

Abstract
Organoids generating major cortical cell types in distinct compartments are used to study cortical development, evolution and disorders. However, the lack of morphogen gradients imparting cortical positional information and topography in current systems hinders the investigation of complex phenotypes. Here, we engineer human cortical assembloids by fusing an organizer-like structure expressing fibroblast growth factor 8 (FGF8) with an elongated organoid to enable the controlled modulation of FGF8 signaling along the longitudinal organoid axis. These polarized cortical assembloids mount a position-dependent transcriptional program that in part matches the in vivo rostrocaudal gene expression patterns and that is lost upon mutation in the FGFR3 gene associated with temporal lobe malformations and intellectual disability. By producing spatially oriented cell populations with signatures related to frontal and temporal area identity within individual assembloids, this model recapitulates in part the early transcriptional divergence embedded in the protomap and enables the study of cortical area-relevant alterations underlying human disorders.
Similar content being viewed by others


Main
The cerebral cortex is the main site for neuronal processing in the human brain, with distinct anatomical, morphological and functional areas1,2,3,4. Although regional circuitry is sharpened by neural projections (protocortex model)5, according to the protomap model6 a certain degree of arealization is specified before these connections arise, by restricted expression of fate-determining transcription factors7,8,9. This pattern is dictated by the relative cellular distance from secondary organizers, groups of cells that secrete diffusible factors known as morphogens, thus generating a signaling gradient in the surrounding tissue that ultimately drives cortical parcellation10,11,12. In the human cortex, various signaling organizers and molecules are involved, such as the anterior neural ridge and the commissural plate at the rostral midline that secrete FGF, the roof plate and cortical hem at the dorsocaudal midline that produce wingless-type (WNT) and bone morphogenetic protein (BMP), and the prechordal plate on the ventral side that secretes sonic hedgehog (SHH)11,12. Although corticogenesis follows similar principles across different species13, the selective expansion of the prefrontal lobe of human brains14,15 suggests a human-specific regulation of cortical area specification. Yet, the precise contribution of individual molecules to human cortical area diversity and potential disruptions in brain disorders are not completely understood.
Human pluripotent stem (hPS) cell-derived brain organoids that recapitulate the transcriptional programs and cellular identities of the fetal brain are extensively used to study the molecular mechanisms underlying human cortical development and disease-related alterations16,17. Although several studies described the ability of various factors to modulate cortical area fates in hPS cell-derived cultures18,19,20, current organoid models lack the cortical topography seen in vivo, as they fail to reproducibly generate and spatially orient cell types with frontal and temporal area-related identities in discrete domains of individual organoids21. Recent attempts to introduce some level of topography in brain organoids described a dorsoventral axis through fusions of independently patterned organoids22,23,24, a corticostriatal axis25, a forebrain-to-hindbrain axis through microfluidic devices that modulate WNT signaling26, and a mosaic inducible SHH (iSHH) spheroid–organoid fusion strategy27. Yet, an in vivo-like rostral-to-caudal configuration within individual cortical organoids has not been achieved. Therefore, we set out to introduce in vitro positional specification along the longitudinal axis of individual organoids mimicking the early transcriptional differences relevant for cortical protomap establishment and to characterize its pathological disruption in achondroplasia, a disease displaying a specific cortical lobe phenotype28.
Results
FGF8 effect on cortical specification in round organoids
To dissect the contribution of various signaling molecules to cortical organoid patterning (Fig. 1a), we administered recombinant morphogens or small molecules to organoid cultures derived from two engineered human embryonic stem (hES) cell reporter lines at 3–5 days in vitro (div; Fig. 1b,c). We applied rostralizing treatments to organoids derived from a SP8 > GFP reporter line carrying an AAVS1 locus knocked-in GFP cassette under the control of an enhancer of SP8 (Supplementary Fig. 1a–c), marked by a high-to-low rostral-to-caudal expression in vivo29. We applied caudalizing treatments to organoids from an EMX1-P2A-mNeon reporter line carrying a knocked-in P2A-mNeon cassette into the EMX1 gene, a gene required for cortical area patterning30 and validated for higher caudal expression in the developing human cortex (Supplementary Fig. 1b–e). To minimize confounding effects on early cortical organoid patterning by exogenous signals31,32, we set up organoid cultures in neural induction (NI) medium and verified low expression of morphogens over time (Supplementary Fig. 1f). Morphological inspection and quantification of organoids at 60 div showed that administration of high and low doses of FGF8 induced the expression of SP8 > GFP and EMX1-P2A-mNeon reporters respectively (Fig. 1c and Supplementary Fig. 2), while FGF3 and CER1 had limited effect and other factors either failed to activate the reporters or restricted organoid growth. Quantitative PCR with reverse transcription (RT–qPCR) of FGF8-treated cultures showed increased levels of the rostral markers LMO4 and CPNE8 (ref. 33), and SP8 transcripts (but not of caudal markers NR2F1, FGFR3 and EMX1), in reporter-positive cells analyzed by fluorescence-activated cell sorting (FACS) that were isolated from treated organoids as compared to negative, untreated, sorted cells (Supplementary Fig. 3), confirming induction of rostral identities by high-dose FGF8. Conversely, the caudal markers NR2F1, FGFR3 and EMX1 were differentially expressed in mNeon+ cells sorted from organoids treated with low-dose FGF8 (Supplementary Fig. 3), confirming induction of caudal identities. These results demonstrate a dose-dependent effect of FGF8 on rostral and caudal marker expression in cortical organoids, thereby suggesting that a gradient of FGF8 in individual organoids might be sufficient for cortical axial patterning.

a, Schematic summarizing factors and gene markers involved in human cortical area specification. b, Overview of the protocol used to treat neural organoids with patterning factors from 3 to 5 div. Diff, differentiation medium ± vitamin A. c, Percentage of organoids with detectable SP8 > GFP expression after treatment with rostralizing factors (left) or EMX1>mNeonGreen expression after treatment with caudalizing factors (right). CHRDL1, chordin like 1; FST, follistatin; SB, SB-431542 dual SMAD inhibitor; CER1, cerberus 1; CHIR, GSK-3β inhibitor CHIR99021; WNT1, Wnt family member 1. Data indicate the mean ± s.d. from three lines per treatment (n = 15 organoids per condition and line; except n = 16 for FGF8 high and CHIR low; n = 20, n = 19 and n = 17 for Chir high, and n = 18, n = 19 and n = 17 for Wnt1 treatment for the three lines). P values from one-way analysis of variance (ANOVA; Tukey’s multiple-comparisons test) for comparisons to untreated conditions are provided. d, Experimental procedure for assembloid generation with the SP8 > GFP line and OrEBs. D, day after EB formation; Diff, differentiation medium ± vitamin A; ULA, ultra-low attachment plate. e, RT–qPCR analysis of FGF8 target genes expressed in the SP8 > GFP EBs severed from OrEB after 1 day of co-culture. Data are the log of expression over TBP, shown as the mean ± s.d. (n = 6 EBs for 0%, n = 5 for 1%, n = 5 for 10% grown from three independent clones). P values are the results of one-way ANOVA. f, GFP intensity of the organoids from g. Whiskers are the minima to maxima, boxes represent the 25th to 75th percentiles (Q1 to Q3) and lines indicate the median (n = 7 organoids for 0%, n = 11 for 1%, n = 19 for 10%, grown from three independent clones). P values are the results of one-way ANOVA. g, Images of organoids generated with the SP8 > GFP transgenic line and co-culture with OrEBs at 60 div (scale bars, 500 µm). ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.1. a.u., arbitrary units.
To create a FGF8 gradient in vitro, we leveraged a spheroid–organoid fusion strategy27 and generated mosaic organizer-like embryoid bodies (OrEBs) by mixing non-fluorescent hPS cells engineered to constitutively produce and secrete FGF8 (CAG > FGF8) with hPS cells expressing tdTomato to enable EB visualization by fluorescence (CAG>tdTOMATO; Fig. 1d and Extended Data Fig. 1). Fusion of mosaic OrEBs containing 1% or 10% CAG > FGF8 cells with EBs aggregated from SP8 > GFP reporter cells (round assembloids) induced expression of the early downstream effector ETV1 in both conditions (Extended Data Fig. 2a), validating active FGF signaling. Furthermore, 10% CAG > FGF8 assembloids induced stronger expression of FGF8 signaling downstream effectors (ETV1, ETV4, DUSP6, SPRY2, SEF, SPRY4) 1 day after fusion, indicating modulation of FGF8 signaling by OrEB composition (Fig. 1e). Analysis of SP8 > GFP expression at 60 div reported an increase in GFP intensity in accordance with the percentage of CAG > FGF8 cells (Fig. 1f,g), demonstrating the ability of OrEBs to induce rostral cortical fates in a composition-dependent manner. However, 1% CAG > FGF8 assembloids exhibited the typical ventricular zone-like rosette morphology as marked by nuclei positioning and rosette immunostaining for the rostral marker LMO4 (Extended Data Fig. 2b–e). RT–PCR analysis of 1% FGF8 fusions confirmed reduced gene expression related to other FGF8-influenced developmental processes, including eye and neural crest development (Extended Data Fig. 2f). Together, these data demonstrate that mosaic OrEBs with 1% CAG > FGF8 cells can induce rostral telencephalic fates while maintaining proper organoid tissue morphology.
Symmetry breaking and pattern formation in elongated assembloids
Because we observed a lack of gradient-like expression of the SP8 > GFP reporter in the round assembloid configuration (1%; Extended Data Fig. 2g) and considering the importance of distance in gradient formation in vivo34, we hypothesized that increasing organoid length could enable position-dependent changes in SP8 > GFP expression. The transcriptome of cerebral organoids at 60 div matches the forebrain identity of the human brain at 8–9 weeks after conception (Carnegie stage 23)35, a stage in which the developing human cortex measures approximately 15 mm36. Because round organoids at 60 div typically reach a maximum diameter of 2 mm, we devised the aggregation of hPS cells inside 15-mm-long polydimethylsiloxane (PDMS) molds to form elongated organoids that could be used in the assembloid configuration (Fig. 2a,b). After 7 div, these were extracted from the molds and immediately embedded in individual Matrigel droplets to maintain their shape, as organoids collapsed into spherical structures when embedding was delayed (Extended Data Fig. 3a,b). Notably, organoids grown individually in six-well plates maintained an elongated shape throughout extended culture (Extended Data Fig. 3c). Furthermore, elongated organoids derived from three hPS cell lines (H9, H1 and 178/5) showed comparable expression trends of neural markers (SOX2, SOX1), rostral cortex (SP8, LMO4), ventral forebrain markers (NKX2.1) and mesodermal genes (SNAI1, TBXT) and similar levels of cell death at multiple growth stages to round counterparts, as revealed by RT–PCR and TUNEL assays (Extended Data Fig. 3d–f), validating cortical specification across lines. In the assembly configuration, tdTomato fluorescence and morphological inspection over time confirmed that the OrEBs remained localized at one pole during assembloid growth, serving as topographical reference (Fig. 2c and Extended Data Fig. 3g).

a, Experimental procedure for elongated assembloids using mosaic OrEBs containing CAG>tdTOMATO and non-fluorescent CAG > FGF8-expressing cells. Diff, differentiation medium ± vitamin A. b, Representative images of elongated cortical assembloids at 1 div in the PDMS molds (indicated by white arrows in (i); scale bar, 500 µm), at 7 div after removal from the molds and before Matrigel embedding (ii) or after embedding in large Matrigel droplets ((iii); scale bar, 5 mm), at 120 div in the six-well plate ((iv); scale bar, 5 mm). c, Position of the OrEB on elongated cortical assembloids length (as a percentage) at 15, 60 and 120 div. Values are the mean ± s.d. (n = 9 organoids for 15 div, n = 11 for 60 div and 120 div grown from three independent clones). P values for comparisons among time points (one-way ANOVA Tukey’s multiple-comparisons test) are: 15 div versus 60 div, P = 0.9455; 15 div versus 120 div, P = 0.9781; 60 div versus 120 div, P > 0.9999. d, Images of elongated cortical assembloids generated with the SP8 > GFP transgenic line and mosaic OrEBs at 60 div (scale bars, 500 µm). Right, SP8 > GFP intensity per segment (P, M and D) in individual assembloids. Each segmented line represents an individual elongated cortical assembloid (n > 2 from at least 2 clones). P values are the results of one-way ANOVA among segments per condition (0%, P = 0.9045; 1%, P < 0.0001 and 10%, P = 0.9828). e,f, Images of proximal and distal CPNE8 (e) or NR2F1 (f) stainings (scale bars, 50 µm). Bottom, fraction of CPNE8+ (e) or NR2F1+ cells (f) normalized to total (DAPI+) cells in proximal and distal insets of controls (conCAs) and polCAs at 60 div. Whiskers are min to max, boxes represent the 25th to 75th percentiles (Q1 to Q3) and lines indicate the median; CPNE8: n = 20 insets for P and D from 3 conCAs, n = 42 insets for P and n = 39 for D from 3 polCAs; NR2F1: n = 60 insets for P and D from 6 conCAs, n = 40 insets for P and D from 4 polCAs. P values from one-way ANOVA (Tukey’s multiple-comparisons test) are: P = 0.9988 in CPNE8 proximal conCA versus distal conCAs, P = 0.8509 in NR2F1 proximal conCAs versus distal conCAs, P < 0.0001 for other comparisons; NS, not significant. g,g′, Images of 60 div polCA immunostained with tdTomato in red, DAPI in blue and NR2F1 in white (g) or intensity rainbow (g′). Scale bar, 500 µm.
Then, we assessed the ability of mosaic OrEBs expressing FGF8 (1% or 10%) to induce a decline of SP8 > GFP expression within individual elongated cortical assembloids at increasing distance from the OrEB. Reporter fluorescence microscopy and morphological inspection of the cultures at 60 div revealed homogeneous SP8 > GFP expression along the entire length of 10% CAG > FGF8 assembloids but high SP8 > GFP expression in proximity of the OrEB in 1% CAG > FGF8 assembloids (Fig. 2d), which are hereafter referred to as polarized cortical assembloids (polCAs). This SP8 > GFP expression trend was not seen when 5-mm-long molds were used (Supplementary Fig. 4). Immunofluorescence staining of longitudinal sections showed that polCAs displayed a higher abundance of cells positive for the rostral markers Cpne8 and Lmo4 (ref. 10) in proximal regions, and higher fractions of cells expressing the caudal marker Nr2f1 (ref. 10) and the deep layer marker Ctip2 in the areas distal to the OrEB (Fig. 2e–g, Extended Data Fig. 3h–j′ and Supplementary Fig. 5). These results indicate major differences in rostral and caudal marker expression and fate acquisition at opposite poles of polCAs. Also, analysis of extracellular signals in polCAs (generated from a transgenic line with a light-sensitive opsin Chrimson, ChrimsonR-tdT, under the human synapsin 1 promoter) revealed a similar number of spontaneous and evoked extracellular units across polCAs and comparable to controls in both proximal and distal areas, suggesting consistent functionality (Supplementary Figs. 6–8 and Supplementary Note).
A rostrocaudal-like transcriptional gradient in polCAs
To examine the emergence of a rostrocaudal gene expression gradient along the longitudinal axis of individual polCAs, we performed RNA-sequencing (RNA-seq) analysis on dissected proximal, medial and distal (P, M and D, respectively) segments of six individual assembloids at 60 div and on consecutive pieces (P1, P2 and P3) from four control elongated organoids (Fig. 3a and Extended Data Fig. 4a,b). We verified forebrain specification in all samples by Voxhunt analysis and by specific marker inspection (Extended Data Fig. 4c,d). Principal component analysis (PCA) showed that, in sharp contrast to control segments that were largely overlapping, segments from polCAs clustered separately (Fig. 3b). In particular, P segments transcriptionally diverged more from M and D regions in the PCA, as also confirmed by differential expression analysis (DEA) and cluster analysis (Extended Data Fig. 4e), indicating the highest difference in proximity to the Fgf8 source. Along PC1, polCA segments separated according to a P-to-D axis and distal segments, although closer, were distinct from controls, possibly reflecting exposure to low FGF8 levels (Fig. 3b). Key genes in PC1 and PC2 included those related to FGF8 signaling and other genes linked to cortical regionalization (FGFR3, LMO3) or neuronal differentiation (NEUROD6, NEUROD2, TBR1, NEUROG2; Fig. 3c). Differential gene expression analysis across polCA domains identified two gene clusters with high-to-low proximal-to-distal expression containing genes implicated in regionalization, anteroposterior pattern specification, cell junction and extracellular matrix organization, and one cluster of genes with low-to-high proximal-to-distal expression associated with neuronal membrane potential regulation and synaptic transmission refinement (Fig. 3d and Supplementary Data 1), indicating transcriptional changes in various cortical processes. Of note, these differential expression patterns were absent in control segments (Extended Data Fig. 4f). We also compared the P-to-D differential gene expression in polCA with in vivo data using an integrative list of human rostral and caudal cortical markers obtained from various fetal datasets8,20,33,37,38,39,40 (Supplementary Data 2). Importantly, we identified many rostral markers that showed the expected high-to-low P-to-D expression and caudal markers with the opposite trend (Fig. 3e), including genes associated with retinoic acid (RA) signaling, recently linked to prefrontal patterning39. Moreover, analysis of expression modality in polCAs identified rostral and caudal genes with steep gradients (including CBLN2, MDK, TENM1, NR2F1, FGFR3) and others with shallow gradients (including DUSP4, POU3F3, FOXP1, BCL11A/CTIP2, NPY; Fig. 3f, Supplementary Fig. 9 and Supplementary Data 3). These data demonstrate that the P-to-D gradient of polCAs reproduces in part the in vivo rostrocaudal gene expression pattern and is defined by different gradient modalities.

a, Overview of sample segmentation for RNA-seq. Control and polCAs were manually cut into three equal pieces and according to their relative position to the OrEB when present. C, control. b, PCA analysis using corrected variance-stabilizing transformation (VST) values of the top 500 most variably expressed genes. Gray lines connect segments belonging to the same organoid. c, Top 30 genes loading from the PCA shown in b. d, Heat map showing k-means clustering of corrected and scaled VST expression of DEGs across polCA segments filtered for adjusted P value < 0.1 and log2FoldChange (FC) ≥ 1 (DESeq2 analysis). The dataset is divided into three clusters; on the right, top Gene Ontology terms for DEGs (log2FC ≥ 1) are shown. e, Corrected and row-centered VST gene expression trends of rostrally or caudally enriched genes ordered by complete linkage clustering. f, Gradient modalities of rostral and caudal genes in polCA datasets derived from Mfuzz analysis.
Attenuated positional effects by a pathogenic FGFR3 mutation
Our transcriptional analysis (Fig. 3 and Extended Data Fig. 4) identified FGFR3 as a caudal gene with a steep low-to-high P-to-D expression trend in polCAs. Gain-of-function mutations in FGFR3 associated with increased Fgfr3 activity cause achondroplasia featuring temporal lobe dysgenesis but normal brain size28,41,42. We hypothesized that these mutations might disrupt the FGF8-dependent signaling gradient in polCAs, particularly affecting transcriptional programs and proliferation in distal domains where FGF8 activity is low. We thus introduced the most common mutation in individuals with achondroplasia42, the c.1138 G > A point mutation which results in a p.Gly380Arg substitution in the transmembrane domain of the protein, in one FGFR3 allele (Supplementary Fig. 10b,c and Fig. 4a). PolCA from these mutated lines showed similar lengths to controls, aligning with observations on patients’ normal brain size28,41,42 (Supplementary Fig. 10d,e). However, hematoxylin and eosin staining revealed that, while in wild-type (WT) polCA ventricular zone-like regions were bigger in P regions compared to D domains, they exhibited similar sizes in these locations in mutant polCA (Fig. 4b). In line with this, EdU labeling and phospho histone 3 (PH3) staining at 15 div revealed increased abundance of cells in the S and M phases in P regions compared to D regions of WT polCAs but not of mutant polCAs (Fig. 4b–d), suggesting loss of position-dependent proliferation effects upon FGFR3 mutation. To assess the mutation’s impact on the transcriptional program, we performed RNA-seq on segments from mutant polCAs at 60 div (Pmut, Mmut, Dmut; Fig. 4e). We verified forebrain specification in all samples by Voxhunt analysis and specific marker inspection (Extended Data Fig. 4c,d). PCA analysis showed that, unlike WT polCA segments, mutant segments did not segregate along the PC1, which was still driven by FGF8 signaling-related and neural differentiation genes (Fig. 4e,f), and clustered instead by sample origin (Supplementary Fig. 10f,g). Particularly, Mmut and Dmut segments belonging to the same assembloid did not separate in PCA (Fig. 4e, Supplementary Fig. 10f and Supplementary Data 4) and all mutant pieces clustered with the proximal segments of WT polCA, suggesting all mutant segments developed proximal-like identities. Clusters of differentially expressed genes (DEGs) and expression patterns of rostrocaudal genes identified in WT polCA were no longer segment specific in mutant polCAs (Fig. 4g, Supplementary Fig. 11 and Supplementary Data 5). Analysis of expression modality also confirmed altered gradient trends of both rostral and caudal markers (Supplementary Fig. 12 and Supplementary Data 3). Together, these data demonstrate that the p.Gly380Arg mutation in FGFR3 attenuates positional effects in polCA, hindering the specification of low FGF activity-related signatures in distal domains.

a, Schematic representing the position of glycine-to-arginine substitution (violet) at codon 380 located in the transmembrane domain (TM) of the Fgfr3 protein. S, sulfur bond, TK1/TK2, intracellular tyrosine kinase domains 1/2. b, Representative images of 15 div conCAs, polCAs and polCAsMUT at proximal and distal positions stained with hematoxylin and eosin on top, or DAPI, EdU and PH3 on the bottom. Scale bars, 50 µm. c,d, Fraction of EdU+ cells (c) and PH3+ cells (d) normalized to total (DAPI+) cells in proximal and distal insets of conCAs, polCAs and polCAsMUT at 15 div. In c, n = 33 insets for proximal conCAs, n = 35 insets for distal conCAs, proximal polCAs and distal polCAsMUT, n = 36 insets for distal polCAs and proximal polCAsMUT. In d, n = 12 insets for proximal and distal conCAs, n = 35 insets for proximal polCAs and distal polCAsMUT, n = 36 insets for distal polCAs and proximal polCAsMUT. Data are from 3 organoids per condition, 2 slices per organoid. P values resulting from one-way ANOVA (Tukey’s multiple-comparisons test) are available (**** P < 0.0001). e, PCA plot of control (CTRL), polCAs and mutant polCAs (polCAMUT) samples using VST expression values of the top 500 most variably expressed genes. The two datasets have been merged with RUVseq batch correction. f, Top loading of the PCA shown in e. g, Heat map showing corrected and row-centered gene expression of the same rostrally or caudally enriched genes from Fig. 3e, across the mutant dataset and ordered by complete linkage clustering.
Cell composition and rostrocaudal-like identities in polCA
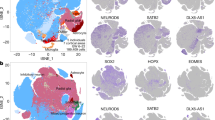
To identify the cell-type composition underlying the rostrocaudal gene expression in polCAs, we profiled dissected polCAs and control segments using single-cell (sc)RNA-seq (10x Genomics Chromium; Fig. 5a). Cell clustering analysis after removing stressed cells revealed a wide diversity of cell types (Fig. 5b–d, Extended Data Fig. 5, Supplementary Data 6 and Supplementary Fig. 13) including cycling radial glial cells (cRGCs; TOP2A, MKI67), two clusters of RGCs (RGC1 and RGC2; SOX2, NES, VIM), two clusters of excitatory neurons (ExN1 and ExN2; DCX, TUBB3), interneuron precursor cells (DLX6-AS1, GAD2), cells expressing stress response markers (stress-related cell; HSPA6, GOLGA4) and retinal progenitor cells (VSX2; RORB). Other populations included two clusters of cilia-bearing cells and BMP-related cells (CB/BRC1 and CB/BRC2; TTR, RSPO2, MSX1, PCP4, NPHP1) and two clusters of endothelial cells (EnC1 and EnC2; COL1A2, DCN, BGN), all of which were previously reported in organoid datasets43. As expected from their relatively young age and neural identity, profiled organoids did not contain clusters of late-born cell types (such as astrocytes, oligodendrocytes) and microglia (Extended Data Fig. 5d,e). Importantly, progenitor clusters (cRGC, RGC1, RGC2) and excitatory neuron clusters (ExN1 and ExN2) correlated with single-cell signatures of counterpart populations from the human developing forebrain44 (Extended Data Fig. 6 and Supplementary Data 7), indicating a forebrain identity. Cell-type composition analysis highlighted differences in segment composition of polCAs but not in control organoids (Fig. 5d). RGC and ExN clusters were more prevalent in P and D segments, while EnC and CB/BRC clusters were notable in M segments. Inspection of cluster composition highlighted differential contribution across polCA segments, with P segments prominently contributing to RGC2 and ExN2 clusters and less so to ExN1 and RGC1 populations compared to D segments and controls (Fig. 5e,f and Extended Data Fig. 7a–c). This distribution was consistent across individual polCA, confirming reproducibility (Extended Data Fig. 7d). Differential abundance analysis between P and D segments formalized the quantitative cell-type differences, particularly in RGC and ExN subtypes (Fig. 5g). To explore identities of differentially represented populations, we performed differential gene expression analysis between the two RGC and ExN clusters (Fig. 5h,i and Extended Data Fig. 7e,f). Top marker genes of RGC2 and ExN2 included the rostral genes TENM1, TENM2 and POUF3F2 and several RA signaling-associated genes preferentially expressed in prefrontal cortex (PFC)45 (GREB1L, MDK, PTX3, GREB1, RBP1), reflecting the emergence of rostral-like signatures in these populations. RGC2 top markers also included rostral genes related to FGF signaling, SPRY1 and FGF13, suggesting a more prominent FGF-dependent signature at the RGC stage. Conversely, RGC1 and ExN1 markers included several caudal genes (EMX2, LHX2, XYLT1, ROBO2, CADM1, EPHA3, BCL11A), pointing toward the emergence of caudal-like signatures in these populations. Of note, some top markers were not previously associated with rostrocaudal cortical patterning (PRTG, TLE4, CTNAA3, PTPRG, ILRAPL1, EBF1, THSD7A, CNRDE, NCKAP5, CALM2, C1ORF61, SAMD3). These results indicate the emergence of area-related transcriptional divergence in RGC and ExN populations. To understand the developmental trajectories and relationships among RGC and ExN subtypes, we analyzed cells along the cRGC-to-ExN trajectory using force-directed graph embeddings for visualization (Fig. 5j,k). We assessed the expression differences between cells from the P-enriched axis (cRGC–RGC2–ExN2; Fig. 5j,k) and others. Pseudotime analysis revealed divergent expression patterns for rostral and caudal genes during differentiation. Known frontal genes confirmed to be upregulated in the P lineage, whereas temporal ones were highly expressed in the non-proximal lineage (Fig. 5l). The maximal difference was observed at the RGC-to-ExN transition (Fig. 5l), emphasizing early transcriptional divergence, in agreement with the in vivo protomap model.

a, Schematic of organoid dissociation for scRNA-seq. b, Uniform manifold approximation and projection (UMAP) embedding for the scRNA-seq dataset containing cells derived from dissections of three segments of control and polarized H9-derived assembloids (8,194 cells from control organoids, 10,628 cells from polarized assembloids). EnCs, endothelial cells; CB/BRCs, cilia-bearing/BMP-related cells; ExN, excitatory neurons; InN, interneurons; RPCs, retinal progenitor cells; SRCs, stress-related cells. c, Correlation map of all clusters. d, Fraction of cells per cluster in individual segments; number of cells is normalized by total cells per segment. e, UMAP embeddings for the scRNA-seq dataset containing cells annotated by organoid type. f, Fraction of cells per cluster; number of cells is normalized by total cells per segment. g, Differential abundance graph where dots represent groups of similar cells; size encodes number of cells in each group; line size shows the number of common cells among groups; color code indicates enrichment for proximal cells versus distal cells (spatial false discovery rate (FDR) < 0.1). The diagram shows differential abundance between proximal and distal segments; red represents enrichment for proximal cells and blue represents enrichment for distal cells. h,i, Clusters are highlighted on the left; on the right, DEA of RGC (h) or ExN (i) clusters is shown. A two-tailed Wilcoxon rank-sum test (Benjamini–Hochberg < 0.05) for RGC1 versus RGC2 or ExN1 versus ExN2 comparisons was used. j, Force-directed graph embeddings of cells along the neurogenic axis, colored by annotated cluster. The red and blue arrows highlight developmental paths undertaken by RGC2 (proximal enriched) and RGC1 (distal enriched), respectively. k, Differential abundance graph along the neurogenic axis (as for g). Color code indicates enrichment for proximal cells versus distal cells (spatial FDR < 0.1). The diagram shows differential abundance between proximal and other segments; red represents enrichment for proximal cells and blue represents depletion for proximal cells. l, Expression of delta along pseudotime between proximal cells from the enriched path and cells from the non-enriched domain (from j and k) for selected significant (Bonferroni < 0.01) rostral markers (solid lines) or temporal markers (dash–dotted lines). Black line at 0 indicates no difference.
Area signatures in spatially segregated cell populations
To evaluate how progenitor and neuronal populations from polCAs recapitulated the transcriptional segregation of in vivo populations from the frontal, medial and caudal areas of the human cortex, we analyzed single-cell transcriptomes of cycling progenitors, non-cycling progenitors and excitatory neurons isolated from the human developing prefrontal cortex (PFC), somatosensory cortex (SS) and temporal cortex (Temp) from the dataset from ref. 37. Different area identities for each main cell type segregated well in PCA (Extended Data Fig. 8a and Supplementary Data 8). We identified key genes showing gradient expression changes across PFC–SS–Temp, ranked by P value and enriched for known markers. This transcriptional analysis allowed us to quantify positional differences among cortical regions along the rostrocaudal axis and assess polCA fidelity in replicating these regions. DEA between segment-derived populations confirmed strong changes between P-derived and D-derived populations and a lower number of DEGs between M and D segments in all three populations analyzed (cycling progenitors, non-cycling progenitors and excitatory neurons; Supplementary Fig. 14 and Data 9), in line with bulk RNA-seq data. We then measured how well polCA segments aligned with fetal cortical areas using the PFC–SS–Temp model. The polCA-derived cell populations from P, M and D segments segregated effectively along the PC1 axis, unlike those from control organoids, demonstrating similar transcriptional identities to in vivo cortical regions (Fig. 6a–b). This pattern was confirmed when both control organoids and polCA were included in the analysis (Extended Data Fig. 8b). A comparison of PC1 gene loadings between polCA and fetal tissues showed alignment at the cell-type level (Fig. 6a,b and Extended Data Fig. 8c). For example, for cycling and progenitor populations, negative loadings defining both proximal and in vivo prefrontal signatures included S100A16, S100A13 and RA-related genes such as RARRES2 and RBP1, while opposite loadings included LHX2, NR2F1, NR2F2 and FGFR3 (Fig. 6a and Extended Data Fig. 8a–c). Proximally driven excitatory neurons shared genes such as LMO3 and FGF12 with PFC-derived neurons, whereas medially/distally driven neurons shared genes like ROBO2, SNTG1, and CADM1 with Temp-derived neurons. Some genes showed the expected gradient in all the populations analyzed (S1100A13, MDK, PRSS23, ROBO2, GABRG1, LHX2 and TENM4), while many showed a cell-type-specific trend (Extended Data Fig. 8d). Notably, the enrichment for prefrontal and temporal identity driver genes (that is, those obtained from the PFC–SS–Temp model that define the rostrocaudal transcriptomics axis, and contribute the most to separate PFC and Temp along the first component of the fetal dataset) is observed in the proximal-to-medial/distal PC1 axis of polCAs for both cycling and non-cycling progenitor populations. In fact, polCAs exhibit a clear enrichment of prefrontal markers among proximal-driving genes, and temporal markers among medial/distal-driving genes, an orientation that is not found in controls (Fig. 6a,b), indicating an arealization gradient in both progenitor populations. In excitatory neurons, while maintaining the separation across segments, the prefrontal driver genes are not fully oriented in the proximal-to-medial/distal PC1 axis (Fig. 6a). On the other hand, the orientation of temporal driver genes is better maintained, albeit to a lesser extent than in the progenitors. These findings indicate that early cell identities in polCAs align closely with PFC-derived and temporally derived signatures.

a,b, Top, PCA embeddings of polCAs (a) or control organoids (b). Each dot represents a metacell using three points to model DEGs in transcriptional space, while the identified distances represent the variability of polCAs in arealization genes defined from the fetal atlas. Top 40 genes (20 up and 20 down) contributing to the relative component variance are shown. Bottom, visualization of gene-set enrichment scores showing the list of prefrontal and temporal genes ranked according to loading values on polCAs PC1 (x axis) and the enrichment score that reflects the degree to which the arealization genes are overrepresented at the extremes (left/negative PC1 or right/positive PC1) of the entire ranked list (y axis).
Discussion
Here, we establish a method to reproduce axial cortical patterning in individual assembloids by fusing a fluorescent organizer-like aggregate expressing FGF8 with an elongated organoid engineered using PDMS molds. This asymmetric source of FGF8 is sufficient to establish in vivo-like rostrocaudal gene expression patterns and spatial cell segregation in individual assembloids. Unlike previous attempts that relied on poorly controlled and low-frequency sources of morphogens resulting in local patterning effects in individual organoids31,45, our approach utilizes a well-controlled source of FGF8 and culture media formulations with minimal exogenous signals to consistently generate cortical polarity along an assembloid’s entire longitudinal axis. Although additional organizers might form, we demonstrate that this polarity depends on finely tuned FGF signaling. In fact, a mutation in FGFR3, linked to temporal lobe malformations in individuals with achondroplasia46, attenuates proliferation and rostrocaudal expression patterns along the proximal-to-distal axis of polCA.
The controlled signaling of the assembloid system enabled us to study the contributions of FGF8 signaling in isolation. While our data align with the role of FGF8 in telencephalic specification in the mouse cortex47, the ability of a single source of morphogen to induce this degree of patterning is remarkable, given the involvement of multiple morphogens and signaling pathways in vivo. We speculate this capability stems from downstream signaling relays and interactions with other pathways, notably Wnt and RA signaling, evidenced by the expression of Wnt receptors FZD4 and FZD9 and RA-related genes RARRES2 and RBP1 in the proximal regions of polCAs. The prominent degree of high-to-low frontal-to-temporal expression patterns in the human cortex48 together with the regulation of the relative size of other patterning centers described in the mouse cortex40 suggest that the anterior neural ridge and its secreted FGF8 may play a primary role in regulating cortical patterning in humans.
Moreover, our cell-type composition and transcriptomic analyses support the acquisition of a position-specific signature along the longitudinal polCA axis, driven by intra-population transcriptomic differences rather than by differences in overall progenitor and neuronal abundance across polCA segments. Our scRNA-seq analysis identified cell populations with signatures related to prefrontal and temporal identities, spatially oriented along the longitudinal axis of individual assembloids, in agreement with the protomap model of cortical progenitor patterning10. The described rostrocaudal gradient in polCAs matches many rostral and caudal gene expression patterns reported in the literature and identifies genes strongly influenced by FGF8 such as CPNE8 and LMO4 among rostral genes and NR2F1, LHX2 and FGFR3 among caudal ones. Our analysis pinpointed RGCs as key drivers of maximal transcriptional divergence and prominent FGF signaling-related signatures, suggesting their crucial role in driving rostrocaudal transcriptional differences in polCAs. Additionally, proximal segments predominantly contributed to rostral-like populations and to a minor extent to caudal-like identities, in contrast to control organoids, which mostly comprised caudal-like populations without showing segment-specific contributions. Together with our observation that rosettes proximal to the OrEBs are more proliferative, these data support a model where FGF8 fosters neural progenitor divisions and patterning49. Conversely, signatures of distal radial glia and distal segments relate to axon guidance and synapse organization and function. We propose that local FGF8 secretion establishes a morphological gradient (proximal–proliferative versus distal–synaptogenic), mirroring the anatomical rostrocaudal characteristics of the developing human brain50. Our characterization of rostrocaudal gene expression modality in polCAs suggests that FGF8 may act through a combination of sharp local responses and more gradual connectivity-related transcriptional changes. Yet, the less prominent differences between medial and distal domains compared to proximal domains suggest that the gradient provided may be limited, and future tissue engineering attempts might be required to expand the FGF8 activation domain.
The observation that the in vitro rostrocaudal progenitors’ identity capturing the PFS-SS-T in vivo axis is not stably maintained in excitatory neurons, which only partially retain the temporal area driver gene signature, suggests interesting avenues for future research. These refinements could enhance in vitro arealization and leverage current limitations to mechanistically dissect long-standing questions on neuronal fate acquisition in humans. While FGF8 signaling is necessary and sufficient to instruct early transcriptomic differences related to the progenitor protomap, the propagation of area-specific identity to neurons (especially for the prefrontal identity) may require further layers of regulation by additional patterning signals39, a transition through an intermediate progenitor protomap as suggested for the mouse cortex51, or external regulation by thalamic innervation5, as in fact postulated by the protocortex hypothesis. Our system could be useful to explore the temporal dynamics of cortical patterning and intermediate progenitor diversity, as well as the effect of RA signaling in combination with thalamic explants on the modulation of area-specific identity during differentiation and activity-derived maturation.
Cortical organoid systems have been used to model diseases related to progenitor abundance and neuronal migration16,17. By combining spatially oriented frontotemporal-related signatures with gene editing of pathogenic mutations, we show that our approach is suitable to study area-related patterning defects underlying human developmental disorders. Indeed, polCAs carrying a recurrent FGFR3 mutation in individuals with achondroplasia failed to mount positional effects. Notably, distal domains displayed early increased proliferation and FGF signaling-related signatures similar to those shown by proximal domains. This could be due to enhanced FGF8 signaling in mutant FGFR3 assembloids, possibly from stabilization of the ligand–receptor complex52, or slower ligand-mediated receptor internalization53. These results indicate that both progenitor overproliferation and impaired establishment of caudal identities could underlie temporal lobe abnormalities in affected individuals. Finally, given that early patterning defects underlying cortical arealization have been implicated in transcriptional dysregulation across cortical areas in the brains of autistic individuals54, our system provides an experimental platform to test this possibility. Thus, polCAs offer a means to connect genetic and environmental alterations relevant to neuropsychiatric disorders with specific early cortical patterning events in individual organoids.
Methods
PDMS mold production
To create the master negative molds, a three-dimensional model was first designed using open-source computer-aided design software (FreeCAD). This master design consists of an array of ten triangular prisms with a length of 15 mm, a base width of 2 mm and a height of 2.5 mm. A toolpath engine (Vectric Cut3D) was used to generate a toolpath as G-code. The master mold was then cut using a micro-milling machining system (Minitech Machinery Corporation) with a 0.1-mm end mill into a block of acrylic glass. The completed master was cleaned with compressed air and stored in a dust-free environment. To create an organoid mold from the master negative mold, PDMS (Sylgard 184, Dow Corning) monomer was mixed with its curing agent at a 10:1 ratio and poured into a Petri dish containing the master. The ensemble was degassed for 30 min in a vacuum bell. A glass coverslip was then positioned above the master’s array of triangular prisms, making contact with the PDMS, to produce a smooth surface in the final mold suitable for imaging. This was then baked at 70 °C overnight, and the cured PDMS mold was carefully removed from the master by hand when finished. The files of master negative mold and polCA mold are provided as Supplementary Data 10.
Stem cell line maintenance
hES cell lines H9 (WA09) and H1 (WA01) were obtained from WiCell. All the reporter and mutant cell lines were derived from the H9 line. The induced pluripotent stem (iPS) cell lines SCCF-176j-clone 1 (female, fibroblast-derived), SCCF-177j-clone 8 (female, fibroblast-derived) and SCCF-178j-clone 5 (male, fibroblast-derived) were generated by the IMBA Stem Cell Core Facility in compliance with the Austrian and European legislation and are part of the IPSC Biobank. iPS cell lines were generated using the Sendai virus (CytoTuneTM-iPS 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific) carrying the Yamanaka reprogramming factors OCT3/OCT4, SOX2, c-MYC and KLF4. All cells were authenticated using a short tandem repeat assay, maintained in a 5% CO2 incubator at 37 °C and routinely tested for mycoplasma. All the cell lines were cultured under feeder-free conditions, seeded onto six-well plates coated with hES-qualified Matrigel (Corning, 354277) and maintained in mTeSR1 medium (STEMCELL Technologies). Cells were fed daily and passaged every 3–4 days using 0.5 mM EDTA solution treatment and mechanical dissociation or archived by freezing in liquid nitrogen with Cell Banker 2 solution (Ambio, 11891).
Generation of reporter and mutant lines
To generate the SP8 > GFP, the SYN1 > CHRIMSONR-tdTomato25,55 and the CAG > FGF8 lines, feeder-free H9 cells were engineered with the TALEN technology as described previously22 to insert the reporter constructs into the AAVS1 safe-harbor locus. Donor plasmids were designed as follows: (i) SA-2A-puro-PA-2xCHS4-CAG > FGF8-WPRE-SV40-2xCHS4; (ii) SA-2A-puro-PA-2xCHS4-hSYN1-INTRON-ChrimsonR-tdTomato-WPRE-SV40-2xCHS4; (iii) SA-2A-puro-PA-2xCHS4-SP8.844-HBBminiP-INTRON-eGFP-WPRE-SV40-2xCHS4. The fragment spanning 7,394 to 6,120 base pairs (bp) upstream of the transcription start site of the human SP8 gene (hs844, Lawrence Berkeley National Laboratory) was selected from https://enhancer.lbl.gov/ (ref. 56) and used as the enhancer. The CAG promoter57 was chosen to drive constitutive expression of FGF8 (NM_006119.4), and the sequence was amplified from GenScript OHu22678. All donor plasmids were verified by sequencing. Nucleofection was carried out with the Amaxa nucleofector (Lonza) and Human Stem Cell Nucleofector Kit 1 solutions containing 0.5 µg of each of the TALEN plasmids and 1 µg of each of the donor plasmids following the manufacturer’s guidelines. Nucleofected cells were grown for 4 days and then selected with 0.5 µg ml−1 puromycin (Jena Bioscience, NU-931-5). The surviving colonies were picked manually and further expanded for genotyping and cryopreservation. DNA was extracted using the QuickExtract DNA Extraction Solution (EpiCentre) for genotyping.
The EMX1-mNeonGreen line was generated by targeting the endogenous locus with CRISPR–Cas9 technology and homologous directed repair (HDR). Briefly, to generate the Emx1 HDR plasmid, the 5′ left arm (746 bp) and 3′ right arm (723 bp) were cloned into a pBluescript vector and a P2A-mNeonGreen cassette was inserted before the EMX1 stop codon. A Puromycin-deltaTK-resistance selection cassette driven by the PGK promoter and flanked by loxP sites was inserted downstream of the P2A-mNEONGreen cassette. In total, 1 × 106 single H9 hES cells were dissociated with Accutase and washed once with PBS−/−. Cells were electroporated with 15 µg of the Emx1 HDR plasmid, 5 µg of Cas9 protein and 12 µg of gRNA targeting the Emx1 site GTCACCTCCAATGACTAGGG58 diluted in cleavage buffer (20 mM Hepes pH 7.5, 150 mM KCl, 0.5 mM dithiothreitol, 10 mM MgCl2, 0.1 mM EDTA) using the Neon Transfection System and Resuspension Buffer R (MPK5000, Thermo Fisher Scientific) according to the manufacturer’s instructions. The gRNA was cloned into a proprietary vector with modified scaffold and in vitro transcribed using AmpliScribe T7-Flash Transcription kit (ASF3257, Lucigen) per the manufacturer’s instructions and Cas9 protein was produced according to Jinek et al.59 by the Protein Technologies Facility at Vienna BioCenter Core Facilities (VBCF), member of the Vienna BioCenter (VBC), Austria. After nucleofection, cells were seeded into Matrigel-coated 10-cm plates in the presence of 2 μM ROCK inhibitor (Y27632, Sigma-Aldrich) for 48 h. After 5 days, cells were selected with Puromycin 0.5 μg ml−1 (P8833, Sigma-Aldrich). About 10 days later, individual clones were picked for genotype analysis. Correctly targeted knock-in clones were confirmed by a genotyping PCR assay based on left arm (LA), right arm (RA), internal piece (IP) and WT probes (Supplementary Fig. 1b). Primer sequences are provided in Supplementary Table 1. Two heterozygous knock-in clones (43 and 51) and one homozygous knock-in clone (65) were used for the experiments shown in this study.
The FGFR3 human isogenic cell line was generated with CRISPR–Cas9 technology. Single-stranded DNA carrying the G > A substitution at codon 380 described in ref. 42 was co-transfected with the selected FGFR3 cutting guide (GGCATCCTCAGCTACGGGGT) and Cas9 protein with the help of IMBA Stem Cell Core Facility. In total, 600 clones were first screened for the acquisition of the Sfc1 restriction site upon mutation G > A, and sequencing was then verified for the selected clones. The CAG>tdTOMATO line was kindly provided by the authors of ref. 60. Targeted insertions and loss of WT alleles in the three selected clones were verified with PCR with the primers included in Supplementary Table 1.
Generation of cerebral organoids
Round and elongated cortical assembloids were generated with a modified version of a previously described protocol61,62. A detailed culture protocol for the generation of polCAs is available in ref. 63. Briefly, on day 0, hES cells and iPS cells were treated with Accutase resulting in a single-cell suspension and transferred to an ultra-low attachment 96-well plate (9,000 cells per well) for round organoids, or to a PMDS mold (5.5 × 106 cells per mold) for elongated organoids. Cells aggregated into EBs in mTeSR1 supplemented with 50 µM Rho-associated protein kinase (ROCK) inhibitor. PMDS molds were sterilized with autoclaving and gamma irradiation, then treated with Pluronic F127 (Sigma-Aldrich, P2443) 1% in PBS and washed with PBS several times before usage. From day 1 onwards, the medium was replaced daily with NI medium containing DMEM-F12 supplemented with 1× N2 supplement, 1 μg ml−1 heparin solution, 1× GlutaMAX, 1× MEM-NEAA and 50 µM ROCK inhibitor. The fusion with the OrEB was carried out at day 3, by simply positioning it on top of one elongated EB extremity and adjusting it with the pipette tip. On day 7, EBs were embedded into droplets of Matrigel and transferred into 6-cm dishes containing Diff-A medium consisting of 50% DMEM-F12, 50% Neurobasal medium, 1× N2, 1× B27–vitamin A, 2.5 µg ml−1 insulin, 0.05 mM betamercaptoethanol (BME), 1× GlutaMAX, 1× MEM-NEAA and 1× antibiotic–antimycotic (Thermo Fisher,15240062). On day 15, the medium was replaced with Diff+A medium consisting of 50% DMEM-F12, 50% Neurobasal medium, 1× N2, 1× B27, 2.5 µg ml−1 insulin, 0.05 mM BME, 1× GlutaMAX, 1× MEM-NEAA and 1× antibiotic–antimycotic, and organoids were moved to an orbital shaker at 52 rpm. The medium was changed every 2–3 days. From day 30 onwards, organoids were fed with Diff+A medium supplemented with 1% (vol/vol) Matrigel basement membrane, brain-derived neurotrophic factor (BDNF, 20 ng ml−1) and glial cell line-derived neurotrophic factor (GDNF, 20 ng ml−1). The patterning treatment was carried out between day 3 and day 5 of the protocol, and the factors were diluted in NI medium as described in Supplementary Table 2.
ELISA
Levels of FGF8 protein in cell extracts and supernatants were measured using an ELISA kit (Cusabio CSB-E15861h) according to the manufacturer’s instruction. Briefly, frozen samples of WT and CAG > FGF8 cells were thawed on ice and centrifuged at 18,000g for 10 min to eliminate debris. Supernatants (3 ml for each sample) were concentrated using a centrifugal filter unit with Ultracel-3 membranes (UFC900308, Merck Millipore). For each sample (50 µl), absorbances at 450 nm and 540 nm were measured in duplicate using a microplate reader (Synergy H1 BioTek). Absorbance at 540 nm was subtracted from absorbance at 450 nm to correct for nonspecific signals. Mean values of absorbance were multiplied for the concentration factor and used to estimate FGF8 amounts (pg ml−1) according to a standard curve. Negative values have been considered as 0 in the graph.
RNA extraction and RT–qPCR analysis
Samples were lysed in 350 µl of RLT buffer supplemented with β-mercaptoethanol, and RNA was extracted using the RNeasy Micro Kit (Qiagen, 74004) following the manufacturer’s protocol. Retrotranscription to cDNA was performed with SuperScript II (Thermo Fisher, 18064071). RT–qPCR reactions were performed using GoTaq qPCR master mix (Promega, A6002) with 384-well (CXF384) Bio-Rad machines using the following reaction protocol: (i) 95 °C for 3 min, (ii) 95 °C for 10 s, (iii) 62 °C for 10 s, (iv) 72 °C for 40 s, (v) go to 2, 40 cycles, (vi) 95 °C for 1 min and (vii) 50 °C for 10 s. Quantification was performed in Excel by calculating the ΔCt value using TBP as a reference gene for normalization. Data are presented as expression (2−ΔCt) or log (expression). Primers used for RT–qPCR are listed in Supplementary Table 3.
PolCA dissociation strategy
To define P, M and D segments for transcriptomics experiments, we first measured the length of individual polCAs with a ruler and separated consecutive pieces of length equal to one-third of the total length using a scalpel under a stereomicroscope.
Bulk RNA-seq and analysis
Individual pieces from seven independent polCAs and four control elongated organoids (without OrEBs) from two experiments (from H9 cells) and three mutant polCAs grown from three independent H9 mutant clones were processed for RNA extraction as described above. Of the seven WT polCA samples analyzed, polCA number 3 was identified as an outlier and, therefore, removed from further analysis. We then assessed RNA concentration and quality with an RNA 6000 Nano Chip (Agilent Technologies) and enriched for mRNA with NEBNext Poly(A) mRNA Magnetic Isolation Module; barcoded samples were then multiplexed and sequenced with 50-bp single-end sequencing on a HiSeq 2500 (Illumina). mRNA sample enrichment, library preparation and sequencing were performed by the VBCF NGS unit (https://www.vbcf.ac.at). Adaptors were clipped with TrimGalore (v0.5.0, https://github.com/felixkrueger/trimgalore)64. Abundant sequences (iGenomes hg38) were removed with Bowtie 2 (v2.3.4.1)65. Cleaned reads were aligned against the genome (GRCh38) with STAR (v2.6.0c)66. Furthermore, reads were subjected to transcripts per million estimations using the Salmon algorithm (v0.11.0)67 and counted toward their corresponding gene (ENSEMBL v94) with featureCounts (v1.6.2)68. RUVseq69 was used to model an additional factor (W_1) of unwanted variation using edgeR residuals (reduced model)69,70. The full model (cellline + batch + W_1 + condition) was used in DESeq2 (v1.18.1) to detect DEGs71 (https://www.R-project.org/). Expression values were corrected with limma using the full model72. PCA analysis of corrected expression values (based on genomic read counts) was carried out on the top 500 most variably expressed genes. Gene Ontology enrichment analysis was performed with clusterProfiler73; soft clustering for gradient analysis was performed with Mfuzz (2.52.0)74,75. Uncertainty of hierarchical clustering on the DESeq2 datasets was assessed with pvclust76; this R package calculates probability values (P values) for each cluster using bootstrap resampling techniques. Two P values are so generated: approximately unbiased (AU) P value and bootstrap probability (BP) value. Multiscale bootstrap resampling is used for the calculation of AU P value, which has superiority in bias over BP value calculated by the ordinary bootstrap resampling. In the figures, we show the Euclidean distance and complete cluster. An integrative list from published datasets was used to subset rostrocaudal genes in Fig. 3 (Supplementary Data 2). The gene VSTs are visualized in a scaled heat map ordered by complete linkage clustering. Analysis of brain organoid region identity was performed with Voxhunt77.
Sample processing for scRNA-seq
Three independent polCAs and three elongated organoids without OrEBs (control, C1–3) were manually dissected at a stereomicroscope and processed for dissociation with gentleMACS Dissociator in trypsin/Accutase solution with TURBO DNase (Thermo, AM2238, 2 U µl−1). After dissociation, DPBS−/− + 10% FBS were gradually added to stop the reaction. Samples were then centrifuged at 400g for 5 min at 4 °C, and supernatant was aspirated without touching the pellet. The pellet was resuspended in an additional 500 µl of DPBS−/− + 10% FBS, filtered through FACS tubes and stained for viability dye DRAQ7 (Biostatus, DR70250, 0.3 mM). Live cells were sorted with a BD FACS Aria III on an Alexa 700 filter and barcoded with unique cholesterol-modified oligos (CMOs) for MULTI-seq analysis. Sequencing was performed on a 10x NovaSeq S4 PE150 XP (single/half lane) platform according to the manufacturer’s indications. Raw reads were processed using the Cell Ranger multi pipeline (v6.1.1) against GRCh38 (refdata 3.0.0 plus construct sequence). Introns were included; expected cells were set to 35,000 (conCAs) and 50,000 (polCAs).
Three-point model DEG generation
Data from ref. 37 were downloaded from the UCSF cell browser and nonrelevant cell types such as vascular cells and microglia were removed. Standard preprocessing was then carried out in accordance with the Scanpy78 workflow. Cells from this dataset were integrated via Harmony79 (with a maximum number of iterations of 20), providing as a covariate the chemistry of the different samples (originally divided in 10x v2 and v3 chemistries). To improve the annotation granularity, we took advantage of the dataset from ref. 80 and the Scanpy ingestion function to combine the original Bhaduri et. al annotation with the one done in Polioudakis et al. In this way, we obtained a finer map of cell-type region, used to more robustly select the relevant cell types that divide cells, radial glia and neurons. For the first two cell types, before selection, we removed the individual marked as ‘GW20’ that was poorly interspersed with other samples in PCA, potentially driven by technical confounders. For the dividing cells, we extracted cells annotated simultaneously as ‘dividing’ in the original annotation, ‘PgS’ or ‘PgG2M’ according to ingestion label transfer, and not marked as being in the G1 phase according to the ‘score cycle genes’ function of scvelo81,82. After integration, we removed two clusters that were exclusive of a single donor or expressing high levels of S100B. Radial glia were selected as cells annotated simultaneously as ‘radial’ from the original annotation and as ‘vRG’ (ventricular radial glia) according to label transfer. Finally, to isolate excitatory neurons, we kept cells annotated as ‘excitatory’ in the original annotation and as ‘maturing excitatory’ from the transferred annotation, and the whole batch 1 was excluded, because it was represented in just one cortical area. We used this curated atlas to define a gene universe strictly dictated by the rostrocaudal expression gradient. We performed the following steps for all three main cell types (dividing, radial glia, excitatory neurons) in parallel, following the same rationale. After aggregating each cortical area in metacells (‘Metacells aggregation’), we used the general linear model implementation in edgeR to find genes displaying incremental or decremental rostrocaudal expression (Bonferroni < 0.001). To refine for high-confidence trending genes, we then ranked them according to P value and fitting an enrichment curve for known rostrocaudal markers. We picked the maximum distance from the fitted curve to a random enrichment as the thresholding point for the P value in order to maximize the true positive rate. To prepare the Polioudakis et al. dataset, count matrices were downloaded from the Gene Expression Omnibus under accession number GSE95023. Only genes expressed in more than 170 cells were retained, counts were normalized using scran83 and selection of highly variable genes was performed via Triku84. Next, PCA was computed using the scanpy.tl.pca function on the top 25 principal components (PCs) and specifying 40 as the local neighborhood size (n).
polCA single-cell data analyses
As for previous datasets, all analyses were performed within the Scanpy framework. Filtered feature matrices were imported from Cell Ranger for each dataset and combined in a single AnnData object78. Droplets displaying higher counts were trimmed according to 10x v3 kit doublet expected value. Only cells with less than 40% and 5% ribosomal and mitochondrial genes counts, respectively, were preserved; cells with less than 200 detected genes and genes detected in less than 3 cells were excluded from the analysis. Counts were then log normalized as for the fetal dataset. After identifying variable genes (‘Highly variable gene selection’), the dataset was scaled with the maximum value set to 20 via the Scanpy function. Finally, we applied Gruffi85 to exclude stressed cells from further analysis.
Highly variable gene selection
A multistep highly variable gene (HVG) detection was adopted to preserve and balance the variability within experiments/conditions. By taking advantage of the three technical replicates for each segment (proximal, medial, distal, piece1, piece2, piece2), we vertically intersected HVGs from technical replicates for each segment and merged the resulting six HVGs sets. Moreover, we intersected HVGs deriving from all possible pairs of technical replicates across contiguous segments to preserve possible variability gradients (proximal–medial, medial–distal, piece1–piece2, piece2–piece3), resulting in four additional HVGs sets. We thus used the union of the ten HVG sets altogether for every further dataset subset iteration.
Dimensionality reduction and clustering
PCA was performed on retrieved HVGs. A neighbors graph was then computed with n = 20 and 20 PCs, and cell types were partitioned using the Leiden algorithm’s Scanpy implementation setting the resolution to 0.3. Unless differently specified, PCA and neighbors graphs were recomputed after each dataset subsetting.
DEA
DEA of single cells was performed via implementation of the Wilcoxon test in Scanpy. Additionally, for RGC1–RGC2, ExN1–ExN2 and among segment comparisons, we also applied metacell-based DEA with edgeR framework, using its normalization and variance estimation, followed by the general linear model test (FDR-corrected P value < 0.05, absolute logFC > 1.5).
Selection of main cell types
After whole-dataset exploration and cell-type annotation, we focused on the three main cell types. We thus isolated cycling progenitors, non-cycling progenitors and excitatory neurons. We considered cycling progenitors, as the cells belonging to progenitor clusters 4 (RGC1), 5 (RGC2) or 6 (cRGC) and simultaneously resulting in G2M or S phases according to scVelo’s ‘score cell cycle genes’ function. Non-cycling progenitors (henceforth, progenitors) were defined as cells belonging to non-proliferative progenitor clusters 4 and 5 and marked as G1 phase from the scVelo’s function. Finally, we considered excitatory neurons the clusters previously annotated as ExN1 and ExN2.
Differential abundance analysis
We used Milo86 to assess the differential abundance between proximal and distal segments across all produced cell types and between proximal segment and other segments/pieces (treated as single group) along the excitatory neuron’s lineage specifically. In both cases, before the test, we downsampled cells to match the number of cells of the two compared groups. Enriched neighbors plots were generated using spatial FDR < 0.1. In the latter case, to isolate cells from proximal-exclusive lineage, we extracted cells belonging to neighbors > 2.5 logFC enrichment between proximal and others.
Patterning markers dynamics
Excitatory neurons lineages were isolated using PAGA87. Diffusion pseudotime (DPT)88 was calculated using Scanpy’s implementation providing as root cells with highest TOP2A expression. We trimmed cells with extreme values of DPT (<1st percentile and >99th percentile) to reduce the noise of the extreme underrepresented DPT segments. Next, after splitting proximal-exclusive cells from others (differential abundance methods) we downsampled each DPT bin (100 bins total) to match the number of cells from the two lineages. Finally, used tradeSeq89 to fit the gene expression dynamics (fitGAM, nknots = 7) on highly variable genes plus known rostrocaudal genes, and test for differences (patternTest). After filtering (Bonferroni < 0.01), we plotted the smoothed expression (fitGAM result) difference (proximal, other) along pseudotime for selected key markers.
Metacell aggregation
For fetal gene gradient extraction, and in-depth characterization of the three main polCA cell types, we decided to aggregate cells into metacells to increase the robustness of observed features90,91. For polCA segment-based metacells after selecting each main cell type, within each segment we randomly sampled cells to result in 10 metacells. The same approach was used for fetal dataset before three-point model creation, targeting 30 metacells per cortical area of derivation for each of the three main cell types.
Assessment of forebrain identity
To assess the forebrain identity of polCAs, we used the recently released atlas of fetal brain development as reference from ref. 44. Specifically, we isolated forebrain (regions originally annotated as telencephalon and diencephalon were merged), midbrain and hindbrain (regions originally annotated as cerebellum, medulla and pons were merged) and gestational week was represented across all three domains (8.5, 11.5, 12, 14). For consistent annotation, we transferred cell-type labels from ref. 80 as for the previous37 atlas curation, and retained vRG, cycling progenitors and maturing excitatory neurons. For similarity assessment, we included scRNA-seq available data from refs. 92,93 as a reference. Data from the Velasco study were downloaded from GSE129519, keeping 3-month-old organoids (the closest polCA differentiation days) from batch one and batch two. Data from the Kanton study were downloaded from E-MTAB-7552, keeping 2-months-old human organoids. Data from the Braun atlas were collected from https://github.com/linnarsson-lab/developing-human-brain/, human_dev-GRCh38-3.0.0.h5ad, removing cells resulting from 10x V2 chemistry kit, that according to the authors caused batch effects. We kept only genes common to all four datasets (Braun, polCA, Velasco and Kanton), and for each cell type and gestation week, we extracted the top five marker genes of each brain domain. The extracted signature (Supplementary Data 7) was used to assess the similarity (Spearman correlation) to forebrain, midbrain and hindbrain of polCA, Velasco and Kanton datasets for any cell type originally annotated as cycling progenitors, radial glia or neurons.
Gradient analysis on single cells
We carried out cell-type wise gene trends clustering via Mfuzz74 using as input the same gene selection as that used for bulk RNA Mfuzz. In this case, data preparation entailed initial normalization of single cells (using normalize_total function from Scanpy, with target = 50,000), sum of counts within each segment and division by number of cells aggregated in that way. Finally, counts across selected genes were scaled to unit variance to make genes with a different magnitude of expression comparable.
Cryo-sectioning, immunostaining and imaging
Alexa Fluor 488-, 568- or 647-conjugated (Invitrogen) secondary donkey antibodies were used at 1:500 dilution. Immunostaining images were captured with Axio Imager confocal microscopes LSM700 and Celldiscoverer 7 (Zeiss), using the ‘tile’, ‘stitching’ and ‘maximum intensity projection’ functions in the Zen software. Live imaging of intact organoids was performed on a widefield microscope (AxioVert.A1, Zeiss) with a SONY Chameleon3 CM3-U3-31S4M CMOS camera (Zeiss). Post-acquisition image adjustments and quantifications were done in Fiji. The human fetal brain sample used in Supplementary Fig. 1e was obtained from Novogenix Laboratories and approved for usage from the IMBA Ethics and Biosafety.
Image analysis
The GFP intensity of live organoids was calculated as the integrated density of the organoid area minus the background mean per area unit multiplied for the area analyzed. Organoids in Fig. 1 are considered GFP+ or mNeon+ when the fluorescence intensity is higher than the threshold (maximum fluorescence intensity of parental WT cell-derived samples in untreated condition). For OrEB distance measurement on the organoid length (Fig. 2c), a Fiji macro developed by T. Lendl from the BioOptics facility at IMP (https://cores.imp.ac.at/biooptics/) was used to straighten the polCA and establish the exact length (Supplementary Code 1). The image was downsampled and a segmented line was drawn manually, following the shape of the organoid. Then the straightening tool of Fiji was applied and a straightened image was created. The fluorescent fusion area was thresholded to find its center, which was then used to measure the distance to the end of the elongated assembloid. For the quantification of SP8 > GFP intensity (Fig. 2d), the exact length measured with the Fiji macro (Supplementary Code 1) was then divided into three equal portions and GFP intensity was calculated per portion. The P portion was identified in each image through the localization of tdTomato-positive cells. For Fig. 2e instead, the total length was divided into two equal portions (P or D) and ten rectangular fields were subsetted from each side with the same region of interest. The elongated assembloid length at different time points was otherwise measured with the freehand line tool in Fiji. For representative images, some of the pictures were clipped with the clipping mask option in Adobe Illustrator to equalize the panel dimension, or the intensity/contrast was adjusted equally in all the panels (for example, for Lmo4 staining, brightness/contrast min–max displayed values were set to 5–51, for DUSP6 to 12–143). The total number of DAPI+, NR2F1+, LMO4+, CPNE8+ and CTIP2+ cells was calculated using the Fiji granulometric filtering plugin ‘GranFilter’ and the watershed tool.
Organoid dissociation and flow cytometry
Two organoids per sample were collected for sorting, washed twice with 1× PBS and dissociated to single cells in a 2:1 solution of Accutase (Sigma-Aldrich, A6964) and trypsin (Gibco, 15400) for 30 min at 37 °C. Specimens were centrifuged at 200g, resuspended in 1 ml Diff+A with 50 µM ROCK inhibitor and filtered through a 35-µm cell strainer. Samples were sorted directly in RLT buffer for subsequent RNA extraction at a SonySorter SH800S. ‘GFP’ on the x axis of flow cytometry graph was ‘laser 488 nm; bandpass 525/50 nm’; ‘autofluorescence’ on the y axis was ‘laser 488 nm; bandpass 720/60 nm’. Flow cytometry plots were generated from the .fcs files with FlowJo software.
Statistics and reproducibility
Statistical analysis was carried out with Prism software using an unpaired t-test or one-way ANOVA. For non-normal distributions, non-parametric tests were used. The threshold for statistical significance was P < 0.05. All details on sample size, the number of replicates, statistical tests and P values for each experiment are provided in the relevant figure legend. Sample sizes of organoid experiments were estimated empirically based on previous experience. Unless stated otherwise, experiments were performed using at least three different cell lines to ensure reproducibility.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data generated in this study have been deposited as SuperSeries at the NCBI Gene Expression Omnibus under the public accession number GSE231320. The data and all unique reagents or biological materials generated in this study are available from the corresponding authors J.K., V.K. and G.T. upon reasonable request and in compliance with material transfer agreements. Source data are provided with this paper.
Code availability
The code used for the single-cell analyses presented in this paper is available at https://github.com/GiuseppeTestaLab/polCAs/.
References
Yeo, B. T. T. et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 106, 1125–1165 (2011).
Triarhou, L. C. Pre-Brodmann pioneers of cortical cytoarchitectonics I: Theodor Meynert, Vladimir Betz and William Bevan-Lewis. Brain Struct. Funct. 226, 49–67 (2021).
Triarhou, L. C. Pre-Brodmann pioneers of cortical cytoarchitectonics II: Carl Hammarberg, Alfred Walter Campbell and Grafton Elliot Smith. Brain Struct. Funct. 225, 2591–2614 (2020).
Loukas, M., Pennell, C., Groat, C., Tubbs, R. S. & Cohen-Gadol, A. A. Korbinian Brodmann (1868–1918) and his contributions to mapping the cerebral cortex. Neurosurgery 68, 6–11 (2011).
Antón-Bolaños, N., Espinosa, A. & López-Bendito, G. Developmental interactions between thalamus and cortex: a true love reciprocal story. Curr. Opin. Neurobiol. 52, 33–41 (2018).
Rakic, P. Specification of cerebral cortical areas. Science 241, 170–176 (1988).
O’Leary, D. D. M., Chou, S. -J. & Sahara, S. Area patterning of the mammalian cortex. Neuron 56, 252–269 (2007).
Johnson, M. B. et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009).
Rubenstein, J. L., Shimamura, K., Martinez, S. & Puelles, L. Regionalization of the prosencephalic neural plate. Annu. Rev. Neurosci. 21, 445–477 (1998).
Cadwell, C. R., Bhaduri, A., Mostajo-Radji, M. A., Keefe, M. G. & Nowakowski, T. J. Development and arealization of the cerebral cortex. Neuron 103, 980–1004 (2019).
Vieira, C. et al. Molecular mechanisms controlling brain development: an overview of neuroepithelial secondary organizers. Int. J. Dev. Biol. 54, 7–20 (2010).
Borello, U. & Pierani, A. Patterning the cerebral cortex: traveling with morphogens. Curr. Opin. Genet. Dev. 20, 408–415 (2010).
Villalba, A., Götz, M. & Borrell, V. The regulation of cortical neurogenesis. Curr. Top. Dev. Biol. 142, 1–66 (2021).
Molnár, Z. et al. New insights into the development of the human cerebral cortex. J. Anat. 235, 432–451 (2019).
Varki, A., Geschwind, D. H. & Eichler, E. E. Explaining human uniqueness: genome interactions with environment, behaviour and culture. Nat. Rev. Genet. 9, 749–763 (2008).
Sidhaye, J. & Knoblich, J. A. Brain organoids: an ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ. 28, 52–67 (2021).
Amin, N. D. & Paşca, S. P. Building models of brain disorders with three-dimensional organoids. Neuron 100, 389–405 (2018).
Imaizumi, K. et al. Rostrocaudal areal patterning of human PSC-derived cortical neurons by FGF8 signaling. eNeuro 5, https://doi.org/10.1523/ENEURO.0368-17.2018 (2018).
Astick, M. & Vanderhaeghen, P. From human pluripotent stem cells to cortical circuits. Curr. Top. Dev. Biol. 129, 67–98 (2018).
Cederquist, G. Y. et al. A multiplex human pluripotent stem cell platform defines molecular and functional subclasses of autism-related genes. Cell Stem Cell 27, 35–49 (2020).
Bhaduri, A. et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020).
Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J. & Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 14, 743–751 (2017).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Xiang, Y. et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398 (2017).
Miura, Y. et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 38, 1421–1430 (2020).
Rifes, P. et al. Modeling neural tube development by differentiation of human embryonic stem cells in a microfluidic WNT gradient. Nat. Biotechnol. 38, 1265–1273 (2020).
Cederquist, G. Y. et al. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 37, 436–444 (2019).
Manikkam, S. A. et al. Temporal lobe malformations in achondroplasia: expanding the brain imaging phenotype associated with FGFR3-related skeletal dysplasias. Am. J. Neuroradiol. 39, 380–384 (2018).
Sahara, S., Kawakami, Y., Izpisua Belmonte, J. C. & O’Leary, D. D. M. Sp8 exhibits reciprocal induction with Fgf8 but has an opposing effect on anterior-posterior cortical area patterning. Neural Dev. 2, 10–22 (2007).
Stocker, A. M. & O’Leary, D. D. M. Emx1 is required for neocortical area patterning. PLoS ONE 11, e0149900 (2016).
Renner, M. et al. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 36, 1316–1329 (2017).
Wataya, T. et al. Minimization of exogenous signals in ES cell culture induces rostral hypothalamic differentiation. Proc. Natl Acad. Sci. USA 105, 11796–11801 (2008).
Lambert, N. et al. Genes expressed in specific areas of the human fetal cerebral cortex display distinct patterns of evolution. PLoS ONE 6, e17753 (2011).
Sagner, A. & Briscoe, J. Morphogen interpretation: concentration, time, competence, and signaling dynamics. Wiley Interdiscip. Rev. Dev. Biol. 6, e271 (2017).
Lancaster, M. A. et al. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 35, 659–666 (2017).
de Bakker, B. S. et al. An interactive three-dimensional digital atlas and quantitative database of human development. Science 354, aag0053 (2016).
Bhaduri, A. et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021).
Ding, S.-L. et al. Cellular resolution anatomical and molecular atlases for prenatal human brains. J. Comp. Neurol. 530, 6–503 (2022).
Shibata, M. et al. Regulation of prefrontal patterning and connectivity by retinoic acid. Nature 598, 483–488 (2021).
Storm, E. E. et al. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831–1844 (2006).
Thomson, R. E. et al. Fgf receptor 3 activation promotes selective growth and expansion of occipitotemporal cortex. Neural Dev. 4, 4 (2009).
Bellus, G. A. et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet 56, 368–373 (1995).
Tanaka, Y., Cakir, B., Xiang, Y., Sullivan, G. J. & Park, I. -H. Synthetic analyses of single-cell transcriptomes from multiple brain organoids and fetal brain. Cell Rep. 30, 1682–1689 (2020).
Braun, E. et al. Comprehensive cell atlas of the first-trimester developing human brain. Science 382, eadf1226 (2023).
Kadoshima, T. et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl Acad. Sci. USA 110, 20284–20289 (2013).
Pugash, D., Lehman, A. M. & Langlois, S. Prenatal ultrasound and MRI findings of temporal and occipital lobe dysplasia in a twin with achondroplasia. Ultrasound Obstet. Gynecol. 44, 365–368 (2014).
Shimogori, T., Banuchi, V., Ng, H. Y., Strauss, J. B. & Grove, E. A. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131, 5639–5647 (2004).
Miller, J. A. et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014).
Furusho, M., Kaga, Y., Ishii, A., Hebert, J. M. & Bansal, R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J. Neurosci. 31, 5055–5066 (2011).
Charvet, C. J., Cahalane, D. J. & Finlay, B. L. Systematic, cross-cortex variation in neuron numbers in rodents and primates. Cereb. Cortex 25, 147–160 (2015).
Elsen, G. E. et al. The protomap is propagated to cortical plate neurons through an Eomes-dependent intermediate map. Proc. Natl Acad. Sci. USA 110, 4081–4086 (2013).
Webster, M. K. & Donoghue, D. J. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 15, 520–527 (1996).
Monsonego-Ornan, E., Adar, R., Feferman, T., Segev, O. & Yayon, A. The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol. Cell. Biol. 20, 516–522 (2000).
Gandal, M. J. et al. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature 611, 532–539 (2022).
Klapoetke, N. C. et al. Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346 (2014).
Visel, A., Minovitsky, S., Dubchak, I. & Pennacchio, L. A. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007).
Niwa, H., Yamamura, K. & Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 (1991).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Wimmer, R. A. et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510 (2019).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Lancaster, M. A. & Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014).
Bosone C., Barilà S.E., Krenn V. and J.A. Knoblich. Detailed culture protocol for the generation of human polarized cortical assembloids (polCA). Protocol.io https://doi.org/10.17504/protocols.io.4r3l2qyz4l1y/v1 (2024).
M Martin, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. https://doi.org/10.14806/ej.17.1.200 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–21 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Yu, G., Wang, L. -G., Han, Y. & He, Q. -Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Kumar, L. & Futschik, M. E. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 (2005).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Fleck, J. S. et al. Resolving organoid brain region identities by mapping single-cell genomic data to reference atlases. Cell Stem Cell 28, 1148–1159 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Polioudakis, D. et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801 (2019).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Lun, A. T. L., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016).
Ascensión, A., Ibáñez-Solé, O., Inza, I., Izeta, A., & Araúzo-Bravo, M. J. Triku: a feature selection method based on nearest neighbors for single-cell data. GigaScience https://doi.org/10.1093/gigascience/giac017 (2022).
Vértesy, Á. et al. Gruffi: an algorithm for computational removal of stressed cells from brain organoid transcriptomic datasets. EMBO J. 41, e111118 (2022).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Van den Berge, K. et al. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 11, 1201 (2020).
Ben-Kiki, O., Bercovich, A., Lifshitz, A. & Tanay, A. Metacell-2: a divide-and-conquer metacell algorithm for scalable scRNA-seq analysis. Genome Biol. 23, 100 (2022).
Persad, S. et al. SEACells infers transcriptional and epigenomic cellular states from single-cell genomics data. Nat. Biotechnol. 41, 1746–1757 (2023).
Velasco, S. et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527 (2019).
Kanton, S. et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019).
Acknowledgements
We thank all members of the laboratory of J.A.K. for technical expertise and feedback, especially T. Kruitwagen, J. Sidhaye and C. Esk for feedback on the manuscript; A. Peer and P. Möseneder for technical help. We are grateful to the IMBA/IMP BioOptics facility for flow cytometry and microscopy services and analysis, IMP/IMBA Bioinformatics for sequencing analysis, the VBCF HistoPathology facility (https://www.viennabiocenter.org/facilities/) for histological services and consultation, the VBCF Sequencing unit (https://www.viennabiocenter.org/facilities/) for sequencing and the IMBA Stem cell facility (https://www.oeaw.ac.at/imba/scientific-facilities/stem-cell-core-facility/). C.B. was a member of the VBC doctoral program. D.C. is a PhD student within the European School of Molecular Medicine (SEMM). We thank K. Chylinski, I. Bilusic Vilagos, the VBCF Protein Technology facility (https://www.viennabiocenter.org/facilities/protein-technologies/) and the Molecular Biology Service Department (http://cores.imp.ac.at/molecular-biology-service/) for technical support. We thank T. Oertner and S. Wieger from the Institute of Synaptic physiology (Hamburg, ZMNH) for providing Chrimson plasmids. Work in V.K.’s laboratory is funded by the Human Technopole Early Career Fellowship Programme (HT-ECF Programme). Work in J.A.K.’s laboratory is supported by the Austrian Academy of Sciences, the Austrian Science Fund (FWF) Special Research Programme F7804-B and Stand-Alone grants P 35680 and P 35369, the Austrian Federal Ministry of Education, Science and Research, and the Simons Foundation Autism Research Initiative (SFARI, 724430).
Author information
Authors and Affiliations
Contributions
C.B., V.K. and J.A.K. conceived the project and wrote the manuscript with comments from all the authors. C.B. performed experiments, collected and analyzed the data with help and supervision from V.K. V.K. conceived, generated and provided the EMX1-mNeonGreen cell line, collected and analyzed the data. T.R.B. performed bioinformatic analysis with input from C.B. and V.K. T.W. conceived and produced the PDMS molds, providing inputs and optimizations, with the help of B.S. S.J.G. performed the electrophysiology experiments and analysis and built the optical and electrical instrumentation required for it. S.B. and J.A.B. conceived, generated and provided the Syn1:ChrimsonR cell line. C.L. set up and helped perform the method for scRNA-seq. C.E.V., D.C., C.C. and N.C. designed the analytical strategy for the scRNA dataset and contributed to scRNA results, discussion and writing. C.E.V., D.C. and G.T. developed the interpretive framework of the study for scRNA-seq data. C.E.V. and G.T. jointly supervised the scRNA-seq data analysis. J.A.K. and V.K. acquired funding.
Corresponding authors
Ethics declarations
Competing interests
J.A.K. is inventor on a patent describing cerebral organoid technology and cofounder and scientific advisory board member of a:head bio AG. J.A.B. is an inventor on a patent describing dorsal–ventral forebrain organoid fusion technology and is the chief scientific officer of a:head bio AG. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Nina Vogt, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Generation of the CAG > FGF8 reporter line.
a) Schematic diagram of CAG > FGF8 reporter (top). Genotyping of the selected clones (bottom) shows the three heterozygotes selected. The PCR was designed to amplify a wild type AAVS1 (WT) amplicon, a left homology arm amplicon (LA), a right homology arm amplicon (RA), and an inner part (IP). b) RT-qPCR analysis of pluripotency markers and differentiation gene T/BRA in the selected CAG > FGF8 expressing clones and parental wild-type cells. Values are mean ± SD (n = 4 biological replicates for wt; n = 3 biological replicates, one for each CAG > FGF8 clone). P-values resulting from one-way ANOVA (Tukey’s multiple comparisons test) among the different lines are: OCT4 wt vs. OCT4 CAG > FGF8, p = 0.9166; NANOG wt vs. NANOG CAG > FGF8, p = 0.9841; SOX2 wt vs. SOX2 CAG > FGF8, p = 0.9997; TBRA wt vs. TBRA CAG > FGF8, p > 0.9999. c) Box plots showing RT-qPCR analysis of FGF8 and WNT1 genes in the selected clones of the CAG > FGF8 line compared to wt. Whiskers are min to max, boxes represent the 25th to 75th percentiles (Q1 to Q3); n = 6 from 3 independent clones. One-way ANOVA analysis is p < 0.0001 for FGF8 expression and p > 0.9999 for WNT1 expression. d) Quantification of FGF8 protein levels in cell lysates (CELLS) and supernatants (SUP) measured by ELISA assay. Values are mean ± SD, n = 6 from 3 independent clones; P-values from two-sided unpaired t-tests are: CELLS wt vs CELLS CAG > FGF8, p = 0.0004; SUP wt vs SUP CAG > FGF8, p = 0.0823.
Extended Data Fig. 2 Further characterization of round assembloids.
a) Representative images of 4 div embryoid bodies co-cultured with OrEBs containing cells expressing FGF8 and tdTomato mixed at different percentage (0%, 1%, 10%), immunostained for the FGF8 downstream target ETV1 (green) (scale bars 50 µm, the SP8 > GFP organoid is outlined with a dashed contour). The experiment has been repeated 4 times with organoids derived from three independent clones. b) Immunostaining of 60 div organoids generated with the SP8 > GFP transgenic line and co-cultured with OrEBs in Fig. 1d–g, stained for the rostral marker LMO4 (scale bars 500 µm in the main panels, 50 µm for the insets). The SP8 > GFP organoid is outlined with a dashed contour. c) Box plots showing the fraction of LMO4+ cells normalized to total DAPI+ cells in 0%, 1% and 10% FGF8 conditions at 60 div. Whiskers are min to max, boxes represent the 25th to 75th percentiles (Q1 to Q3); n = 8 organoids. P-values from one-way ANOVA (Tukey’s multiple comparisons tests) are: 0% vs 1%, p = 0.0019, 0% vs 10%, p < 0.0001, 1% vs 10%, p < 0.0001. d) Immunostaining of organoids generated with the SP8 > GFP transgenic line and co-cultured with OrEBs as described in Fig. 1d–g for 60 days, stained for the neural marker Sox1 and DAPI. Scale bars 500 µm. e) Quantification of the number of Sox1+ cells over total (DAPI + ) cells. Data show mean ± SD (n = 2 assembloids for 0%, n = 4 assembloids for 1%, n = 3 assembloids for 10%). P-values for each comparison resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: 0% vs 1%, p = 0.113; 0% vs 10%, p = 0.2171; 1% vs 10%, p = 0.8893. f) RT-qPCR analysis of anterior (PAX6, SIX3) and optic cup (SNAI2, PAX3, OTX2, LHX2, RAX) markers from SP8 > GFP organoids severed from the co-culture experiment at 60 div. Data is Log of expression over TBP, shown as mean ± SD; n = 6 for 0% condition, n = 5 for 1% and 10% condition, from 3 independent clones. P-values from one-way ANOVA (Tukey’s multiple comparisons tests) are: RAX 0% vs. RAX 1%, p = 0.0718; RAX 0% vs. RAX10%, p = 0.0003; RAX 1% vs. RAX 10%, p = 0.0263; all the other comparisons are p < 0.0001. g) GFP intensity per segment (P, proximal; M, medial; D, distal) in the 1% FGF8 condition from experiments shown in panel b. Each segmented line represents an individual organoid. n = 7 from 3 clones; ns, p = 0.5783 one-way ANOVA. Ns, non-significant. a.u., arbitrary units.
Extended Data Fig. 3 Further characterization of elongated cortical assembloids.
a-b) Representative images of elongated assembloids (8 div) properly embedded maintaining elongated shape (a) compared to elongated assembloids from the same batch without matrigel, shrinking without spatial constrain (b). c) Length of elongated assembloids after different days in vitro (div, div1 n = 8, div7 n = 6, div30 n = 4, div60 n = 24, div120 n = 22 assembloids grown from 3 independent clones). P-values resulting from one-way ANOVA Tukey’s multiple comparisons tests) among the different lines are: div1 vs div7, p = 0.0045; div1 vs div30, p = 0.9999; div7 vs div30, p = 0.0366; div7 vs div60, p = 0.9998; div7 vs div120, p = 0.51; div30 vs div60, p = 0.0049; div60 vs div120, p = 0.1825; div1 vs div60, div1 vs div120, div30 vs div120, p < 0.0001. d) RT-qPCR analysis of neural markers (SOX2, SOX1, SP8 and LMO4), ventral marker NXK2.1 and differentiation markers SNAI1 and T-BRA, in elongated assembloids (eOrg) compared to round organoids (rOrg), after treatment with high FGF8 as described in Fig. 1. Values are mean ± SD, n = 3 in three lines (hESC H9, H1 and iPSC 178/5). P-values resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: eORGSOX2 d10 vs. rORGSOX2 d10, p = 0.0024; eORGSOX2 d30 vs. rORGSOX2 d30, p = 0.3144; eORGSOX2 d60 vs. rORGSOX2 d60, p = 0.9434; eORGSOX1 d10 vs. rORGSOX1 d10, p = 0.0009; eORGSOX1 d30 vs. rORGSOX1 d30, p = 0.1573; eORGSOX1 d60 vs. rORGSOX1 d60, p = 0.7266; eORGSP8 d10 vs. rORGSP8 d10, p = 0.0006; eORGSP8 d30 vs. rORGSP8 d30, p = 0.1891; eORGSP8 d60 vs. rORGSP8 d60, p = 0.8004; eORGLMO4 d10 vs. rORGLMO4 d10, p = 0.0114; eORGLMO4 d30 vs. rORGLMO4 d30, p = 0.8378; eORGLMO4 d60 vs. rORGLMO4 d60, p = 0.9821; eORGNKX2.1 d10 vs. rORGNKX2.1 d10, p = 0.0008; eORGNKX2.1 d30 vs. rORGNKX2.1 d30, p = 0.1345; eORGNKX2.1 d60 vs. rORGNKX2.1 d60, p = 0.9036; eORGSNAI1 d10 vs. rORGSNAI1 d10, p = 0.0026; eORGSNAI1 d30 vs. rORGSNAI1 d30, p = 0.6125; eORGSNAI1 d60 vs. rORGSNAI1 d60, p = 0.96; eORGT-BRA d10 vs. rORGT-BRA d10, p = 0.6815; eORGT-BRA d30 vs. rORGT-BRA d30, p = 0.3026; eORGT-BRA d60 vs. rORGT-BRA d60, p = 0.1297. e) Fraction of TUNEL+ cells normalized to total DAPI+ cells in elongated versus conventional round organoids (div, days in vitro). N = 36 insets for 4 div elongated, n = 33 for 4 div Round, n = 122 for 7 div elongated, n = 37 for 7 div Round, n = 81 for 30 div elongated, n = 82 for 30 div Round organoids from 3 lines per condition. P-values resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: 4 div eOrg vs. 4 div rOrg, p < 0.0001; 7 div eOrg vs. 7 div rOrg, p = 0.31; 30 div eOrg vs. 30 div rOrg, p = 0.0115. f) Box plots showing the fraction of TUNEL+ cells normalized to total DAPI+ cells in proximal and distal insets of elongated assembloids (n = 18 insets for 4 div Proximal and Distal, n = 32 for 7 div Proximal, n = 33 for 7 div Distal, n = 40 for 30 div Proximal, n = 42 for 30 div Distal; organoids are grown from 3 lines per condition). P-values resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: 4 div proximal vs. 4 div distal, p = 0.6097; 7 div proximal vs. 7 div distal, p = 0.6559; 30 div proximal vs. 30 div distal, p = 0.9897. g) Representative images of the OrEB localization throughout elongated assembloids growth quantified in Fig. 2c (scale bars 500 µm). h) Top, representative images of proximal and distal CTIP2 staining (scale bars 50 µm). Bottom, fraction of CTIP2+ cells normalized to total DAPI+ cells in proximal and distal insets of controls (conCA) and polCAs at 60 div (n = 50 insets for P and D conCAs, n = 40 insets for P and D polCAs from 3 organoids). P-values resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: proximal conCA vs. distal conCA, p = 0.8204; proximal polCA vs. distal polCA, p < 0.0001. i) Top, representative images of proximal and distal LMO4 staining (scale bars 50 µm). Bottom, fraction of LMO4+ cells normalized to total DAPI+ cells in proximal and distal insets of control(tdt) and polCAs at 60 div (n = 61 insets for P and D from 6 conCAs, n = 51 insets for P and D from 4 polCA). P-values resulting from one-way ANOVA (Tukey’s multiple comparisons tests) are: proximal conCA vs. distal conCA, p > 0.9999; proximal polCA vs. distal polCA, p < 0.0001. ns, non-significant. All whiskers are min to max, boxes represent the 25th to 75th percentiles (Q1 to Q3) and lines indicate median. j-j’) Images of immunostained longitudinal sections of elongated assembloids at 60 div (scale bars 500 µm).
Extended Data Fig. 4 Additional data related to RNA-seq transcriptome.
a) PCA analysis using RUVseq corrected variance-stabilizing transformations (VST) expression values of top 500 most variably expressed genes; proximal and medial segment of the outlier polCA are labelled in black. b) Matrix reporting the intersection among differentially expressed genes with Log fold change (LFC) absolute value of ≥0 from pairwise comparison. Adjusted p-value is < 0.05 (DESeq2 pipeline). c) Voxhunt analysis shows comparison with different brain regions of mouse fetal brain at E13 per polCA or control segment. d) Expression data from RNA-seq from control segments (Piece1-3, as P1-P3), proximal (P), medial (M) and Distal (D) segments from wild type polCA, and mutant polCA segments (PMut, MMut and DMut) are shown. Individual samples represent segments from control elongated organoids (n = 4), wild type polCA (n = 6) and mutant polCA (n = 3) grown from three independent mutant clones. TPM, transcripts per Million. Whiskers are min to max, boxes represent the 25th to 75th percentiles (Q1 to Q3) and lines indicate median. e) Hierarchical clustering of samples with uncertainty estimates of pvclust (bootstrapped clustering). Approximately unbiased (AU, red) and bootstrap (BP, green) p-values are shown for each cluster. Edge hierarchical number is indicated in grey; distance is correlation and cluster method is average. f) Heatmap showing gene expression of the DE genes in polCAs segments from Fig. 3d, across the control samples (genes are plotted in the same order as in Fig. 3d for comparison).
Extended Data Fig. 5 Further analysis of polCA cell composition.
a) UMAP embedding for the scRNA-seq dataset containing cells derived from dissections of three segments of polCA and control counterparts annotated by stress levels based on Gruffi analysis. b) Sankey plot showing similar mapping of cells from individual segments of both control and polCA to stressed cells according to Gruffi’s results. c) Box plots showing the number of cells expressing FGF8 normalized by the total number of cells per segment for the three replicates. The box displays the median, the inter-quartile range, the minimum and maximum values for each segment. d) Dot plot for the top five markers for each cluster, genes with logFC> 3 are shown. LogFCs colors are scaled by gene. e) UMAP embedding plots colored by expression levels of markers of radial glia (NES, SOX2, VIM), cycling radial glia (TOP2A, MKI67), Cajal-Retzious cells (RELN), neurons (DCX, TUBB3), interneuron progenitor cells (DLX6-AS1, GAD2), LGE-derived progenitors (GSX2), MGE-derived progenitors (NKX2.1), retinal progenitor cells (RORB, VSX2), stress responsive cells (GOLGA4), endothelial cells (DCN, BGN, COL1A2), BMP responsive cells (TTR, RSPO2, LMX1A, MSX1), cilium bearing cells (PCP4, NPHP1), oligodendrocytes (OLIG1, OLIG2), astrocytes (GFAP, AQP4) and microglia (AIF1, CD68).
Extended Data Fig. 6 Correlation analysis with fetal brain region-specific brain data.
a) Correlation plots of various cell populations found in cortical organoids generated by Velasco et al. (ref. 92) or Kanton et al. (ref. 93) against signatures extracted from cycling cells, ventricular radial glia (vRG) and excitatory maturing neurons (ExM) derived from forebrain, midbrain and hindbrain single cell RNA-seq datasets of human developing brains at various gestational weeks (gw, Supplementary Data 7). b) Correlation plots of various cell populations profiled in the current study against the in vivo signatures validated in a. CFuPN, corticofugal projection neurons; CPN, cortical projection neurons; cN, cortical neurons; IP, intermediate progenitors.
Extended Data Fig. 7 Further analysis of RGC and ExN composition.
a) UMAP embedding of cells colored by organoid type. b) Fraction of cells per annotated Leiden cluster. Number of cells is normalized by total cells in each organoid type. c) Fraction of cells per annotated Leiden cluster. Number of cells is normalized both by total cells in each segment/piece. d) UMAP embedding of cells colored by organoid. e-f) Top 15 GO terms associated to differentially expressed genes in RGC (e) or ExN (f). Ranked p-values were calculated using EnrichR.
Extended Data Fig. 8 Description of the PFC-SS-T model.
a-b) PCA embeddings of metacells of the three cell populations extracted from prefrontal cortex (PFC), somatosensory cortex (SS) and temporal cortex (Temp) of the human fetal brain37 (a) or from distinct segments of organoids (b) showing segregation of domain-associated identities along the PC1. PCA was computed on highly variable genes. PCA data are oriented to have prefrontal (a) or proximal (b) embeddings on the negative loading side (left side) of PC1. Each dot is a metacell calculated from cycling progenitors (left panels), progenitors (middle panels) and excitatory neurons (right panels). c) Number of coherent genes between fetal and organoid populations among the top 50 negative and positive PC1 loadings, where PFC or proximal metacells, and Temp or medial-distal metacells are located respectively. Data are shown per individual population. d) Genes following P low < M < D high trend (top) or P high > M > D low trend (bottom) for each cell type. Empty dot: no trend, Filled dot: expected trend. In bold, genes with a similar trend in all three cell types.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15, Supplementary Note and Supplementary Tables 1–4.
Supplementary Data 1
Differential expression of polCA and control segments in bulk transcriptome.
Supplementary Data 2
List of rostral and caudal markers.
Supplementary Data 3
Expression modality of rostral and caudal genes in bulk transcriptome (source data related to Supplementary Figs. 9 and 12).
Supplementary Data 4
Source data related to Supplementary Fig. 10f.
Supplementary Data 5
Source data related to Supplementary Fig. 11.
Supplementary Data 6
Cell-type markers. Markers for each Leiden cluster obtained through two-tailed Wilcoxon rank-sum test (logFC > 3, Benjamini–Hochberg < 0.05).
Supplementary Data 7
Regional signatures per cell type and age showing top markers for five genes distinguishing forebrain, midbrain and hindbrain for each of the three cell types analyzed (cycling progenitors, vRG and excitatory neurons) at gestational weeks 8.5, 11.5, 12 and 14.5 from ref. 45.
Supplementary Data 8
a, PFC–SS–Temp gradient genes from three-point model for fetal cycling progenitors (likelihood-ratio test Bonferroni < 0.001). b, PFC–SS–Temp gradient genes from three-point model for fetal progenitors (likelihood-ratio test Bonferroni < 0.001). c, PFC–SS–Temp gradient genes from three-point model for fetal neurons (likelihood-ratio test Bonferroni < 0.001).
Supplementary Data 9
Pseudobulk-based differential expression for proximal versus distal, and medial versus distal segments in cycling radial glia (a), non-cycling radial glia (RG, b) and excitatory neurons (ExN; likelihood-ratio test, FDR < 0.05, absolute logFC > 1.5. Source Data related to Supplementary Fig. 14.
Supplementary Data 10
Files for polCA and master negative molds for the fabrication of 15-mm polCA molds.
Supplementary Code 1
Fiji macro code for polCA straightening.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data and microscopy image.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig./Table 1
Statistical source data.
Source Data Extended Data Fig./Table 2
Statistical source data.
Source Data Extended Data Fig./Table 3
Statistical source data and unprocessed images.
Source Data Extended Data Fig./Table 4
Statistical source data and code.
Source Data Extended Data Fig./Table 5
Statistical source data.
Source Data Extended Data Fig./Table 6
Statistical source data.
Source Data Extended Data Fig./Table 7
Statistical source data.
Source Data Extended Data Fig./Table 8
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bosone, C., Castaldi, D., Burkard, T.R. et al. A polarized FGF8 source specifies frontotemporal signatures in spatially oriented cell populations of cortical assembloids. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02412-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41592-024-02412-5
- Springer Nature America, Inc.