

Abstract
Multiple myeloma is a treatable, but currently incurable, hematological malignancy of plasma cells characterized by diverse and complex tumor genetics for which precision medicine approaches to treatment are lacking. The Multiple Myeloma Research Foundation’s Relating Clinical Outcomes in Multiple Myeloma to Personal Assessment of Genetic Profile study (NCT01454297) is a longitudinal, observational clinical study of newly diagnosed patients with multiple myeloma (n = 1,143) where tumor samples are characterized using whole-genome sequencing, whole-exome sequencing and RNA sequencing at diagnosis and progression, and clinical data are collected every 3 months. Analyses of the baseline cohort identified genes that are the target of recurrent gain-of-function and loss-of-function events. Consensus clustering identified 8 and 12 unique copy number and expression subtypes of myeloma, respectively, identifying high-risk genetic subtypes and elucidating many of the molecular underpinnings of these unique biological groups. Analysis of serial samples showed that 25.5% of patients transition to a high-risk expression subtype at progression. We observed robust expression of immunotherapy targets in this subtype, suggesting a potential therapeutic option.
Similar content being viewed by others

Main
Multiple myeloma is a treatable, but currently incurable, hematological malignancy of plasma cells (PCs). The incorporation of new treatment modalities over the last two decades has vastly improved outcomes of patients with myeloma; however, patients still relapse, and some have poor outcomes. Despite substantial efforts to understand the molecular basis of the disease, predicting patient outcomes and identifying high-risk patients remains a challenge.
Multiple myeloma is a genetically heterogeneous disease with two broad karyotypic groups. A hyperdiploid (HRD) phenotype, with characteristic trisomies of chromosomes (chr) 3, 5, 7, 9, 11, 15, 19 and 21, is present in 50–60% of tumors1,2,3. The remaining non-HRD (NHRD) tumors typically have an immunoglobulin translocation dysregulating NSD2/WHSC1/MMSET, MYC, CCND1 or MAF transcription factors4,5,6,7,8,9. Tumors harbor many other genetic aberrations, including nonimmunoglobulin structural abnormalities and mutations10,11,12,13. Although previous genomic studies were instrumental in deconvoluting the genetic heterogeneity of myeloma, they are mostly limited by small cohort sizes, the number and types of assays performed, a lack of longitudinal sampling, clinical follow-up and biased inclusion of heavily pretreated patients, limiting our comprehensive understanding of the disease.
To understand the impact of tumor genetic profile on patient outcomes and treatment response, the Multiple Myeloma Research Foundation (MMRF) sponsored the Relating Clinical Outcomes in Multiple Myeloma to Personal Assessment of Genetic Profile (CoMMpass) study (NCT01454297). CoMMpass was a prospective, longitudinal, observational clinical study that accrued 1,143 newly diagnosed, previously untreated patients with multiple myeloma from sites throughout the United States, Canada, Spain and Italy between 2011 and 2016. Tumor samples collected at diagnosis and each progression event were profiled using whole-genome sequencing (WGS), whole-exome sequencing (WES) and RNA sequencing (RNA-seq). Clinical parameters were collected every 3 months through the 8-year observation period.
We present a molecular analysis of the complete baseline cohort, with a median follow-up of 4 years, identifying recurrent loss-of-function (LOF) and gain-of-function (GOF) events and distinct copy number and gene expression subtypes of myeloma. The comprehensive nature of this dataset and our integrated analysis framework define both the overall frequency of gene alterations in myeloma and the genetic basis of a high-risk patient population that does not benefit from current therapies.
Results
Cohort description
The demographic and clinical parameters of the cohort follow expected distributions with a median diagnostic age of 63 years (range = 27–93 years), the expected over-representation of males (60.4%) and an international staging system (ISS) distribution of 35.1% ISSI, 35.1% ISSII and 27.2% ISSIII14 (Table 1 and Supplementary Table 1). This cohort is primarily from the United States, and, unlike most clinical trials, the distribution of self-reported ancestry reflects US Census Bureau statistics with 80.6% Caucasian, 17.5% Black and 1.9% Asian.
Each patient’s cytogenetic phenotype was defined by WGS with 57.2% HRD and 42.8% NHRD, 24.3% del(1p22), 35.2% gain(1q21), 52.0% del(13q14) and 12.5% del(17p13). Translocations involving common target genes from any of the three immunoglobulin loci occurred at the following frequencies: 20.0% CCND1, 1.2% CCND2, 1.8% CCND3, 4.0% MAF, 0.7% MAFA, 1.3% MAFB, 14.3% MYC and 12.8% WHSC1. Of these events, 83.0% involved the IgH locus, while 5.3% and 11.7% involved the κ and λ loci, respectively.
Irrespective of treatment, the cohort median time to second-line therapy was 38.1 months, and the median overall survival (OS) was 103.6 months (Extended Data Fig. 1a,b). The median OS for ISSIII patients was 53.9 months, while the median OS for ISSI and ISSII patients could not be confidently predicted (Extended Data Fig. 1c,d). Patients with at least one high-risk cytogenetic feature had worse OS outcomes, even with uniform usage of new agents (Supplementary Fig. 1)15.
Integrated analysis for gain and LOF genes
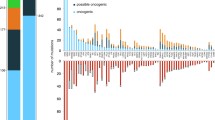
To comprehensively identify LOF and GOF events in patients with myeloma, an integrated model was developed to overcome the limitations of analyzing any one data type by combining measurements from WES, WGS and RNA-seq to assign a functional state to each gene (Supplementary Tables 2–6). In the LOF model, a single event in a gene was designated as partial LOF, whereas genes with two or more events were designated as complete LOF. At diagnosis, 592 patients had all three sequencing assays performed and were included in the analysis (Supplementary Fig. 2). We identified 70 genes where a complete LOF event was identified in ≥5 patients (Fig. 1a). Complete LOF was observed in 12 genes in >2% of the cohort, including TRAF3 (10.1%), DIS3 (6.9%), TENT5C/FAM46C (5.1%), CYLD (4.7%), TP53 (4.1%), MAX (3.5%), RB1 (3.2%), WWOX (3.2%), HUWE1 (2.7%), PVT1 (2.5%), CDC42BPB (2.0%) and MAGEC1 (2.0%). However, CDC42BPB is in a contiguous gene region on chr14 with TRAF3, which was previously shown to be the target of bi-allelic loss in this region16.

The location and proximity of individual genes are shown next to each gene with the alternating gray and black bars illustrating when the chromosomal location changes, while black bars directly to the right denote contiguous genes. Each tick along the x axis represents a patient with the corresponding event. a, Complete LOF was observed in 53 autosomally located genes. b, Genes on chr13q that were the target of complete LOF events in at least five patients in the baseline cohort. c, GOF events were detected in 27 autosomal genes due to amplification, gene fusions, mutations or over expression associated with a structural variant (SV).
The target gene(s) of chr13 loss continues to be controversial. WGS data detected 13q14 deletion in 52.0% of patients, while LOF analysis identified that 26.5% of patients had complete LOF of one or more genes on chr13. The commonly assumed target, RB1, showed complete LOF in 3.2% of patients, while DIS3 complete LOF was detected in 6.9% of patients; however, a striking number of additional genes were independently knocked out in myeloma (Fig. 1b). The following two contiguous gene regions with complete LOF were identified: the first comprising MPHOSPH8 (1.4%), PSPC1 (1.5%), ZMYM5 (1.4%), ZMYM2 (1.0%), and the second comprising TGDS (1.9%) and GPR180 (0.8%) where the minimal region of deletion and LOF frequency suggest that the targets are PSPC1 and TGDS, respectively. Additional complete LOF events were identified targeting LATS2 (1.4%), BRCA2 (1.2%), PARP4 (1.0%), MYCBP2 (1.0%), TPP2 (1%), CDK8 (0.8%), TSC22D1 (0.8%) and ARHGEF7 (0.8%). These results highlight that monosomy 13 is associated with multiple independent gene inactivation events.
The GOF analysis identified an event in 92% of patients at diagnosis and 27 genes where a GOF event was identified in five or more patients (Fig. 1c). There were seven genes in which a GOF event was identified in greater than 2% of the cohort, including KRAS (23.6%), NRAS (21.6%), WHSC1 (10.3%), BRAF (7.1%), FGFR3 (4.9%), HIST1H1E (3.2%) and EGR1 (2.5%).
Identification of copy number subtypes of multiple myeloma
To discover potential underlying phenotypes of myeloma beyond the known dichotomy of HRD and NHRD karyotypes, unsupervised consensus clustering was performed on copy number data from 871 patients (Extended Data Fig. 2). Three independent trials were highly consistent and identified eight subtypes as the optimal solution (Supplementary Figs. 3 and 4).
The copy number subtypes consisted of five HRD and three NHRD subtypes and were annotated based on defining features (Fig. 2a). The HRD classic subtype had gains of classic HRD chromosomes, and the remaining HRD subtypes were annotated based on deviations from this phenotype. The subtype designated as HRD, ++15 exhibited tetrasomy of chr15 (Supplementary Fig. 5), while two subtypes (HRD, diploid 7 and HRD, diploid 3, 7) were defined by the absence of chr7 and chr3 trisomies. Finally, the complex HRD, +1q, diploid 11, −13 subtype lacked chr11 trisomy but harbored gain of chr1q and loss of chr13. Of the NHRD subtypes, the diploid subtype was mostly devoid of copy number events and highly associated with translocations targeting a D-type cyclin (71.3%). The remaining two NHRD groups were strongly associated with canonical immunoglobulin translocations (71.1%) and were defined by chr13 loss. The −13 subtype contained a subpopulation of patients with chr14 loss, while the +1q, −13 subtype had gains of 1q.

a, Consensus clustering of WGS copy number data identified eight unique copy number subtypes, comprising five HRD and three NHRD clusters that were annotated based on common copy number features. b, Pointwise OS estimates are shown by the respective lines for each copy number subtype. Median OS was met for the HRD, +1q, diploid 11, −13 (55.7 months, 95% CI = 31.7–NA (not available)), +1q, −13 (69.3 months, 95% CI = 53.0–97.4), HRD, diploid 3, 7 (95.3 months, 95% CI = 43.6–NA) and −13 (103.9 months, 95% CI = 79.2–NA) subtypes. Pairwise outcome comparisons identified eight significantly different subtypes by the log-rank test after multiple testing corrections using the Benjamini–Hochberg method. The significant differences were between the HRD, +1q, diploid 11, −13 subtype and HRD, ++15 (P = 0.0015), HRD, classic (P = 0.0373), diploid (P = 0.0373) or −13 (P = 0.0373); between the +1q, −13 subtype and HRD, ++15 (P = 0.0015), HRD, classic (P = 0.0373) or diploid (P = 0.0448); and between HRD, ++15 and HRD, diploid 3, 7 (P = 0.03733). c, Pointwise OS estimates are shown by the respective lines, and pairwise outcomes were compared by the log-rank test, which showed a significant difference (P = 9.5 × 10−6) between patients in the +1q, −13 and HRD, +1q, diploid 11, −13 groups (median = 67.2 months, 95% CI = 53.0–83.2) versus patients in other copy number subtypes (median = 103.9, 95% CI = 103.9–NA).
There was no difference in outcomes between HRD and NHRD patients (Supplementary Fig. 6). However, the HRD and NHRD subtypes with both 1q gain and chr13 loss had inferior OS outcomes when compared to patients in other copy number subtypes (Fig. 2b), suggesting that HRD patients should not be universally considered as a group with good outcomes. Combining these two subtypes identified a group with inferior outcomes as compared to patients with other genetic backgrounds (Fig. 2c; hazard ratio (HR) = 1.732, 95% confidence interval (CI) = 1.354–2.215, P < 0.001). The 35-month difference in OS outcomes between NHRD patients in the +1q, −13 subtype and the −13 subtype suggested 1q gain, rather than 13q loss, is the predictor of poor outcome; however, a Cox proportional hazard model examining the contribution of 13q14 and 1q21 copy numbers on OS outcomes did not identify gain(1q21) as an independent feature (Extended Data Fig. 3).
RNA subtypes of multiple myeloma
Consensus clustering was performed on RNA-seq results from 714 baseline samples to identify subtypes of myeloma defined by gene expression (Extended Data Fig. 4). Three independent replicates identified 12 clusters as the optimum solution; two were identical, and a third had 20 (2.8%) patients assigned to different classes (Supplementary Figs. 7 and 8). Many of the observed subtypes were associated with known immunoglobulin translocations and copy number states (Fig. 3a and Extended Data Fig. 5), and there were clear relationships with subtypes identified in previous studies (Supplementary Figs. 9 and 10)17,18. Four subtypes were identified across all studies, including MMSET expressing (MS) (characterized by t(4;14) patients), MAF family transcription factor expressing (MAF) (primarily characterized by t(14;16) patients), cyclin D expressing group 1 (CD1) (characterized by t(11;14) patients) and proliferation (PR) (characterized by patients with a high proliferation index). To maintain consistency across studies, we used subtype names from previous studies when appropriate but otherwise assigned names based on common molecular features.

a, Consensus clustering of RNA-seq data revealed 12 RNA subtypes of multiple myeloma. The MYC STR flag indicates the detection of a MYC translocation (Ig or non-Ig) or intrachromosomal deletion centromeric of MYC. b, Pointwise OS estimates are shown by the respective lines for each RNA subtype. Median OS was reached for the PR (21.3 months, 95% CI = 15.0–55.3); HRD, low TP53 (55.4 months, 95% CI = 36.7–NA); MS (79.2 months, 95% CI = 57.3–97.4) and MAF (103.9 months, 95% CI = 34.4–NA) subtypes. Pairwise outcome comparisons identified ten significantly different subtypes by the log-rank test after multiple testing corrections using the Benjamini–Hochberg method. All significantly different pairs were compared against the PR subtype, with P values ranging from 0.03305 compared to MAF and 1.3 × 10−6 compared to HRD, ++15. The only PR pairwise comparison that was not significant was against HRD, low TP53, the subtype with the second lowest median OS. c, Pointwise OS estimates of patients in the PR (21.3 months, 95% CI = 15.0–55.3) versus non-PR (median = 103.9 months, 95% CI = 97.4–NA) subtype at diagnosis (P = 1.1 × 10−10), HR = 3.16 (95% CI = 2.19–4.57). d,e, Expressed (median transcripts per million (TPM) > 1 in at least one group) checkpoint inhibitor (d) and immunotherapy (e) targets in independent non-PR (n = 663) versus PR (n = 51) patients. Significant differences in median expression between the two groups were determined using a two-sided unpaired Wilcoxon rank sum test and are indicated when significant (*P < 0.05, **P < 0.01 and ***P < 0.001).
The MS subtype comprised 10.6% of patients for whom a t(4;14)-WHSC1 was detected in 62 of 67 (92.5%) samples by WGS. One patient had a t(2;4) bringing the κ enhancer close to WHSC1, while two others had fusion transcripts between WHSC1 and the highly expressed genes FUT8 or CXCR4. The MAF subtype included 6.4% of patients, of whom 38 of 41 (92.7%) patients had translocations (27 t(14;16)-MAF, 4 t(8;14)-MAFA, 6 t(14;20)-MAFB and 1 t(20;22)-MAFB). All three patients with undetectable immunoglobulin translocations had high expression of an MAF family gene. One had a t(1;16) juxtaposing the FAM46C super-enhancer with MAF19,20. Another had an atypical insertion of a class-switch circle telomeric of MAF. One patient had both a t(14;16)-MAF and a t(4;14)-WHSC1 yet strongly associated with the MAF subtype, suggesting the MAF expression signature overpowers the MS signature. Immunoglobulin translocations targeting MAF family transcription factors are associated with higher mutation load21, and in this cohort, 8 of 10 patients with high tumor mutation burden (>10 mutations per Mb) were in the MAF subtype and could qualify to receive a checkpoint inhibitor.
Three subtypes were highly associated with overexpression of a D-type cyclin caused by t(11;14)-CCND1, t(12;14)-CCND2, or t(6;14)-CCND3 (Supplementary Fig. 11a–c). The CD1 subtype included 4.3% of patients, of whom 24 of 25 (96%) patients had a D-type cyclin targeting translocation. One patient had a t(9;14), resulting in the overexpression of the B-cell master regulator PAX5 (Supplementary Fig. 11d). Unlike previous studies that identified a single cyclin D expressing group 2 (CD2) subtype, we identified two related subtypes designated as CD2a and CD2b. The CD2a subtype comprised 7.8% of patients, of whom 40 of 47 (85.1%) patients had a detected D-type cyclin IgH translocation. The CD2b subtype included 8.0% of patients, of whom 51 of 56 (91.1%) patients had a detected D-type cyclin targeting translocation. Both the CD2a and CD2b subtypes were associated with cell surface expression of CD20, which is largely absent in other RNA subtypes, including CD1. Compared to CD2a, patients in the CD2b subtype had higher proliferative index scores (P < 0.005), but there is not a significant difference in OS or time to second-line therapy between these groups, and unexpectedly, CD2a patients start second-line therapy 8.4 months earlier.
The PR subtype contained 7.1% of patients with an admixture of classic genetic subtypes and very poor clinical outcomes, with a median OS of 21 months (Fig. 3b,c). High proliferation index scores were also concentrated in this subtype (Extended Data Fig. 6). Clearly, current treatment regimens are ineffective for these patients. We compared the expression of current checkpoint and immunotherapy targets in non-PR versus PR patients and observed that all five checkpoint targets (CD200, CD86, CD274/PDL1, TNFRSF14/HVEM and LGALS9/GAL9) were decreased in the PR group, whereas TNFRSF17/BCMA was increased, and the other four immunotherapy targets (CD38, SLAMF7/CS1, GPRC5D and FCRL5/FCRH5) showed no difference in expression in PR patients (Fig. 3d,e).
A subtype representing 11.1% of patients most closely resembled the previously defined low bone subtype17 (Supplementary Fig. 9); however, there was no decrease in bone lesions (Supplementary Fig. 12). This subtype comprised an admixture of 59.2% HRD and 40.8% NHRD patients, but 74.0% had a gain of chr1q with 26.0% having ≥4 copies and was thus termed the 1q gain subtype.
Four of the RNA subtypes were associated with HRD (Extended Data Fig. 5) and either did not uniquely associate with a subtype from a previous study, or the original name could not be justified. Two HRD subtypes associated closely with the previously identified HY subtype, but differed due to an enrichment of tetrasomy 15, observed in 58.7% and 60.8% of patients. Because structural events involving MYC are associated with HRD karyotypes22, we investigated the association between these two groups and MYC rearrangements. We identified 37 of 49 (75.5%) versus 23 of 76 (30.3%) patients who had MYC rearrangements, and thus these subtypes were named HRD, ++15, MYC and HRD, ++15, respectively. A third HRD subtype comprising 8.3% of patients most closely associated with the PRL3 subtype18; however, the signature was elevated in four subtypes (Supplementary Fig. 10). A MYC structural event was identified in 35 of 49 (71.4%) of these patients, and this group was also distinguished from all others except PR in having a low nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) index (Extended Data Fig. 7) and was thus named HRD MYC, low NF-kB. The smallest HRD group contained 4.6% of patients and was associated with the previously defined NF-kB subtype18; however, no clear association existed with the NF-kB index used to define the subtype (Supplementary Fig. 10 and Extended Data Fig. 7). One of the predictors of this RNA subtype was overexpression of NINJ1 (Supplementary Table 7 and Supplementary Fig. 13a), which inhibits translation of TP53 (ref. 23). TP53 was also found to be underexpressed, exhibiting the lowest median expression in this subtype as compared to all other RNA subtypes (Supplementary Fig. 13b). Taken together, this subtype was termed HRD, low TP53. The final subtype, termed low purity, with 12.2% of patients correlated with the previously defined myeloid group but was dominated by lower purity samples (Supplementary Fig. 10 and Extended Data Fig. 8)18.
Clinical and molecular associations with RNA subtypes
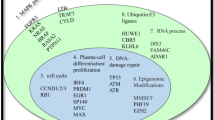
To identify additional defining features of each RNA subtype, we tested for significant associations between clinical and molecular features, including complete LOF and GOF events. Overall, 21 genes with complete LOF or GOF were identified to have a significant association with one or more RNA subtypes (Fig. 4). As expected, GOF was detected in the translocation target genes associated with the MAF, MS, and CD subtypes. Although the loss of one WWOX allele is expected in t(14;16), we frequently detected complete LOF of WWOX (P < 0.001), supporting a possible role of WWOX in myeloma. Both the MS and 1q gain subtypes were diminished for NRAS GOF, and the latter was enriched for TRAF3 LOF. The CD2a subtype was enriched for GOF events in NRAS (P < 0.005) and IRF4 (P < 0.005), while the CD2b subtype was enriched for GOF events in IRF4 (P < 0.005) and EFTUD2 (P < 0.01), representing potential subtype-specific therapeutic targets. In general, the HRD RNA subtypes were not enriched for any GOF or LOF events aside from the HRD, ++15, MYC subtype, which was enriched for LOF events in FAM46C (P < 0.001).

The location and proximity of individual genes are shown next to each gene with the alternating gray and black bars illustrating when the chromosomal location changes, while black bars directly to the right denote contiguous genes. LOF and GOF features that are significantly enriched or depleted within a specific RNA subtype are highlighted. Enrichment analysis was conducted using a two-tailed Fishers test, and the Benjamini–Hochberg test correction was used for multiple testing to compute a positive false discovery rate (pFDR) value. Genes are only included when pFDR ≤ 0.1, P < 0.05.
The PR subtype was enriched for LOF of RB1 (P < 0.001) and MAX (P < 0.01), gain(1q21) (P < 0.001), del(13q14) (P < 0.001) and ISSIII patients (P < 0.001). Interestingly, 50% of PR patients were ISSIII, while 22% and 28% of PR patients were ISSI and ISSII, respectively, highlighting that ISS underestimates disease severity in half of these high-risk patients. Complete loss of RB1 typically involves a one-copy deletion of 13q coupled with a second molecular event (Supplementary Fig. 14). Identifying LOF of RB1 and MAX represents defining genetic features of the high-risk PR phenotype.
Association of G1/S checkpoint with transition to PR subtype
We developed a predictive model to assign the serial samples to the subtypes with the highest class probability (Supplementary Table 7). Overall, 71 patients were assigned a subtype at two or more time points, with 55 patients assigned a subtype other than low purity for at least two time points. At diagnosis, five serial patients were classified as low purity; however, at progression, they all had a subtype other than low purity (one each CD1; 1q gain; PR; HRD, ++15 and HRD, ++15, MYC), further supporting that this phenotype is driven by relative sample purity rather than distinct disease biology (Extended Data Fig. 9). Although most patients remained in the same subtype throughout their disease course, 13 of 49 (26.5%) patients who were not classified as low purity or PR at baseline, transitioned into the PR subtype at progression (Fig. 5a). Patients who transitioned to the PR subtype rapidly succumbed to their disease (Fig. 5b), with a median OS after the detected progression of 88 days (Supplementary Fig. 15), and had inferior outcomes compared to other patients who also progressed (Fig. 5c).

a, Node size reflects the relative number of patients in each RNA subtype at each time point, while edge width reflects the relative number of patients remaining in, or transitioning to, a particular RNA subtype, with the thinnest line and thickest line representing one and seven patients, respectively. b, Swimmers plot of patients in the PR subtype at either baseline or progression. Vertical breaks indicate visits with available RNA-seq data for RNA subtype prediction. Fill color indicates RNA subtype between visits. Asterisks denote OS events. c, Pointwise OS outcomes for serial patients who transition to the PR subtype at progression (median = 27.9 months, 95% CI = 18.9–68.1) versus those that do not (median = 81.5 months, 95% CI = 67.3–NA) are shown. The pairwise outcomes were significantly different (P = 0.0081) by the log-rank test.
To identify molecular events potentially driving the transition of patients to the PR subtype, gene functional status was compared at the PR and prior non-PR time points. Molecular data were available for comparison at both time points for 9 of 13 patients who transitioned to PR. Despite the prevalence at baseline, none of the patients transitioning to PR acquired complete LOF of RB1. However, three patients (33%) had complete LOF of a cyclin-dependent kinase inhibitor at progression. Two patients had complete LOF of CDKN2C at progression due to homozygous deletions (Extended Data Fig. 10). One patient acquired two independent deletions at progression, while the other had one clonal deletion at diagnosis, and the second increased from 26% to 100% at progression, suggesting this aggressive clone existed before treatment. One patient acquired complete loss of CDKN1B at progression from a pre-existing deletion, and a clonal frameshift mutation was detected only at progression (Supplementary Fig. 16). Similar to the baseline observations, there are multiple genetic defects in G1/S checkpoint genes that can result in the PR phenotype.
Discussion
The MMRF CoMMpass study represents—to the best of our knowledge—the largest single sequencing study of patients with multiple myeloma undertaken to date based on the number of enrolled patients and the total number of sequencing assays performed. The cohort has facilitated the identification of distinct copy number and expression subtypes of myeloma, as well as both recurrent and rare molecular events that occur at frequencies that would not be detected in smaller patient cohorts.
A diverse array of genetic events can contribute to the development or progression of cancer, with individual genes often being affected by multiple types of alterations; however, these diverse processes are generally summarized in isolation, thus underrepresenting a gene’s contribution. To accurately summarize the frequency of these changes, we integrated seven different data formats extracted from WES, WGS and RNA-seq data and identified 70 LOF and 27 GOF genes occurring in five or more patients. Differentiating between partial and complete LOF is pertinent for the accurate identification of high-risk patient populations. For TP53, solitary deletions or mutations have been associated with poor prognosis; however, only patients with complete LOF of TP53 have poor outcomes, suggesting that only one-third of patients with del(17p13) identified by clinical cytogenetic assays are true high-risk patients12,24,25,26. Finally, there is a long-standing interest in determining the gene associated with monosomy 13 in myeloma, detected in 44.9% of patients. Our analysis not only identified recurrent complete loss events in RB1 and DIS3 but also identified independent complete loss events in PSPC1, TGDS, LATS2, BRCA2, PARP4, MYCBP2, TPP2, CDK8, TSC22D1 and ARHGEF7, suggesting that multiple genes on chr13 can independently contribute to myelomagenesis. Oddly, DIS3 LOF events almost always maintain a full-length polypeptide, suggesting the dual function of this gene in PCs. Notably, no complete loss event was identified in nearly half of the patients (48.1%) with monosomy 13, thus haploinsufficiency, as observed in Mir15a/Mir16-1, may also be a contributing factor27.
We identified eight distinct copy number subtypes, including five HRD and three NHRD subtypes. Although previous studies have shown that HRD patients have favorable outcomes compared to NHRD patients, in CoMMpass there was no difference in outcomes. This large cohort analysis revealed a number of seemingly interrelated events, such as 1q gains and monosomy 13, HRD patients lacking both trisomy 3 and trisomy 7, and groups with a classic HRD phenotype defined by trisomy or tetrasomy 15. Interestingly, the HRD subtype with 1q gain and chr13 loss lacks the classic trisomy 11, suggesting that the combination of these events can phenocopy the benefits of trisomy 11. Although patients with 1q gain and 13 loss represent poor outcome copy number subtypes, the median OS of these patients was between 56 and 69 months, comparable to the 54 months observed for ISSIII, but not the 44-month OS associated with R-ISSIII, which includes patients with high-risk clinical and cytogenetic features15. Taken together, this highlights that copy number features alone are insufficient to distinguish the subset of ultra-high-risk patients.
Previous studies clustering microarray gene expression data identified eight to ten unique subtypes, many of which were consistent among studies including the MS, MAF, CD1, CD2 and PR subtypes17,18. Our consensus clustering of RNA-seq data identified 12 unique RNA subtypes. Some previously identified subtypes were further subdivided, while others were renamed to better reflect the underlying biology, aided by incorporating the diverse data types in this study. For instance, several studies have sought to identify treatment strategies for CD20-positive patients, but based on the distribution within CD2a and CD2b, it may be pertinent to consider that these patients originate from two unique populations. In the context of precision medicine, the strong link between t(11;14) and the CD subtypes leads to the question of whether one of the CD subtypes better predicts response to venetoclax than t(11;14)28.
The PR RNA subtype defined a group of patients with extremely poor OS, high proliferative index scores and nearly ubiquitous 1q gains. This group has remained controversial because of the competing supervised patient segregation TC classification models29,30 that argue to group patients by defined genetic features, while this subtype has a mixture of those genetic events. Through our integrated analysis, we identified LOF of RB1 or MAX as common genetic events in baseline PR patients, providing a genomic link to this gene expression phenotype. Loss of MAX was recently associated with transformation and increased proliferation in small cell lung cancer31, and thus, LOF of RB1 or MAX likely contributes to the highly proliferative phenotype observed in PR patients. PR patients exhibited lower median expression of checkpoint targets but equivalent or higher expression of most immunotherapy targets when compared to non-PR patients, suggesting that immunotherapies may represent a viable therapeutic option in these high-risk patients and highlighting the importance of identifying these patients in future clinical studies.
There was also a strong tendency for patients to transition to the PR subtype at progression, with 26.5% of serial patients in a non-PR subtype at diagnosis transitioning to PR, similar to recent results32. Patients who transitioned to the PR subtype had extremely poor outcomes after the transition, with a median survival of less than 3 months after their progression visit. An acquired complete loss of a cyclin-dependent kinase inhibitor, such as CDKN2C or CDKN1C, was observed in 33% of patients transitioning to the PR subtype at progression, suggesting that transition to the PR phenotype at progression is associated with genetic events disrupting cell cycle control.
These findings demonstrate that advanced molecular diagnostics such as WGS and RNA-seq are better predictors of disease behavior than current staging systems based on clinical laboratory, conventional cytogenetic and fluorescence in situ hybridization (FISH) data. These assays also identify therapeutic targets, including RAS, BRAFV600E and FGFR3 mutations, that are actionable with agents already approved for other cancer indications. In fact, early CoMMpass findings drove the development of a number of clinical trials, notably MMRC-085 Myeloma-Developing Regimens Using Genomics (NCT03732703), an umbrella trial using targeted exome and transcriptome sequencing to stratify subjects into subprotocols with approved targeted agents. These findings favor the adoption of advanced molecular diagnostics into the routine care of myeloma, especially as the breadth of clinical impact broadens and costs decrease.
Comprehensive molecular analyses of the baseline CoMMpass cohort have permitted a more thorough understanding of the genetic diversity and subtypes of the disease. This approach clearly defined the primary molecular features driving different subtypes of multiple myeloma and identified high-risk patients at both diagnosis and progression. Innovative clinical trials targeting this high-risk population are needed given the current poor outcomes with therapies that are otherwise highly effective in other subtypes. Given that patients frequently transition to the high-risk PR subtype at progression, it will be important to know the percentage of PR patients in clinical trial populations, particularly in the relapse/refractory setting, to understand if the arms are balanced and if there is a difference in response between these groups. The identification of unique subtypes and the frequency of target gene dysregulation, via our integrated analysis, provide a solid foundation to prioritize targets for precision medicine approaches in multiple myeloma.
Methods
Sample collection and biobanking
Patients were enrolled in the CoMMpass study (NCT01454297), sponsored by the MMRF in accordance with the Declaration of Helsinki. The study was approved by panel 2 of the Western Institutional Review Board and the Internal Review Board at each participating site, and the patients were not compensated for participation and were only enrolled after providing written informed consent. All samples analyzed originate from interim analysis 14, and clinical data analyzed in this study were collected as of interim analysis 22. The MMRF CoMMpass study accrued patients from clinical sites in Canada, Italy, Spain and the United States. All patient samples were shipped to one of the following three biobanking operations: Van Andel Research Institute (VARI) in Grand Rapids, Michigan, for all samples collected in Canada or the United States; University Hospital of Salamanca for samples collected in Spain; or University of Torino for samples collected in Italy. In North America and Spain, potassium ethylenediaminetetraacetic acid (K2-EDTA) tubes were used for the collection of peripheral blood (PB), and sodium heparin tubes were used for the collection of bone marrow (BM) aspirates. These samples were shipped to their respective biobanks using CoMMpass study kits that maintained samples at 7–12 °C. In Italy, clinical sites participating in the FORTE clinical trial (NCT02203643) collected BM and PB sample aspirates in sodium citrate vacutainers. Samples collected at sites in Italy were shipped at ambient temperature to the biorepository site.
At VARI, the received BM and PB specimens were first quality controlled by flow cytometry to determine the percentage of PCs in the PB and BM specimens. Patients were only included in the study when the submitted BM contained at least 1% PCs. If the PB showed less than 1% circulating PCs, white blood cells were used as the constitutional DNA source; however, when >1% circulating PCs were observed, enriched CD3-positive T cells were used. Whole BM PC enrichment, or PB PC enrichment when >5% circulating PCs were detected, was performed using the Miltenyi autoMACS Pro Separator using anti-CD138 microbeads. The purity of the enriched samples was assessed using a three-color slide-based immunofluorescence assay that identified cells with DAPI and the presence or absence of κ or λ light chains. Clinically eligible baseline patients with tumor samples with greater than 250,000 cells recovered after CD138 enrichment and monoclonal purity greater than or equal to 80% moved forward for nucleic acid extraction. For progression samples, the cell recovery requirement was 200,000 cells. To minimize nucleic acid isolation failures, the first 750,000 cells were used exclusively for DNA isolation. When more than 750,000 cells were recovered, the sample was split 50/50 for DNA and RNA isolations. When more than 4 million cells were recovered, multiple aliquots were stored for future use. Cells destined for DNA isolation were stored as snap-frozen pellets, while samples for RNA extraction were lysed in QIAzol before long-term storage at −80 °C.
Samples from clinical sites in Spain were collected and shipped using the provided CoMMpass collection kits. Red blood cells were removed from the PB and BM specimens using a red cell lysis buffer, and, following a PBS wash, the remaining white blood cells were counted. After red cell lysis, the isolated cells were quality controlled using flow cytometry to determine the percentage of PCs in the PB and BM specimens. If the PB showed less than 1% circulating PCs, white blood cells were used as the constitutional DNA source; however, when >1% circulating PCs were observed, enriched CD3-positive T cells were used. The isolated PB cells (1–5 million cells) were snap-frozen as dry pellets for constitutional DNA isolation. The isolated BM cells were stained with anti-CD138 microbeads, and PCs were enriched using a Miltenyi autoMACS Pro Separator. The enriched CD138+ cells were stored as snap-frozen dry pellets, and, when possible, a separate aliquot was lysed with QIAzol and stored at −80 °C until shipped on dry ice to VARI for isolation. The purity of the CD138-enriched PC fractions was determined using flow cytometry with antibodies against CD38, CD138 and CD45.
Samples collected in Italy were treated with red cell lysis buffer. After washing the remaining WBC with PBS, the cells were counted. The isolated PB cells (1–5 million cells) were snap-frozen as dry pellets for constitutional DNA isolation. The BM WBC were stained with anti-CD38 magnetic beads, and PCs were enriched using a Miltenyi autoMACS Pro Separator. After sorting, the purity of the enriched specimens was assessed by flow cytometry using a fluorescent anti-CD38 antibody. Cells destined for DNA isolation were stored as snap-frozen pellets, while samples for RNA extraction were lysed in QIAzol before long-term storage at −80 °C.
Flow cytometry phenotyping and quality control process
All samples received at VARI were tested by flow cytometry to phenotype and quality control the received specimens. The antigens and corresponding commercial antibodies used in the flow cytometry assays are as follows: CD38 (BD Biosciences, 340677), CD45/PTPRC (BD Biosciences, 340665), CD138/SDC1 (BD Biosciences, 347205), CD319/SLAMF7 (Invitrogen/eBioscience, 12-2229-42), CD13/ANPEP (BD Biosciences, 340686), CD19 (BD Biosciences, 340720), CD20/MS4A1 (BD Biosciences, 346581), CD27 (BD Biosciences, 654665), CD28 (BD Biosciences, 348047), CD33 (BD Biosciences, 340679), CD52 (Life Technologies, MHCD5204), CD56/NCAM1 (BD Biosciences, 340724), CD117/KIT (BD Biosciences, 340867), FGFR3/CD333 (R&D Systems, FAB766P), κ (BD Biosciences, 643774) and λ (Life Technologies, MH10614). Flow panels performed included CD38 × CD45 × CD138 × CD56 (initial BM and PB screening panel 1), CD38 × CD45 × CD138 × CD319 (updated screening panel 1 after the introduction of daratumumab), CD38 × CD45 × cytoplasmic κ × cytoplasmic λ (BM and PB screening panel 2), CD38 × CD45 × CD138 × either CD13, CD19, CD20, CD27, CD28, CD33, CD52, CD117 or FGFR3, and propidium iodide-stained nuclei to determine the DNA content of each tumor.
Nucleic acid isolation
All nucleic acid isolations were performed at VARI. DNA was extracted from the dry cell pellets with the Qiagen Gentra Puregene Tissue Kit (Qiagen, 158667) with isolated DNA suspended in Qiagen buffer ATE and stored at −20 °C. DNA was extracted from PB samples using the Qiagen QIAsymphony, which uses magnetic beads for automated sample processing. Blood tubes were either processed immediately upon receipt or frozen at −20 °C and processed in batches. QIAsymphony extractions were performed using the DSP DNA Midi Kit (Qiagen, 937255). DNA was eluted in Qiagen buffer ATE and stored at −20 °C. DNA was quantified by NanoDrop spectrophotometric analysis as well as by fluorescence using Qubit 2.0 to determine dsDNA content. Sample quality was determined by agarose gel or Agilent TapeStation Genomic ScreenTape. Samples with at least 250 ng of dsDNA were submitted to TGen for analysis.
Tumor cells designated for RNA extraction were dissolved in QIAzol Lysis Reagent (Qiagen, 79306), stored at −80 °C and extracted with the Qiagen RNeasy Plus Universal Mini Kit (Qiagen, 73404). RNA was eluted in nuclease-free water and stored at −80 °C. RNA was quantified by NanoDrop spectrophotometric analysis, and RNA quality was evaluated using the Agilent Bioanalyzer 2100. Samples with a RNA integrity number (RIN) ≥ 6.0 and at least 200 ng of RNA were submitted to TGen for analysis.
Sequencing library construction
Details on the specific procedures used for genome, exome and RNA-seq library construction and Illumina-based sequencing are provided in the accompanying Supplementary Information.
Primary sequencing data analysis
Analysis of all sequencing data was performed at TGen on a high-performance computing system using an internally developed analysis pipeline (Medusa Subversion, https://github.com/tgen/medusaPipe) and the MMRF CoMMpass-specific TGen05 recipe. This recipe is based on the hs37d5 version of the GRCh37 reference genome used by the 1000 Genomes Project, with gene and transcript models from Ensembl v74. Additional automated CoMMpass-specific primary processing was also performed (https://github.com/tgen/Post_Medusa_Processing). Code for the creation of the reference genome and gene models used, as well as secondary and tertiary analysis methods, are available on GitHub (https://github.com/tgen/MMRF_CoMMpass).
The paired-end fastq files generated in the long-insert whole genome sequencing (LI-WGS) and WES assays from each sequencing lane were aligned with bwa (v0.7.8-r455). The output SAM file was converted to a BAM file and sorted using SAMtools (v0.1.19-44428cd), after which base recalibration was performed using GATK (3.1-1-g07a4bf8). When multiple lanes existed, they were merged into a single BAM file, duplicate reads were marked using Picard (v1.111(1901)) and joint indel realignment was performed using GATK to produce the final BAM files used for analysis. The quality of each assay was determined using multiple Picard and SAMtools quality control metrics. To be included in the analysis, both the tumor and constitutional sample needed to meet the following criteria for genomes: physical coverage of ≥25×, chimera read rate of <3% and derivative log ratio spread (DLRs) of ≤0.2. To be included in the analysis, both the tumor and constitutional sample needed to meet the following criteria for exomes: >90% target bases at 20× coverage and chimera read rate of <3%. Somatic mutations were identified using Seurat (v2.6, https://github.com/tgen/seurat), Strelka (v1.0.13) and MuTect (v2.2-25-g2a68eab), and their outputs were combined to identify somatic events called by at least two callers. The coding effect of each mutation was determined using snpEFF (v4.2 (build 2015-12-05)), and additional annotations were added using snpSIFT. Somatic structural abnormalities were detected using Delly (v0.7.6), to which additional filtering fields were added to ensure that informative read pairs spanned at least a 100 bp window on both breakends. The MYC STR flag was derived from Delly structural calls and indicates the detection of either (1) a MYC translocation (Ig or non-Ig) or (2) an intrachromosomal deletion with a break end of <3 Mb centromeric of, but not spanning, MYC. Somatic copy number abnormalities were identified with a CoMMpass-specific implementation of tCoNut (https://github.com/tgen/MMRF_CoMMpass).
Paired-end fastq files from the RNA-seq assays were aligned using STAR (v2.3.1z, 24 January 2013), and the output SAM file was converted to a BAM file and sorted using SAMtools followed by duplicate marking with Picard. For RNA-seq to be included in the analyses, we required at least 50 million read pairs (100 million reads) generated from each library, a 5′ bias ratio ≥0.5 and a 5′/3′ bias ratio ≥0.75. Gene expression estimates were determined using multiple tools. Counts were extracted from the unsorted SAM file using HtSeq (v0.6.0). TPMs were estimated with Salmon (0.7.2) using the fastq reads as input for quasi-alignment to a transcriptome defined by the GTF gene model. To correct for the variable level of immunoglobulin transcription between samples that compress the TPM values, we removed plasma-cell specific transcripts, including immunoglobulin, mitochondrial, rRNA and chrY genes, from the final TPM calculation (https://github.com/tgen/Post_Medusa_Processing). Gene fusion events were identified using the TopHat-Fusion workflow in TopHat2 (2.0.8b) followed by independent cross-validation that an associated genomic event existed in the matched LI-WGS assay.
The genotypes of each result file were compared using SNPsniffer (v5; https://github.com/tgen/snpSniffer) to ensure that the constitutional DNA, tumor DNA and tumor RNA were from the same individual and that each patient was uniquely represented. To ensure accurate alignment with clinical data, molecular predicted sex was required to match the clinically recorded sex, and, when available, the clinical immunoglobulin isotype was confirmed to match the isotype defined by flow cytometry and RNA-seq. All constitutional DNA samples were manually reviewed to ensure that they represented a diploid genome. Potential low-level cross-contamination of the tumor DNA specimens was determined by comparing the number of high-confidence mutations detected versus the percentage of those mutations that exist in dbSNP.
Survival analyses
Survival curves were computed using the Kaplan–Meier method as implemented in R by the survfit function from the survival (v3.1-8) package and plotted using the ggsurvplot function from the survminer (v0.4.6) package. Pairwise comparisons of survival curves were calculated using pairwise_survdiff from the survminer package. Time to second-line survival estimates were computed using the TTSLT_Censor_Flag (censt2line_curated) and Time_To_Second_Line_Therapy (ttct2line_curated) fields (Supplementary Table 1). OS estimates were computed using OS_Censor_Flag (censos) and Time_To_Censored_OS (ttcos) fields. Univariate and multivariate Cox proportional hazards models were calculated using the coxph function from the survival package.
Secondary sequencing data analysis
Details on the specific secondary analysis procedures used for the integrated analysis, consensus clustering, along with clinical and molecular associations with subtypes are outlined in detail in the accompanying Supplementary Note.
Statistics and reproducibility
No statistical method was used to predetermine sample sizes, and each experiment used all samples meeting the outline quality control requirements. The investigators were not blinded to allocation during experiments, as subset allocation was based on observed features and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MMRF CoMMpass study was funded in part by a precompetitive consortium of pharmaceutical companies that had early access to each data release. This body of work uses data from Interim Analysis 22, the most recent nonembargoed dataset available at this time, which includes the full 8 years of planned observation for 963 of 1,143 (84.3%) patients. All of the data needed to reproduce the presented observations are fully accessible. These data have been available on two platforms for the majority of this study. The nonidentifiable clinical data and processed datasets are on the MMRF researcher gateway (https://research.themmrf.org/), where data are freely accessible for all academic researchers who register. For-profit entities can contact the MMRF to negotiate access. Identifiable raw DNA and RNA-seq read data are deposited in dbGAP under accession phs000748. All sequencing read data in dbGAP are available under the General Research Use consent group and can be freely accessed through the standard dbGAP data access request process. As part of a data-sharing agreement with the National Institutes of Health—National Cancer Institute, the IA11 dataset was added to the Genomic Data Commons (GDC; https://gdc.cancer.gov/). Nonidentifiable clinical data and processed sequencing data from that release are available from the GDC, where the processed data originates from their harmonized data processing pipeline. Going forward, the clinical and molecular datasets will also be available at https://www.mmrfvirtuallab.org/ and can be accessed through an email to mmrfcommpass@themmrf.org. Supplementary Tables 1–7 referenced in this manuscript are also freely available on Zenodo (https://doi.org/10.5281/zenodo.10608273).
Code availability
The complete analysis pipeline used to process all sequencing result files is publicly available on the TGen GitHub repository (https://github.com/tgen). The pipeline code base, individual repositories and software version are noted in the Methods.
References
Lewis, J. P. & Mackenzie, M. R. Non‐random chromosomal aberrations associated with multiple myeloma. Hematol. Oncol. 2, 307–317 (1984).
Avet-Loiseau, H. et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 27, 4585–4590 (2009).
Carrasco, D. R. et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell 9, 313–325 (2006).
Venti, G., Mecucci, C., Donti, E. & Tabilio, A. Translocation t(11;14) and trisomy 11q13-qter in multiple myeloma. Ann. Genet. 27, 53–55 (1984).
Bergsagel, P. L. et al. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc. Natl Acad. Sci. USA 93, 13931–13936 (1996).
Shaughnessy, J. et al. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood 98, 217–223 (2001).
Avet-Loiseau, H. et al. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood 99, 2185–2191 (2002).
Boersma‐Vreugdenhil, G. R. et al. The recurrent translocation t(14;20)(q32;q12) in multiple myeloma results in aberrant expression of MAFB: a molecular and genetic analysis of the chromosomal breakpoint. Br. J. Haematol. 126, 355–363 (2004).
Fonseca, R. et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood 102, 2562–2567 (2003).
Bolli, N. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 5, 2997 (2014).
Lohr, J. G. et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 25, 91–101 (2014).
Walker, B. A. et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 33, 3911–3920 (2015).
Chapman, M. A. et al. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 (2011).
Greipp, P. R. et al. International staging system for multiple myeloma. J. Clin. Oncol. 23, 3412–3420 (2005).
Palumbo, A. et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J. Clin. Oncol. 33, 2863–2869 (2015).
Keats, J. J. et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 12, 131–144 (2007).
Zhan, F. The molecular classification of multiple myeloma. Blood 108, 2020–2028 (2006).
Broyl, A. et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 116, 2543–2553 (2010).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Davis, C. A. et al. The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res. 46, gkx1081 (2017).
Walker, B. A. et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 6, 6997 (2015).
Misund, K. et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 34, 322–326 (2020).
Cho, S.-J. et al. Ninjurin1, a target of p53, regulates p53 expression and p53-dependent cell survival, senescence, and radiation-induced mortality. Proc. Natl Acad. Sci. USA 110, 9362–9367 (2013).
Chng, W. J. et al. Clinical significance of TP53 mutation in myeloma. Leukemia 21, 582–584 (2007).
Boyd, K. D. et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia 26, 349–355 (2012).
Walker, B. A. et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 33, 159–170 (2019).
Chesi, M. et al. Monosomic loss of MIR15A/MIR16-1 is a driver of multiple myeloma proliferation and disease progression. Blood Cancer Discov. 1, 68–81 (2020).
Kumar, S. et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 130, 2401–2409 (2017).
Bergsagel, P. L. et al. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 106, 296–303 (2005).
Stein, C. K. et al. The varied distribution and impact of RAS codon and other key DNA alterations across the translocation cyclin D subgroups in multiple myeloma. Oncotarget 8, 27854–27867 (2017).
Augert, A. et al. MAX functions as a tumor suppressor and rewires metabolism in small cell lung cancer. Cancer Cell 38, 97–114 (2020).
Boyle, E. M. et al. High‐risk transcriptional profiles in multiple myeloma are an acquired feature that can occur in any subtype and more frequently with each subsequent relapse. Br. J. Haematol. 195, 283–286 (2021).
Annunziata, C. M. et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130 (2007).
Demchenko, Y. N. et al. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 115, 3541–3552 (2010).
Acknowledgements
The CoMMpass study was funded by a grant from the MMRF (to J.J.K., J.C., S.D.J., P.K. and S.L.). The authors would like to thank all of the MMRF CoMMpass study participants and their families for making this research possible. The authors would also like to thank all of the laboratory staff at the Spectrum Health Advanced Technology Laboratory who assisted with sample processing and flow cytometry analysis.
Author information
Authors and Affiliations
Consortia
Contributions
D.A., J.C., S.L., K.C.A. and J.J.K. conceptualized the project. S.S., D.P., A.W.C., S.N., M.B., S.K., K.S., D.C., W.S.L. and J.J.K. developed the methodology. S.S., D.P., A.W.C., S.N., J.L.A., C.L., B.B., C.M., B.T., A.K., M.W., V.Y., M.T., K.M., D.C. and J.J.K. conducted software analysis. S.S., A.W.C., M.T., K.M. and J.K. performed validation. S.S., D.P., A.W.C., S.N., J.L.A., C.L., B.B., C.M., B.T., A.K., M.W., V.Y. and J.J.K. carried out the formal analysis. A.C., S.J., D.S.S., R.V., G.O., A.J., R.N., D.L., J.B., M.L., J.W., S.Z.U., The MMRF CoMMpass Network, J.R.A., L.C., M.B., A.H., S.K., J.M., R.R., K.S., E.T., A.B., B.L., M.K., M.D.A., M.G., K.C., J.S., P.K., A.D., B.Z., M.T., K.M., S.D.J., H.J.C., S.L. and J.J.K. conducted the investigation. A.C., S.J., D.S.S., R.V., G.O., A.J., R.N., D.L., J.B., M.L., J.W., S.Z.U., The MMRF CoMMpass Network, H.J.C. and S.L. arranged the resources. S.S., D.P., A.W.C., S.N., J.L.A., C.L., D.C.R., M.D.A., M.G., A.D., B.Z., M.T., K.M., M.D., J.R., S.D.J., H.J.C., D.A. and J.J.K. curated the data. S.S. and J.J.K. wrote the original draft. S.S., A.C., D.S.S., S.Z.U., B.B., N.C.G., K.C.A., H.J.C., S.L. and J.J.K. did the writing, reviewing and editing of the manuscript. S.S., D.P., A.W.C., S.N., J.L.A. and J.J.K. handled visualization. S.S., K.C., J.S., P.K., J.R., D.C., W.S.L., D.A., S.D.J., J.C., S.L. and J.J.K. provided supervision. D.C.R., K.C., A.D., B.Z., K.M., M.D., J.R., N.C.G., S.D.J., J.C., D.A., S.L. and J.J.K. managed project administration. P.K., S.D.J., J.C., S.L. and J.J.K. secured funding.
Corresponding authors
Ethics declarations
Competing interests
S.S. is currently employed by Janssen. A.C. has received research funding from Amgen, BMS, Janssen, Seattle Genetics and Takeda, and is a speaker/consultant/advisory board member for Abbvie, Amgen, Antengene, BMS, Genentech, GSK, Janssen, Karyopharm, Sanofi, Seattle Genetics, Secura Bio, Shattuck Labs and Takeda. S.J. is a speaker/consultant/advisory board member for BMS, Caribou Biosciences, DMC, Janssen, Regeneron, Sanofi and Takeda. D.S.S. has received research funding from Amgen, BMS, Janssen, Karyopharm, Novartis and Takeda, and is a speaker/consultant/advisory board member for Amgen, BMS, Celularity Scientific, Janssen, Karyopharm, Novartis and Takeda. R.V. has received research funding from BMS, Sanofi and Takeda, and is a speaker/consultant/advisory board member for BMS, Harpoon, Janssen Karyopharm, Legend Biotech, Pfizer, Sanofi and Takeda. A.J. is a speaker/consultant/advisory board member for Abbvie, BMS, Amgen, GSK, Janssen and Sanofi. R.N. has received research funding from BMS, GSK, Janssen, Karyopharm, Pfizer, Regeneron and Takeda, and is a speaker/consultant/advisory board member for BMS, GSK, Janssen, Karyopharm, Pfizer, Regeneron, Sanofi and Takeda. D.L. has received research funding from Annexon Biosciences, Alpine Immune, Abbvie, Astex Pharmaceuticals, Baxalta, BeiGene, Bioverativ, CSL Behring, BMS, Delta-Fly Pharma, Exact Sciences, Forma Therapeutics, Global Blood Therapeutics, Immunovant, Incycte, Janssen, NeoImmuneTech, Novartis, Novo Nordisk, Partner Therapeutics, Pharm-Olam, Principia Biopharma, Salix Pharmaceuticals, Sanofi, Takeda and Vifor Pharma. J.B. has received research funding from AbbVie, Acetylon, Amgen, BMS, C4 Therapeutics, CARSgen, Cartesian, Celularity, CRISPR Therapeutics, EMD Serono, Fate Therapeutics, Genentech, GSK, Ichnos Sciences, Incyte, Janssen, Karyopharm, Lilly, Novartis, Poseida, Sanofi, Takeda, Teva and 2seventy bio, and is a speaker/consultant/advisory board member for BMS, CRISPR Therapeutics, Janssen, Kite Pharma, Legend Biotech, Roche and Takeda. M.L. is a speaker/consultant/advisory board member for Abbvie, Amgen, AstraZeneca, BeiGene, BMS, Gilead, Genmab, Janssen, Jazz, Karyopharm, Morphosys, Novartis, Seagen, Sellas, Sobi, Sanofi and Takeda. S.Z.U. has received research funding from AbbVie, Amgen, Array Biopharma, BMS, EdoPharma, Gilead Sciences, GSK, Janssen, K36 Therapeutics, Merck, Moderna, Pharmacyclics, Sanofi, Seattle Genetics, SkylineDX and Takeda, and is a speaker/consultant/advisory board member for BMS, Genentech, Gilead Sciences, GSK, Janssen, Novartis, Oncopeptides, Sanofi, Seattle Genetics, Secura Bio, SkylineDX, Takeda and TeneoBio. M.D’A. has received research funding from Janssen and is a speaker/consultant/advisory board member for Adaptive Biotechnology, BMS, GSK, Janssen and Sanofi. M.D. is currently employed by the funder (MMRF). J.R. is currently employed by the funder (MMRF). K.C.A. is a speaker/consultant/advisory board member for Amgen, AstraZeneca, Daewoong, Dynamic Cell Therapies, Janssen, Pfizer, Mana Therapeutics, Oncopep, Precision Biosciences, Starton and Window Therapeutics, and is an equity holder in C4 Therapeutics, Dynamic Cell Therapies, NextRNA, Oncopep, Raqia, Starton and Window Therapeutics. H.J.C. is currently employed by the funder (MMRF) and has received research funding from BMS and Takeda. D.A. was previously employed by the funder (MMRF) and is currently employed by AstraZeneca. S.L. has received research funding from BMS, Janssen, Novartis and Takeda, and is a speaker/consultant/advisory board member for AbbVie, Amgen, BMS, Genentech, GSK, Janssen, Novartis, Pfizer, TG Therapeutics and Takeda. J.J.K. has received research funding from Amgen, Genentech and Janssen, and is a speaker/consultant/advisory board member for Janssen. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Stephen Nutt, P. Leif Bergsagel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Survival outcomes of the cohort.
Pointwise survival estimates are shown by the respective dark lines, while the 95% confidence interval is shown by the matching shaded bands. (a) Time to second line therapy (median 38.1 months, 95% CI = 35.2–40.6 months) and (b) overall survival (OS; median 103.6 months, 95% CI = 92.7–not met). The median of the CoMMpass cohort has been met; however, as of the IA22 release, there is insufficient follow-up to accurately report the upper limit of the OS 95% confidence interval. As expected, ISS stage stratified patients into three clinically distinct classes. (c) Time to second line therapy outcomes for patients classified as ISSI (53.7 months, 95% CI = 44.9–63.3), ISSII (35.7 months, 95% CI = 31.5–42.2) and ISSIII (24.4 months, 95% CI = 20.6–28.5) at diagnosis are clearly different. Pairwise outcomes were compared by the log-rank test after multiple testing corrections using the Benjamini–Hochberg method. Significant differences in outcomes were observed ISSI vs ISS2 (P = 7.6e−06), ISS1 vs ISS3 (P < 2e−16) and ISS2 vs ISS3 (P = 2.9e−05). (d) OS outcomes for patients classified as ISSI (103.9 months, 95% CI = NA–NA), ISSII (median not met, 95% CI = 91.3–NA) and ISSIII (53.9 months, 95% CI = 43.3–59.6) at diagnosis are compared with significant differences in outcomes observed for ISSI vs ISS2 (P = 0.00023), ISS1 vs ISS3 (P < 2e−16) and ISS2 vs ISS3 (P = 2.2e−09).
Extended Data Fig. 2 Copy number consensus clustering matrix.
Consensus clustering matrix with an optimal clustering solution of K = 8. The M3C (Monte Carlo reference-based) consensus clustering algorithm was applied to the CN measurements of 26,771 (100 Kb) intervals across the GRCh37 reference genome for 871 WGS BM-derived baseline samples. Five of the eight subtypes include only samples classified as hyperdiploid.
Extended Data Fig. 3 Survival outcomes for patients with gain(1q21) and del(13q14).
(a) Time to second line therapy, and (b) OS outcomes for CoMMpass patients with gain(1q21) and del(13q14) (both), gain(1q21) alone, del(13q14) alone and those with neither event. gain(1q21) was defined as a gain of 1 or more copies of 1q21, whereas del(13q14) was defined as a loss of one copy of 13q14. There is a significant difference in time to second line therapy and OS for all groups compared to the group with neither event (p < 0.05); however, there is no significant difference between the groups with gain(1q21) and/or del(13q14). The median time to second line therapy for gain(1q21), del(13q14) patients was 29.3 months (95% CI = 24.4–33.7), while gain(1q21) was 35.3 months (95% CI = 23.6–49.2), del(13q14) was 35.7 months (95% CI = 31.4–40.6), and for those with neither, it was 51.4 months (95% CI = 42.0–55.9). The median OS for gain(1q21), del(13q14) patients was 69.2 months (95% CI = 55.7–97.4), while gain(1q21) was 83.2 months (95% CI = 56.0–not met), del(13q14) was 92.7 months (95% CI = 72.7–not met), conversely the median has not been reached for those with neither. (c) In a univariate Cox proportional hazards model, both gain(1q21) (P = 8.2e−05) and del(13q14) (P = 0.0022) were found to significantly impact OS outcome using a Wald test. (d) In a multivariate model, both gain(1q21) (n = 307) and del(13q14) (n = 453) were found to have a significant impact on outcome within the full cohort of patients with CN data (n = 871). The box represents the hazard ratio, and the error bars represent the 95% confidence interval.
Extended Data Fig. 4 RNA-seq consensus clustering matrix.
Consensus matrix showing the consistency of class assignment for K = 12 clustering of RNA-seq data derived from 714 BM baseline samples and 4811 feature-selected genes.
Extended Data Fig. 5 RNA subtypes and association with copy number.
Copy number states for patients by RNA subtype are shown. Diploid copy number is represented as 2 (white), copy loss is shaded in blue and copy gain is shaded in red. Rare copy number values exceeding 4 are represented as a copy number value of 4 to maintain uniformity in the heatmap scales for gain and loss.
Extended Data Fig. 6 Relationship between proliferation index and RNA subtypes.
The association with an RNA-seq-defined proliferation index and CoMMpass subtypes is shown (n = 714). The Bergsagel proliferation index27 for each sample was determined by calculating the geometric mean expression of 12 genes (TYMS, TK1, CCNB1, MKI67, KIAA101, KIAA0186, CKS1B, TOP2A, UBE2C, ZWINT, TRIP13 and KIF11). The PR subtype had the highest median proliferation index score. The index range is shown as a boxplot with the upper and lower bounds of the box representing the 25th and 75th percentile, while the center line indicates the median and whiskers represent the highest and lowest value within 1.5 (IQR).
Extended Data Fig. 7 NF-kB index distribution by RNA subtype.
The association with an RNA-seq-defined NF-kB index and the CoMMpass subtypes is shown (n = 714). The NFKB(11) index for each sample was determined by calculating the geometric mean expression of 11 genes (BIRC3, TNFAIP3, NFKB2, IL2RG, NFKB1, RELB, NFKBIA, CD74, PLEK, MALT1 and WNT10A)33,34. The index range is shown as a boxplot with the upper and lower bounds of the box representing the 25th and 75th percentile, while the center line indicates the median and whiskers represent the highest and lowest value within 1.5 (IQR).
Extended Data Fig. 8 Low-purity RNA subtype association with low-purity metrics.
The low-purity RNA subtype was defined based on an association of the samples in this category with multiple independent measures of sample purity. (a) An index associated with genes expressed in non-B-cell tissues was used to identify samples with contamination of non-B lineage cells in the CD138+-enriched cell fractions (n = 714). (b) Tumor purity was estimated from the exome copy number or mutation data based on the absolute allele frequency of constitutional variants in deletion regions or somatic SNV allele frequency in diploid regions of the genome when no usable deletions were detected in the tumor (n = 593). The range of estimated contamination (a) and purity (b) is shown as a boxplot with the upper and lower bounds of the box representing the 25th and 75th percentile, while the center line indicates the median and whiskers represent the highest and lowest value within 1.5 (IQR). (c) The full distribution of observed somatic SNV allele frequencies (n = 593) is shown as a violin plot, where the median is indicated by the horizontal line and the population frequency of the value is indicated by the width of the plot.
Extended Data Fig. 9 Change in RNA subtype probabilities over time.
RNA subtype probabilities for the 71 serial patients with RNA-seq data at two or more time points. All patients classified in the low-purity subtype at baseline have a discernable RNA subtype other than low purity at progression, supporting the observation that this subtype is driven by sample purity. Shifts from a non-PR baseline subtype to a largely PR subtype or partial population of PR cells are evident.
Extended Data Fig. 10 Deletion of CDKN2C in patients who transitioned to PR.
Two patients that transitioned to the PR subtype at progression acquired complete loss of function of CDKN2C due to overlapping deletion. Panels show long-insert WGS reads from tumor samples for patients MMRF_2523 (a) and MMRF_1269 (b) at baseline (non-PR) and progression (PR). (a) At baseline, patient MMRF_2523 was diploid (log2 CN = −0.0747) with no evidence of a deletion spanning CDKN2C; however, at progression, the patient had a 2-copy deletion of CDKN2C (blue bar, log2 CN = −3.3505) due to two unique deletions (red bars) spanning CDKN2C (green box). (b) At baseline, patient MMRF_1269 had a 1 copy loss of CDKN2C (light blue bar, log2 CN = −0.3511) due to a larger deletion on chr1. There is also read evidence supporting a deletion involving CDKN2C/FAF1, suggesting that a subclonal population with complete loss of CDKN2C was present at diagnosis in this patient. At progression, when the patient transitioned to PR, the patient’s tumor had a 2-copy deletion of CDKN2C (dark blue bar, log2 CN = −4.6212). In this patient, the minor clone harboring the CDKN2C deletion at baseline constitutes the bulk of the tumor population at progression.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16 and Supplementary Note.
Supplementary Table 1
Individual patient features.
Supplementary Table 2
Observed somatic SNV and INDEL mutations.
Supplementary Table 3
Observed somatic structural events (deletions, insertions, duplications, inversions and translocations).
Supplementary Table 4
Per specimen gene expression matrix and inframe fusion events.
Supplementary Table 5
Per specimen copy number and alternate allele frequency matrices.
Supplementary Table 6
Per specimen gene level LOF and GOF status.
Supplementary Table 7
Gene expression subtype classifier results and model coefficients.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Skerget, S., Penaherrera, D., Chari, A. et al. Comprehensive molecular profiling of multiple myeloma identifies refined copy number and expression subtypes. Nat Genet 56, 1878–1889 (2024). https://doi.org/10.1038/s41588-024-01853-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-01853-0
- Springer Nature America, Inc.