cells with ice-cold 1% FBS/PBS. The embryos, from which the zona pellucida was removed by acidic Tyrode’s solution, were transferred into 1% FBS/PBS under the microscope with a mouth pipette. The embryos were then placed onto an APS-coated glass slide (Matsunami) with less than 1 μl of 1% FBS/PBS. Then, 20 μl of spreading buffer (0.5% SDS, 200 mM Tris-HCl (pH 7.5), 50 mM EDTA, 100 mM NaCl) (NaCl was added for longer fibre recovery63) was added onto the embryos on the glass slides, which were incubated at room temperature for 6 min (ref. 9). The slides were then gently tilted 20° from horizontal to stretch the DNA fibres. The DNA fibre slides were dried at room temperature for at least 1 h and the slides were fixed (methanol:acetic acid, 3:1) for 2 min. DNA fibres on the fixed slides were denatured with 1 M NaOH for 22 min, neutralized by five washes with PBS, and blocked with 1% BSA/PBST (0.05% Tween-20). Immunostaining of labelled DNA was performed with mouse anti-BrdU antibody (1:5; recognizes IdU and CldU64; BD, 347580) and rat anti-BrdU antibody (1:25; recognizes CldU64; Abcam, 6326) at 37 °C for 45 min followed by incubation with Alexa Fluor 488 donkey anti-mouse IgG (H+L) secondary antibody (1:200, Invitrogen, A21202) and Alexa Fluor 594 donkey anti-rat IgG (H+L) secondary antibody (1:200, Invitrogen, A21209) at 37 °C for 30 min. After these antibody incubation steps for IdU/CldU detection, the slides were further incubated with mouse monoclonal antibody against ssDNA (1:100, Millipore, MAB3034, 16-19) for 30 min at 37 °C and Alexa Fluor 647 goat anti-mouse IgG2a secondary antibody (1:50, Invitrogen, A21241) for 30 min at 37 °C to avoid cross-reaction65. Finally, the samples were mounted with ProLong Diamond (Invitrogen, P36970), and photographs were taken with the DeltaVision Elite microscope using a ×60 Plan/Apo NA1.42 oil-immersion objective at 2,048 × 2,048 pixels. The excitation and emission band-pass filter sets used were 542/27 and 594/45 nm, respectively (TRITC), or 632/22 and 679/34 nm, respectively (Cy5), to avoid signal overlap between Alexa Fluor 594 and 647. Using λDNA as a control, we estimated the extension rate to be around 4.4 kb per μm. As the length of the smallest individual dots (30 min labelled DNA) that we could observe by imaging using the DeltaVision microscope was approximately 0.3 μm (~1.3 kb), the maximum resolution of our microscopy analysis was about 44 bp per min (around 1.3 kb per 30 min).
Categorization of replicated DNA fibres and the measurement of IOD and fork speed
Images of DNA fibres were subjected to auto-thresholding with the ‘minimum’ or ‘default’ algorithm in Fiji software. Individual DNA fibres were identified as a series of linearly arranged Alexa Fluor 647 (ssDNA) ‘dot’ signals (Fig. 2h). The thresholded fibres containing both Alexa Fluor 488 (IdU+CldU) and Alexa Fluor 594 (CldU) signals were manually categorized into those with ‘immobile’, ‘intermediate’ and ‘mobile’ class forks. The immobile class forks were defined as those with single dot signals of IdU + CldU or CldU with gaps between dots, reflecting extremely slow fork movement. Here, gaps were defined as those with at least one dot of Alexa Fluor 647 ssDNA signal (Fig. 2h (immobile)). The mobile class forks were defined as those with a series of dot signals (≥2 dots) of IdU + CldU that contain a series of dot signals (≥2 dots) of CldU on either side of the same DNA fibre (Fig. 2h (mobile)). The intermediate class forks were defined as those with an intermediate character between the two other classes; these fibres also contained dot signals of IdU + CldU and CldU with gaps between dots but also contained some IdU + CldU single colour signals (that is, IdU-only regions) in between these dot signals. The IOD measurement method on the mobile fork class fibres is provided in Extended Data Fig. 5b. The IOD between the immobile forks was calculated by measuring the distance between the brightest pixels in the centre of the dots using the Fiji software. To determine the fork speed of the mobile forks, CldU tracks flanked by IdU tracks were identified, their lengths were measured, and were divided by the duration of the second pulse (30 min). CldU tracks that had no ssDNA signals ahead of them were excluded from the fork speed measurement as these fibres may have been broken in the middle of the CldU track. For the immobile fork speed measurement, the details are provided in Supplementary Note 2.
Intra-S-phase checkpoint analysis
For the experiments described in Extended Data Fig. 5d–i, MC12 cells were cultured on a glass-bottomed dish, treated with 0.5 μM nocodazole for 17 h to synchronize in prometaphase and subjected to 3 μg ml−1 aphidicolin treatment for 3 h. After synchronization at the G1/S-phase border, the cells were further cultured in the presence of aphidicolin for 15 h with or without 10 mM 2-aminopurine (2-AP; an intra-S-phase checkpoint inhibitor). After removing aphidicolin or 2-AP, the cells were labelled with 20 μM EdU for 60 min and stained with EdU. The C57BL/6 strain 4-cell and 8-cell embryos within 1–2 h after the 2-to-4-cell or 4-to-8-cell division (that is, in early S phase) were treated with 3 μg ml−1 aphidicolin with or without 2-AP for 5.5 h followed by reagent removal and EdU staining.
Live-cell imaging
After linearization of the template plasmid, mRNA was synthesized using the mMESSAGE mMACHINE KIT (Ambion). Synthesized RNAs were stored at −80 °C until use. In vitro-transcribed mRNAs (0.9 pl of 150 ng µl−1 mEGFP-SLX4, 0.9 pl of 150 ng µl−1 mEGFP-PCNA, 0.9 pl of 150 ng µl−1 Major-satellite-mClover and 0.9 pl of 35 ng µl−1 H2B-mCherry) were microinjected into 1-cell embryos. Live-cell imaging was performed as previously described66 with some modifications. In brief, a Zeiss LSM710, LSM780 or LSM880 confocal microscope equipped with a 40× C-Apochromat 1.2NA water-immersion objective lens (Carl Zeiss) was controlled by a multi-position autofocus macro67 for Zen Software (Carl Zeiss). For major-satellite imaging, 17 confocal z sections (every 1.5 µm) of 512 × 512 pixel xy images covering a total volume of 30.30 × 30.30 × 24.00 µm were acquired at 2 min 15 s intervals for at least 3 h just after nuclear envelope breakdown. For SLX4 imaging, 17 confocal z sections (every 2 µm) of 512 × 512 pixel xy images covering a total volume of 30.30 × 30.30 × 32.00 µm were acquired at 3 min intervals for at least 10 h. For PCNA imaging, 29 confocal z sections (every 3 µm) of 512 × 512 pixel xy images covering a total volume of 84.85 × 84.85 × 84.00 µm were acquired at 5 min intervals from the 1-cell to 16-cell stages. In Figs. 3c,g and 4j and Extended Data Fig. 7a, to achieve high-resolution live imaging while minimizing phototoxicity, we selected and imaged blastomeres (cells) that were just entering M phase and close to the objective lens, up to two blastomeres (cells) per embryo.
3D imaging analysis
To detect chromosome aberration using live imaging (Fig. 3c), we analysed images of embryos expressing major-satellite-mClover and H2B-mCherry using Imaris software (Bitplane). Chromosome bridges were detected at anaphase timepoints. To analyse DNA repair foci at M phase (Fig. 3f), we used images of embryos expressing mEGFP–SLX4 and H2B–mCherry. To detect DNA repair foci, images at 6 min after nuclear envelope breakdown were processed using the 3D Spots detection function in Imaris software with a threshold of 2.0 times the cytoplasmic signal intensity. The detected spots were manually checked for quality and tracked over time through prometaphase until they disappeared. At each timepoint, the number of SLX4 foci on chromosomes was counted. To measure the cell cycle progression from the 1- to 8-cell stage (Fig. 4 and Extended Data Fig. 7), we analysed images of embryos expressing mEGFP–PCNA and H2B–mCherry with Imaris. To determine the timing of late S, images were processed using the ‘3D Spots’ function in Imaris with a threshold of 6.0 times the cytoplasmic intensity. The detected spots were manually checked for quality. The timing when the first PCNA spot appeared was defined as the onset of late S, whereas the timing when the last spot disappeared was defined as the onset of G2. We tracked all cells in an embryo while detecting chromosome bridges at each cell division. In Fig. 4, we defined error cells as those that exhibited chromosome bridges for the first time during the 4-to-8-cell division.
scRepli-seq data analysis
In brief, after NGS, the raw scRepli-seq FASTQ files were processed for adapter trimming of both Illumina and SEQXE adapters, mapped to the mm9 reference genome and we filtered out the duplicated reads and reads that overlapped with the mm9 blacklists as described previously16. For quality control of scRepli-seq data, we applied MAD-score-based screening to filter out cells with problematic data (MAD scores of 0 or >1.0). More than 90% of cells in each sample passed these criteria. To generate log2[median] single-cell RT profiles, we counted the reads in sliding windows of 200 kb at 40 kb intervals after normalizing S-phase data with AneuFinder’s correctMappability command based on G1 control without karyotype defects in each strain. The binarization using 80 kb or 400 kb (haplotype-resolved analysis) windows was performed using the findCNVs command in AneuFinder as described previously16. For 4-cell embryos, we applied the 1-somy mode for early-S-phase cells and the 2-somy mode for mid/late-S-phase cells. For 1- and 2-cell embryos, we used the 2-somy mode for binarization (that is, the default copy number is ‘replicated’). As such, the overall binary profile will become blue (replicated) if there is no copy-number variation; likewise, if we use the ‘1-somy’ mode for the analysis of 1- and 2-cell embryos, it will be all yellow (unreplicated) instead of blue (Extended Data Fig. 2c). If we use a third colour to describe the peculiar replication regulation of the 1- and 2-cell S phase, we thought that it would be confusing. Thus, to highlight the binarization failure and the unconventional replication regulation in 1- and 2-cell-stage embryos, we decided to use the ‘2-somy’ mode and blue (replicated) to describe the copy-number state of the majority of bins (Figs. 1d and 2f and Extended Data Figs. 1g, 2e,h and 4a). The percentage replication scores (that is, the percentages of all of the genomic bins that have completed their replication) were calculated from binarized scRepli-seq data (excluding chromosome X) as described previously16. Averaged scRepli-seq profiles shown in Fig. 1b and Extended Data Fig. 1d were calculated from cells with 30–70% replication scores (excluding chromosome X). To identify the heterogeneously late-replicating domains described in Fig. 2f and Extended Data Figs. 1g and 2h, scRepli-seq profiles obtained from cells throughout the S phase were used to calculate the averaged scRepli-seq profile. The tag-density profile was generated in sliding windows of 200 kb at 40 kb intervals using AneuFinder as described previously16. t-SNE clustering analysis of scRepli-seq data (excluding chromosome X) was performed using RtSNE. For RtSNE, log2[median] RT scores of mid-S cells (cells with 30–70% replication scores) were used that were obtained from 4-, 8-, 16-cell embryos, ICM and TE.
Hi-C data analysis
Principal component 1 (PC1; A/B compartment profile) of Hi-C data in 200 kb bins was computed from the .hic file using published mapped Hi-C datasets of sperm68 and cumulus60 cells as described previously16. The genomic coordinates of PC1 profiles were converted from mm10 to mm9 using the liftover tool (UCSC Genome Browser). In Extended Data Figs. 2k and 4c, we defined the four PC1 (A/B compartment) categories, A1, A2, B2 and B1, as those containing 25% of all PC1 values (200 kb bins) each from the highest (strongest A) to lowest (strongest B) without an overlap.
Chromosome aberration analysis by scRepli-seq
To analyse chromosome aberration, we used the findCNVs command in AneuFinder with a 500 kb bin size as described previously16 (6-HMM options: method=“HMM”, max.iter=3000, states=c(“zero-inflation”, “0-somy”, “1-somy”, “2-somy”, “3-somy”, “4-somy”, “5-somy”, “6-somy”), eps=0.01). Using scRepli-seq data of 4-, 8- and 16-cell-stage embryos, we determined the cell division that produced de novo chromosome errors as follows. When a pair of cells within an embryo was found to exhibit a 3:1 copy-number ratio (3-somy:1-somy) in a complementary manner for a particular chromosome or a chromosomal region, these cells were judged to be a pair of sister cells that experienced a chromosome segregation error for the first time in the last cell division (shown in Fig. 3b as de novo chromosome aberrations). When identical chromosomal abnormalities were commonly found in multiple pairs of cells within an embryo, these cells were judged to have experienced errors in divisions preceding the last division. Details of all of the detected chromosome aberrations are shown in Extended Data Fig. 6b and Supplementary Table 1. As we pooled embryos from multiple females before single-embryo sampling, our dataset is not suitable for female-to-female variability assessment.
Statistics and reproducibility
Statistical analyses were performed with GraphPad Prism v.7.02 using one-way ANOVA with Dunn’s multiple-comparison test (Figs. 2j–l, 3g–i and 4h,I); two-tailed unpaired Student’s t-tests (Fig. 4d); two-tailed unpaired χ2 tests (Fig. 4f); two-tailed Mann–Whitney U-tests (Fig. 4g); and two-tailed Fisher’s exact tests (Fig. 4j).
Experimental reproducibility was demonstrated as follows: Fig. 1g, two independent experiments; Fig. 2d, two (2-cell) and three (1-cell, 4-cell) independent experiments; Fig. 2l, two independent experiments; Fig. 3b, two independent experiments; Fig. 3c, four independent experiments; Fig. 3g, two (2-to-4-cell and 16-to-32-cell) and three (4-to-8-cell and 8-to-16-cell) independent experiments; Fig. 3h, two independent experiments; Fig. 3i, two independent experiments; Fig. 4a–d, two independent experiments; Fig. 4h, two independent experiments; Fig. 4i, two independent experiments; and Fig. 4j, four independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
References
Hu, Y. & Stillman, B. Origins of DNA replication in eukaryotes. Mol. Cell 83, 352–372 (2023).
Fragkos, M., Ganier, O., Coulombe, P. & Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 16, 360–374 (2015).
Gilbert, D. M. Making sense of eukaryotic DNA replication origins. Science 294, 96–100 (2001).
Alver, R. C., Chadha, G. S. & Blow, J. J. The contribution of dormant origins to genome stability: from cell biology to human genetics. DNA Repair 19, 182–189 (2014).
Kermi, C., Aze, A. & Maiorano, D. Preserving genome integrity during the early embryonic DNA replication cycles. Genes 10, 398 (2019).
Mashiko, D. et al. Chromosome segregation error during early cleavage in mouse pre-implantation embryo does not necessarily cause developmental failure after blastocyst stage. Sci. Rep. 10, 854 (2020).
Vázquez-Diez, C. & FitzHarris, G. Causes and consequences of chromosome segregation error in preimplantation embryos. Reproduction 155, R63–R76 (2018).
Vanneste, E. et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 15, 577–583 (2009).
Palmerola, K. L. et al. Replication stress impairs chromosome segregation and preimplantation development in human embryos. Cell 185, 2988–3007.e20 (2022).
Takahashi, S. et al. Genome-wide stability of the DNA replication program in single mammalian cells. Nat. Genet. 51, 529–540 (2019).
Dileep, V. & Gilbert, D. M. Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat. Commun. 9, 427 (2018).
Vouzas, A. E. & Gilbert, D. M. Mammalian DNA replication timing. Cold Spring Harb. Perspect. Biol. 13, a040162 (2021).
Hiratani, I. & Takahashi, S. DNA replication timing enters the single-cell era. Genes 10, 221 (2019).
Ryba, T. et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 20, 761–770 (2010).
Miura, H. et al. Single-cell DNA replication profiling identifies spatiotemporal developmental dynamics of chromosome organization. Nat. Genet. 51, 1356–1368 (2019).
Miura, H. et al. Mapping replication timing domains genome wide in single mammalian cells with single-cell DNA replication sequencing. Nat. Protoc. 15, 4058–4100 (2020).
Du, Z. et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235 (2017).
Ke, Y. et al. 3D Chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170, 367–381 (2017).
Flyamer, I. M. et al. Single-cell Hi-C reveals unique chromatin reorganization at oocyte-tozygote transition. Nature 544, 110–114 (2017).
Dileep, V. et al. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication timing program. Genome Res. 25, 1104–1113 (2015).
Ladstätter, S. & Tachibana, K. Genomic insights into chromatin reprogramming to totipotency in embryos. J. Cell Biol. 218, 70–82 (2019).
Fu, X., Zhang, C. & Zhang, Y. Epigenetic regulation of mouse preimplantation embryo development. Curr. Opin. Genet. Dev. 64, 13–20 (2020).
Martin, C. et al. Genome restructuring in mouse embryos during reprogramming and early development. Dev. Biol. 292, 317–332 (2006).
Chen, M. et al. Chromatin architecture reorganization in murine somatic cell nuclear transfer embryos. Nat. Commun. 11, 1813 (2020).
Conti, C. et al. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 18, 3059–3067 (2007).
Takebayashi, S.-I. et al. The temporal order of DNA replication shaped by mammalian DNA methyltransferases. Cells 10, 266 (2021).
Nakatani, T. et al. DNA replication fork speed underlies cell fate changes and promotes reprogramming. Nat. Genet. 54, 318–327 (2022).
Cremer, T. & Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001).
Dimitrova, D. S. & Gilbert, D. M. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat. Cell Biol. 2, 686–694 (2000).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Dehé, P.-M. & Gaillard, P.-H. L. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 18, 315–330 (2017).
Chagin, V. O. et al. Processive DNA synthesis is associated with localized decompaction of constitutive heterochromatin at the sites of DNA replication and repair. Nucleus 10, 231–253 (2019).
Bester, A. C. et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 (2011).
Halliwell, J. A. et al. Nucleosides rescue replication-mediated genome instability of human pluripotent stem cells. Stem Cell Rep. 14, 1009–1017 (2020).
Yuan, K., Seller, C. A., Shermoen, A. W. & O’Farrell, P. H. Timing the Drosophila mid-blastula transition: a cell cycle-centered view. Trends Genet. 32, 496–507 (2016).
Hörmanseder, E., Tischer, T. & Mayer, T. U. Modulation of cell cycle control during oocyte-to-embryo transitions. EMBO J. 32, 2191–2203 (2013).
Langley, A. R., Smith, J. C., Stemple, D. L. & Harvey, S. A. New insights into the maternal to zygotic transition. Development 141, 3834–3841 (2014).
Farrell, J. A. & O’Farrell, P. H. From egg to gastrula: how the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu. Rev. Genet. 48, 269–294 (2014).
McCleland, M. L., Shermoen, A. W. & O’Farrell, P. H. DNA replication times the cell cycle and contributes to the mid-blastula transition in Drosophila embryos. J. Cell Biol. 187, 7–14 (2009).
Hyrien, O. & Méchali, M. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J. 12, 4511–4520 (1993).
Hyrien, O., Maric, C. & Méchali, M. Transition in specification of embryonic metazoan DNA replication origins. Science 270, 994–997 (1995).
Sasaki, T., Sawado, T., Yamaguchi, M. & Shinomiya, T. Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolα-dE2F locus of Drosophila melanogaster. Mol. Cell. Biol. 19, 547–555 (1999).
Vallot, A. & Tachibana, K. The emergence of genome architecture and zygotic genome activation. Curr. Opin. Cell Biol. 64, 50–57 (2020).
Hug, C. B., Grimaldi, A. G., Kruse, K. & Vaquerizas, J. M. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228 (2017).
Cho, C.-Y., Seller, C. A. & O’Farrell, P. H. Temporal control of late replication and coordination of origin firing by self-stabilizing Rif1-PP1 hubs in Drosophila. Proc. Natl Acad. Sci. USA 119, e2200780119 (2022).
Seller, C. A. & O’Farrell, P. H. Rif1 prolongs the embryonic S phase at the Drosophila mid-blastula transition. PLoS Biol. 16, e2005687 (2018).
Niu, L. et al. Three-dimensional folding dynamics of the Xenopus tropicalis genome. Nat. Genet. 53, 1075–1087 (2021).
Ferreira, J. & Carmo-Fonseca, M. Genome replication in early mouse embryos follows a defined temporal and spatial order. J. Cell Sci. 110, 889–897 (1997).
Aoki, E. & Schultz, R. M. DNA replication in the 1-cell mouse embryo: stimulatory effect of histone acetylation. Zygote 7, 165–172 (1999).
Wike, C. L. et al. Chromatin architecture transitions from zebrafish sperm through early embryogenesis. Genome Res. 31, 981–994 (2021).
Nakamura, R. et al. CTCF looping is established during gastrulation in medaka embryos. Genome Res. 31, 968–980 (2021).
Siefert, J. C., Georgescu, C., Wren, J. D., Koren, A. & Sansam, C. L. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 27, 1406–1416 (2017).
Kaaij, L. J. T., van der Weide, R. H., Ketting, R. F. & de Wit, E. Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. 24, 1–10 (2018).
Ottolini, C. S. et al. Tripolar mitosis and partitioning of the genome arrests human preimplantation development in vitro. Sci. Rep. 7, 9744 (2017).
Capalbo, A. et al. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and trophoblast: insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum. Reprod. 28, 509–518 (2013).
McCoy, R. C. et al. Meiotic and mitotic aneuploidies drive arrest of in vitro fertilized human preimplantation embryos. Genome Med. 15, 77 (2023).
Borsos, M. et al. Genome-lamina interactions are established de novo in the early mouse embryo. Nature 569, 729–733 (2019).
Nakatani, T. et al. Emergence of replication timing during early mammalian development. Nature 625, 401–409 (2024).
Bakker, B. et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 17, 115 (2016).
Zhang, K. et al. Analysis of genome architecture during SCNT reveals a role of cohesin in impeding minor ZGA. Mol. Cell 79, 234–250 (2020).
Kyogoku, H., Wakayama, T., Kitajima, T. S. & Miyano, T. Single nucleolus precursor body formation in the pronucleus of mouse zygotes and SCNT embryos. PLoS ONE 13, e0202663 (2018).
Xu, J. et al. A simple and effective method for the isolation of inner cell mass samples from human blastocysts for gene expression analysis in vitro. Cell. Dev. Biol. Anim. 50, 232–236 (2014).
Kaykov, A., Taillefumier, T., Bensimon, A. & Nurse, P. Molecular combing of single DNA molecules on the 10 megabase scale. Sci. Rep. 6, 19636 (2016).
Jackson, D. A. & Pombo, A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 140, 1285–1295 (1998).
Bianco, J. N. et al. Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods 57, 149–157 (2012).
Kyogoku, H. & Kitajima, T. S. Large cytoplasm is linked to the error-prone nature of oocytes. Dev. Cell 41, 287–298 (2017).
Rabut, G. & Ellenberg, J. Automatic real-time three-dimensional cell tracking by fluorescence microscopy. J. Microsc. 216, 131–137 (2004).
Alavattam, K. G. et al. Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat. Struct. Mol. Biol. 26, 175–184 (2019).
Acknowledgements
We thank T. Ichinose, A. Tanigawa and Y. Wu for technical assistance; K. Inoue and H. Kiyonari for providing the frozen embryos; S. Wakana for providing the MSM mice; R. Nakamura for discussion on pre-ZGA zebrafish embryos; R. Cerbus, H. Hamada and S. Kuratani for comments on the manuscript; and J. Ellenberg for providing the imaging macro. This work was supported by RIKEN BDR intramural grants, RIKEN Pioneering Projects ‘Genome Building from TADs’ and ‘Long-timescale Molecular Chronobiology’, JST CREST grant number JPMJCR20S5, MEXT KAKENHI grant number 18H05530 and JSPS KAKENHI grant number 20K20582 to I.H.; JSPS KAKENHI grant numbers 23H04948, 21H02407 and 18H05549 to T.S.K.; JSPS KAKENHI grant number 18K14681 and the Mochida Memorial Foundation for Medical and Pharmaceutical Research to S.T.; and JST PRESTO grant number JPMJPR20K4 and JSPS KAKENHI grant number 22H04674 to H.K.
Author information
Authors and Affiliations
Contributions
S.T., H.K., T.S.K. and I.H. conceived the project. S.T. performed the metaphase spread and intra-S-phase checkpoint experiments. H.K. performed the preparation of all embryos and collection of cells for scRepli-seq, live-cell imaging and EdU staining. S.T. and H.K. performed scRepli-seq. S.T., H.K. and H.M. performed the bioinformatic analyses. S.T., A.O., Y.K., T.H. and S.-i.T. collected the embryos, performed DNA combing and analysed data. S.T., H.K., T.S.K. and I.H. wrote the manuscript with comments from H.M., A.O. and S.-i.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Jan Ellenberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Evaluation of scRepli-seq resolution and the analysis of scRepli-seq profiles of pre-implantation mouse embryos.
(a) NGS tag density plots of mid-S mESC scRepli-seq data (three individual cells) with 4–6 million (M) (top panel) and ~20 M (bottom panel) total NGS reads per cell. Density was calculated using sliding windows of 200 kb at 40-kb intervals, 80 kb at 16-kb intervals, 40 kb at 8-kb intervals, or 20 kb at 4-kb intervals. While the bimodal distribution is evident when using 200-kb and 80-kb windows, it becomes obscure when using 40-kb windows. For the ~20 M read samples, bimodality is barely visible at 40-kb. (b) scRepli-seq NGS read density plots of representative mid-S mESC single-cell data (Extended Data Fig. 1a, MidS#1) with 4.6 M and 18.4 M reads with the indicated bin size settings. Binarization calling outputs are also shown. Note that the valid read count is not necessarily proportional to the total read count, owing to an increase in duplicate reads, suggesting the presence of redundancy due to the limit of the scRepli-seq library complexity. The dotted line shows the lower read count limit of 20 that we set for the unreplicated bins (see Supplementary Note 1). (c) Pearson correlation matrix of averaged mid-S cell scRepli-seq data (excluding chrX). (d) Average scRepli-seq RT (Ave. scRT) profiles calculated from mid-S cells with 30–70% replication scores derived from 8-cell embryos and ICM. Chr8 is shown (80-kb bins). RT class definitions are from Dileep et al.20 (CE, constitutively early; CL, constitutively late; D, developmentally regulated). RT-switching regions were defined as regions with ∆ave. scRT (ICM–8-cell) values of >1 or <–1. (e) Types and frequency of RT switching (from the 8-cell stage to ICM) and their RT classes (defined in (c)). LtoE and EtoL represent RT changes from late-to-early and early-to-late S, respectively. (f) Pearson correlation matrix of averaged scRepli-seq data of ICM and TE derived from mid-S cells (30–70% replication score, excluding chrX). (g) Haplotype-resolved binarized and log2median scRepli-seq profiles of 1/2/4-cell B6MSM embryos along with BrdU-IP population RT profiles of mESCs. For binarization by AneuFinder59, 2-somy mode was applied to 1-, 2- and mid/late 4-cell blastomeres, and 1-somy mode to early 4-cell blastomeres. 1- and 2-cell scRepli-seq profiles are ordered based on their sampling order, as we were unable to calculate their percentage replication score values. The resolution of binarized RT profiles and log2median RT profiles of non-haplotype-resolved B6MSM data are 80 kb (non-overlapping windows) and 200 kb (sliding windows at 40-kb intervals), respectively. For B6 or MSM (haplotype-resolved) data, it was 400 kb (non-overlapping windows) and 1 Mb (sliding windows at 40-kb intervals), respectively. Heterogeneously late RT regions shown beneath the 1-cell scRepli-seq profiles were defined as regions with an average scRT of <0.75 on the MSM (paternal) chromosome. Asterisks, data reflecting gradual and uniform replication (see Fig. 1d legend for details).
Extended Data Fig. 2 Detailed analyses of scRepli-seq profiles of 1-, 2-, 4-cell embryos.
(a,b) (left) NGS tag density plots of mid-S scRepli-seq data (three individual cells) from 1-cell (a) and 2-cell embryos (b) with >30 M total NGS reads per cell. Density was calculated using sliding windows of 200 kb at 40-kb intervals, 80 kb at 16-kb intervals, or 40 kb at 8-kb intervals. In all cases, bimodality is not visible. (right) scRepli-seq NGS read density plots of representative mid-S single-cell data from 1-cell (a) and 2-cell embryos (b) with 78.3 M and 45.9 M reads, respectively, with the indicated bin size settings. Binarization calling outputs are also shown, which revealed the lack of bimodal distribution regardless of the binarization modes chosen. The dotted line shows the lower read count limit of 20 that we set for the unreplicated bins (see Supplementary Note 1). (c) Binarized mid-S scRepli-seq profiles derived from mESCs, 1-cell embryos, and 2-cell embryos with high read counts (18.4–19.9, 30.7–78.3, 34.5–46.6 M reads respectively). The indicated bin size settings and binarization modes were chosen. Unlike mESCs, 1- and 2-cell embryos lacked bin-to-bin variation, showing either seemingly totally ‘unreplicated’ or totally ‘replicated’ profiles depending on the binarization mode (1-somy or 2-somy; see Methods). (d) NGS tag density plots of representative 1/2/4-cell and mESC scRepli-seq profiles during cell cycle at G1 and S (S1–S4), which are indicated in (e). Tag densities were calculated for 200-kb sliding windows at 40-kb intervals. For (d–k), we used scRepli-seq data with 4–6 M total reads (see Supplementary Note 1). (e) Binarized scRepli-seq profiles of 1/2/4-cell B6MSM embryos on chr15 (80-kb bins). For binarization by AneuFinder59, 2-somy mode was applied to 1-, 2- and mid/late 4-cell blastomeres, and 1-somy mode to early 4-cell blastomeres (see Methods for more details). 1- and 2-cell scRepli-seq profiles are ordered based on their sampling order, as we were unable to calculate their percentage replication score. Asterisks, data reflecting gradual and uniform replication (see Fig. 1d legend for details). (f) MAD score distribution of 1/2/4-cell blastomeres analysed by scRepli-seq with 40-kb, 80-kb, and 500-kb bins. MAD scores were constant during S-phase in 1- and 2-cell embryos, in sharp contrast to 4-cell embryos, which exhibited inverted V-shape patterns with the MAD score peaking at mid-S regardless of bin size settings (40, 80, and 500-kb bins). (g) Haplotype-resolved MAD score distribution in 1- and 2-cell embryos. Relatively high scores were detected on the 1-cell paternal genome, although they were lower than somatic cells. (h) Binarized 1-cell S-phase scRepli-seq profiles of the MSM (paternal) chromosome, along with BrdU-IP population RT profiles of mESCs. The averaged scRepli-seq profile (Ave. scRT) was generated by calculating the mean of scRepli-seq data derived from S-phase cells on the MSM (paternal) chromosome. The sperm A/B compartment profile (Hi-C PC1) was calculated from published Hi-C data68. RT class definition is based on Dileep et al.20 (CE, constitutively early; CL, constitutively late; D, developmentally regulated). Asterisks, data reflecting gradual and uniform replication (see Fig. 1d legend for details). (i) Percentages of heterogeneously late RT (Ave. scRT<0.75) regions (chrX excluded) on the paternal and maternal genome in 1- and 2-cell embryos. They were observed specifically on the 1-cell paternal genome. (j) RT class distribution of all genomic bins and heterogeneously late RT regions on the 1-cell paternal genome (chrX excluded from analysis). The majority of the heterogeneously late RT regions were classified as constitutively late-replicating (CL). (k) A/B compartment (Hi-C PC1) category distribution of all genomic bins and heterogeneously late RT regions on the 1-cell paternal genome. A1 (strongest A), A2, B2, and B1 (strongest B) each contain 25 % of all PC1 values (200-kb bins) based on a sperm Hi-C data68. The majority of the heterogeneously late RT regions were classified as B compartment (heterochromatin), B1 and B2.
Extended Data Fig. 3 Replication foci and H3K9me2 dynamics during early cleavage stages.
(a) (top) Experimental scheme. (left) A cell-cycle fluorescence-activated cell sorting (FACS) profile of mESCs. Cells were stained with Hoechst 33342, and the six gates (S1 through S6) used to collect cells throughout S-phase are shown. (right) Replication foci pattern dynamics throughout S-phase (this figure is identical to Fig. 2d). Replication foci patterns were categorized into the following classes: G1, no signal; I, uniform (weak signal); II, uniform (strong signal); III, nuclear periphery + internal foci; IV, internal foci. (b, c) scRepli-seq profiling after replication foci image analysis. (left) Replication foci pattern classification based on the definition provided in (a) of 4-cell (b) and 8-cell (c) embryos. M, mitosis. (right) Binarized scRepli-seq profiles of the 20 (b) or 10 (c) EdU-stained nuclei shown on the left. Our data provide direct, single-cell-level evidence that the spatial patterns of replication foci are a good indicator of S-phase time. Chr2 is shown. For experimental details, see Methods under ‘Single-cell DNA replication profiling (scRepli-seq) using mouse embryos’. (d) Top panel shows the representative images of H3K9me2 (green) staining. Histone H3 was stained for reference (magenta). Bottom panel shows the heatmap images of the H3K9me2 signal. (e) H3K9me2 levels were increased after 4-cell stage. One-way ANOVA using Dunn’s multiple comparisons test was performed. Error bars represent the mean and SD. (f) The spatial pattern of H3K9me2 shifted at the transition from 2-cell to 4-cell stage. Number of nuclei from two independent experiments are shown in (e) and (f).
Extended Data Fig. 4 In-depth scRepli-seq data analysis of SCNT embryos.
(a) Binarized and log2median scRepli-seq profiles of SCNT 1- and 2-cell embryos (80-kb bins), along with BrdU-IP population RT profiles of mESCs. 2-somy mode was applied for binarization. Chr3 is shown. Asterisks, data reflecting gradual and uniform replication (see Fig. 1d legend for details). (b) RT class distribution of all genomic bins and heterogeneously late RT regions (defined as regions with an average scRT of <0.50 from the analysis of S-phase cells) in SCNT 2-cell embryos. The majority of the heterogeneously late RT regions were classified as constitutively late-replicating (CL). RT class definitions are from Dileep et al.20 (CE, constitutively early; CL, constitutively late; D, developmentally regulated). (c) A/B compartment (Hi-C PC1) category distribution of all genomic bins and heterogeneously late RT regions on the SCNT 2-cell genome. A1 (strongest A), A2, B2, and B1 (strongest B) each contain 25 % of all PC1 values (200-kb bins) based on a cumulus cell Hi-C data60. More than 80% of the heterogeneously late RT regions were classified as B (heterochromatin) compartment, B1 and B2. (d) Averaged S-phase scRepli-seq profiles and heterogeneously late RT regions on the 1-cell paternal genome and in SCNT 2-cell embryos, along with BrdU-IP population RT profiles of mESCs and RT class distribution (see also Fig. 2f and Extended Data Fig. 2h). There is a substantial overlap between the heterogeneously late RT regions on the 1-cell paternal genome and in SCNT 2-cell embryos just by visual inspection. Chr12 and chr4 are shown. (e) A Venn diagram showing the degree of overlap between the heterogeneously late RT regions on the 1-cell paternal genome and the SCNT 2-cell genome (80-kb bins). (f) MAD score distribution16 of SCNT 1- and 2-cell embryos analysed by scRepli-seq. SCNT 1-cell embryos showed constant MAD scores throughout S-phase as in control 1- and 2-cell embryos (Fig. 1e and Extended Data Fig. 2f,g). In contrast, while the SCNT 2-cell embryos did not show inverted V-shape patterns observed in mESCs and control 4-cell embryos (Fig. 1e), they clearly showed fluctuation during S-phase, indicative of the co-existence of ~2 (nearly replicated) and ~1 copy (under-replicated) genomic bins.
Extended Data Fig. 5 Analysis of replication forks in early cleavage stages and re-analysis of replication states in 4-cell embryos.
(a) Representative replicated DNA fibres in 1/2/4/8-cell stage embryos. Scale = 4.55 μm (20 kb). (b) Schematic diagram of IOD measurement on the ‘immobile’ and ‘mobile’ class fibres (see also Fig. 2j). Scale = 4.55 μm (20 kb). (c) Tag density plots of scRepli-seq data derived from individual cells in 4- and 8-cell embryos in comparison to mESCs at the indicated time points (% replication scores) during S-phase. During early S (30–40% S; in the middle of the first half of S, equivalent to 3.3–4.4 hr in S assuming a 11-h S-phase), the bimodal distribution of the 4-cell data was less prominent with an unclear ‘replicated’ peak compared to 8-cell embryos and mESCs (see arrowheads and highlighted areas), suggesting the presence of incompletely replicated genomic bins. Such features disappeared at mid-S. (d–i) Analysis of intra-S-phase checkpoint. (d) Scheme of the intra-S-phase checkpoint experiment using somatic cells (MC12 cells). Briefly, synchronized cells were released from G1/S border arrest into EdU-containing medium after 0-h or after 15-h in the presence of either aphidicolin (15-h Aph) or aphidicolin + 2-aminopurine (2-AP; an intra-S-phase checkpoint inhibitor) (15-h Aph+2AP), and their replication foci patterns were evaluated. (e) Replication foci patterns of MC12 cells. While the majority of 0-h and 15-h Aph samples exhibited the early-S pattern, the majority of 15-h Aph+2AP exhibited the mid-S pattern. This indicated that suppression of intra-S-phase checkpoint by 2-AP caused replication initiation of mid-S RT domains in the absence of early-S RT domain replication, as expected29. (f, g) Scheme of the intra-S-phase checkpoint experiment using 4-cell (f) and 8-cell (g) embryos. Briefly, cells were released from G1/Early-S into EdU-containing medium after 0-h, 5.5-h, 5.5-h in the presence of aphidicolin (5.5-h Aph), or 5.5-h in the presence of aphidicolin + 2-AP (5.5-h Aph+2AP), and their replication foci patterns were analysed. (h, i) Replication foci patterns of 4-cell (h) and 8-cell (i) embryos. While the majority of 0-h and 5.5-h Aph samples exhibited the early-S pattern, the majority of 5.5-h Aph+2AP exhibited the mid-S pattern as in 5.5-h samples. Thus, 2-AP treatment caused replication initiation of mid-S RT domains in the absence of early-S RT domain replication, indicating that intra-S-phase checkpoint was already functional in 4-cell embryos.
Extended Data Fig. 6 Chromosome aberrations detected by scRepli-seq and live cell imaging.
(a) (left) Schematic diagram of chromosome aberration types (whole, whole chromosome gain/loss; break, chromosome breakage). (right) Shown are representative karyograms of three pairs of sister cells within an 8-cell mouse embryo generated by scRepli-seq. Aberrant chromosome pairs (partial (‘break’) or non-partial (‘whole’) trisomy/monosomy) are highlighted in red and by the red rectangles. (b) All chromosome breaks identified by scRepli-seq in in vitro and in vivo 8-cell embryos. Chromosomal abnormalities appear as large segments of high (blue) and low (yellow) copy numbers that are complementary between a pair of sister cells (red rectangles). The breakpoint position coordinates are shown on the right. As a reference, the averaged scRepli-seq profile of 4-cell embryos is shown at the top. (c) Live imaging of chromosome segregation at each cell division. Maximum z-projection images of Major-satellite-mClover (centromeres, green) and H2B-mCherry (chromosomes, magenta). The cell shown as “4-to-8-cell, Error” is identical to the one shown in Fig. 3c (acentric fragment). No chromosome alignment defects were observed at the late metaphase of 4-to-8-cell divisions (0/59 divisions), including those preceding chromosome bridges (0/11 divisions). Time after nuclear envelope breakdown is shown (mm:sec). (d) Three representative chromosome breakpoints in early, mid, and late-S RT regions are shown, along with the log2median scRepli-seq data of sister cell pairs with complementary errors. Note that the three regions are identical to those shown in Fig. 3d.
Extended Data Fig. 7 Aphidicolin-induced chromosome aberrations and single-cell tracking throughout early embryonic development by live imaging.
(a) Chromosome aberrations after aphidicolin treatment. Embryos were treated with aphidicolin at G1 of each embryonic stage. Chromosome aberrations at the subsequent cell division were detected and categorized by live imaging with Major-satellite-mClover (centromeres, green) and H2B-mCherry (chromosomes, magenta). Anaphase bridges consisting of one or two chromosomes were categorized as ‘mild’, whereas those consisting of three or more chromosomes were categorized as ‘severe’ chromosome aberrations. Representative snapshots of ‘mild’ and ‘severe’ chromosome aberrations at the indicated time after anaphase onset (mm:sec) are shown. Arrowheads, chromosome bridges and fragments. Number (n) of cell division from at least two independent experiments analysed live. Each circle shows the data from one experiment. Fisher’s exact test was performed using the pooled data. (b) Late-replicating regions are prone to chromosome breaks during the 4-to-8-cell division after treatment with aphidicolin. Error bars represent the mean and SD. Number (n) of chromosome breaks is shown. (c) Live imaging of cell cycle progression from 1- to 8-cell stages. 3D-reconstructed images of mEGFP-PCNA (S-phase marker, green) and H2B-mCherry (chromosomes, magenta) with signal interpolation in z. The cell lineage that exhibited a chromosome segregation error at the 4-to-8-cell division is indicated by asterisks. The mEGFP-PCNA signals of this cell linage is magnified and shown in inverted greyscale in the lower panel. Note that PCNA foci mark late S-phase (arrows). Time after the first M-phase in shown (hr:mm). (d) Durations of each cell cycle phase were measured at 2-, 4-, and 8-cell stages. (e) Total cell cycle length of 2-, 4- and 8-cell stages. For (d) and (e), One-way ANOVA using Dunn’s multiple comparisons test was performed. Error bars represent the mean and SD. Number (n) of cells from 26 embryos in two independent experiments. (f) Average cell cycle profiles of 2-, 4- and 8-cell stages. (g) Cell lineage trees from 1- to 8-cell stages. All embryos that exhibited de novo chromosome segregation errors during the 4-to-8-cell division are shown. Grey lines indicate the cells that were located outside of the images.
Supplementary information
Supplementary Information
Supplementary Notes 1–3, Supplementary Table 1 and the legends for Supplementary Videos 1 and 2.
Supplementary Video 1
Live imaging of chromosome segregation at each cell.
Supplementary Video 2
Live imaging of cell cycle progression from 1 to 8 cell.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Takahashi, S., Kyogoku, H., Hayakawa, T. et al. Embryonic genome instability upon DNA replication timing program emergence. Nature 633, 686–694 (2024). https://doi.org/10.1038/s41586-024-07841-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07841-y
- Springer Nature Limited