
Abstract
Indigenous chickens predominate poultry production in Africa. Although preferred for backyard farming because of their adaptability to harsh tropical environments, these populations suffer from relatively low productivity compared to commercial lines. Genome analyses can unravel the genetic potential of improvement of these birds for both production and resilience traits for the benefit of African poultry farming systems. Here we report whole-genome sequences of 234 indigenous chickens from 24 Ethiopian populations distributed under diverse agro-climatic conditions. The data represents over eight terabytes of paired-end sequences from the Ilumina HiSeqX platform with an average coverage of about 57X. Almost 99% of the sequence reads could be mapped against the chicken reference genome (GRCg6a), confirming the high quality of the data. Variant calling detected around 15 million SNPs, of which about 86% are known variants (i.e., present in public databases), providing further confidence on the data quality. The dataset provides an excellent resource for investigating genetic diversity and local environmental adaptations with important implications for breed improvement and conservation purposes.
Measurement(s) | genome |
Technology Type(s) | DNA sequencing |
Factor Type(s) | animal population |
Sample Characteristic - Organism | Gallus gallus |
Sample Characteristic - Location | Ethiopia |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.16999891
Similar content being viewed by others

Background & Summary
Poultry farming constitutes an important economic activity across Africa, providing a livelihood for millions of people. However, the lion’s share of the poultry production in most countries still comes from smallholder backyard indigenous poultry reared under scavenging or semi-scavenging conditions, with no or limited human intervention (e.g., secured sheltering at night, supplementary feeding, or vaccination)1,2. Ethiopia is one of the sub-Saharan African countries where chicken farming plays a crucial role in the country’s sociocultural context and economy, with ~97% of the production still coming from “extensive” farming practice of local birds.
Domestic chickens were originally introduced into Ethiopia from Asia from around 3000 years ago3,4. Since their introduction, chicken populations have been dispersed throughout the country and, over time, have adapted to thrive in its diverse agro-ecologies. These birds, now considered as indigenous, show greater resistance to various local poultry diseases and parasites compared to exotic and commercially improved chickens. Due to their superior adaptability to local tropical environmental conditions as well as their foraging ability and broodiness, these indigenous birds are often preferred by smallholder farmers for backyard rearing2,5,6. However, in the absence of proper management practices or any systematic selection efforts, local birds generally show poorer productivity but higher survivability compared to the commercial counterparts. Their untapped genetic potential can be utilized for improving their performance.
Genome analyses can unravel the genetic diversity of indigenous chicken populations and provide the basis for genetic improvements for better production and performance. Moreover, genome analysis of populations from different agro-ecological zones can elucidate the genetic basis of local environmental adaptation. Resilient genotypes, identified from such studies, can then be selected for or introgressed in improved productive breeds for superior performance under local climate. The Ethiopian landscape can be considered a microcosm of different agro-ecologies encountered in Africa due to extreme variations in its altitudinal topography and rainfall pattern. This has given rise to diverse agro-climate zones in the country, ranging from hot-arid and hot-humid to cold-humid and cold-arid7. Therefore, genomic analysis of Ethiopian chicken populations is particularly pertinent for elucidating their local adaptation.
This article reports whole genome sequencing data from hundreds of indigenous chickens (n = 234), sampled from 24 different Ethiopian villages or populations distributed under diverse agro-ecological and climatic conditions [Table 1; also see Fig. 1A,B and supplementary Table S1 in the study by Gheyas et al.8]. The study also reports about 15 million Single Nucleotide Polymorphisms (SNPs) detected by mapping the sequencing data against the chicken reference genome (GRCg6a; https://www.ncbi.nlm.nih.gov/assembly/?term=GCA_000002315.5). Sequencing has been performed at a very high coverage (average 57X), increasing the power and resolution of genomic analyses. Although most of the reported variants are already known (only 14% are novel), the associated VCF file (submitted to European Variant Archive) shows genotype data for individual samples; therefore it offers an excellent resource for a variety of population genetics analyses. Some of these sequences and variant data have been used in a recent study to elucidate the genome-environmental adaptation in Ethiopian chickens8.

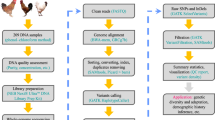
Overview of the sequence mapping, variant calling and variant filtration pipeline. The pipeline follows GATK best practice protocol for germline short variant discovery18.
The data are expected to have many utilities, ranging from exploring genetic diversity, identifying signatures of positive selection, analysing genome-environment associations, finding genetic variants from regions of interests (e.g., within or near candidate genes or QTLs associated with disease and production traits), exploring different types of genetic variants (e.g., small insertions/deletions, structural variants, avian retroviral elements), and for developing tools for genomic analysis (e.g., high or low density SNP genotyping arrays for use in breeding programmes). Furthermore, the data represent the largest number of indigenous chicken samples sequenced from an African country. Only a few studies have previously reported such large scale sequencing of chicken samples but none generated such large scale African data9,10,11,12. These data are therefore a rich addition to global chicken genome sequence databases and can be used in conjunction with sequencing data from other countries/regions around the globe for studying demographic and domestication histories in chicken.
Methods
Chicken sampling
Chicken sampling considered different agro-climatic conditions and geographic regions of Ethiopia. Sampling of local foraging chickens was performed from 24 villages or ‘kebeles’ from across six regional states – Afar, Amhara, Gumuz, Oromia, SNNPR (Southern Nations, Nationalities and Peoples’ Region), and Tigray, representing diverse agro-climatic and ecological conditions observed in Ethiopia. Each village was considered as a separate population. To capture genetic diversity within populations, 8 to 10 chicken samples were collected from each village (Table 1). Sampling was performed by drawing blood (50–250 µl) from the wing vein of each bird with syringes using cryotubes filled with 1.5 ml absolute ethanol (100%) following the guidelines available at https://www.sheffield.ac.uk/nbaf-s/protocols_list. The samples consisted of 146 female and 88 male birds (total 234) and varied in their age (4–30 months; average 10.3 months) and body weight (0.6–2.6 kg, average 1.27 kg). The samples were collected with the logistical support and agreement of the Ethiopian Ministry of Agriculture and Ethiopian Institute of Agricultural Research (EIAR). All animal works were approved by the Institutional Animal Care and Use Committee of the International Livestock Research Institute (IREC2017-26). The sample information has been submitted to the European Nucleotide Archive (ENA) under the study accession PRJEB3927513 (see Online-only Table 1 for details about the samples).
Genomic DNA isolation and quality control
All the collected blood samples were processed for DNA extraction at the BecA-ILRI Hub facility, Nairobi, Kenya (http://hub.africabiosciences.org/) using the Qiagen DNeasy blood and tissue kit protocol (https://www.qiagen.com/ca/resources/download.aspx?id=63e22fd7-6eed-4bcb-8097-7ec77bcd4de6&lang=en). DNA concentration was evaluated by spectrophotometry (Thermo Scientific NanoDrop spectrophotometer 2000c) and the integrity of DNA was confirmed by agarose gel electrophoresis. The genomic DNA (gDNA) from each sample was then normalized to a final volume of 100 µl and final concentration of 50 ng/µl and was sent to Edinburgh Genomics, UK for whole genome sequencing (WGS). At Edinburgh Genomics, gDNA samples were re-evaluated for quantity and quality using an AATI Fragment Analyzer and the DNF-487 Standard Sensitivity Genomic DNA Analysis Kit https://www.agilent.com/cs/library/usermanuals/public/quick-guide-dnf-487-genomic-dna-kit-SD-AT000137.pdf. The AATI ProSize 2.0 software (https://dna.biotech.iastate.edu/fragmentanalyzer.html) provided a quantification value and a quality (integrity) score for each gDNA sample. Samples with a score >7 passed quality control. Based on the quantification results, gDNA samples were pre-normalised to fall within the acceptable range for library preparation.
Sequence library preparation and quality control
Next Generation sequencing libraries were prepared using Illumina SeqLab specific TruSeq Nano High Throughput Library preparation kits in conjunction with the Hamilton MicroLab STAR and Clarity LIMS X Edition. The normalized gDNA samples were sheared to a 450 bp mean insert size using a Covaris LE220 focused-ultrasonicator. The inserts were ligated with blunt ended, A-tailed, size selected TruSeq adapters and enriched using eight cycles of PCR amplification. The libraries were evaluated for mean peak size and quantity using the Caliper GX Touch with a HT DNA 1k/12 K/HI SENS LabChip and HT DNA HI SENS Reagent Kit. The libraries were normalised to 5 nM using the GX data and the actual concentration was established using a Roche LightCycler 480 and a Kapa Illumina Library Quantification kit and Standards (https://rochesequencingstore.com/wp-content/uploads/2017/10/KAPA-Lib-Quant-ILMN_9.17-IfU_1.pdf).
Sequencing
The libraries were denatured, and pooled in groups of eight for clustering and sequencing using a Hamilton MicroLab STAR with Genologics Clarity LIMS X Edition. Libraries were clustered onto HiSeqX Flow cells v2.5 on cBot2s and the clustered flow cells were transferred to a HiSeqX for sequencing using a HiSeqX Ten Reagent kit v2.5. Sequencing was performed in paired-end mode with read length of 150 bp.
Sequencing data processing, mapping and variant calling
Demultiplexing was performed using bcl2fastq (v2.17.1.14)14, allowing a single mismatch when assigning reads to barcodes. Adapters (Read1: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA, Read2: AGATCGGAAGA GCGTCGTGTAGGGAAAGAGTGT) were trimmed during the demultiplexing process. Sequencing data quality was checked using the FASTQC package (v0.11.5)15. FASTQC reports for all samples were aggregated in a single report by the MultiQC package16 for easy review of sequence quality. No quality-based trimming was performed on the sequence reads prior to mapping and sequencing data from all samples were processed.
Sequence reads were mapped against the latest version of chicken reference genome (GCA_000002315.5_GRCg6a) using the BWA-mem (v0.7.15) algorithm17. The resulting SAM/BAM files from the mapping step underwent a series of further processing steps, including coordinate sorting (using the SortSam function in Picard v2.9.2), duplicate reads marking (using MarkDuplicates function in Picard) and Base Quality Score Recalibration (BQSR) using GTAK v3.8-0. The final recalibrated BAM files were then used for variant calling. Figure 1 shows an overview of the mapping and variant calling steps.
SNP calling was performed following the GATK best practice protocol for “Germline short variant discovery”18 using the HaplotypeCaller function on individual samples followed by joint genotyping (using GenotypeGVCFs function) of the samples. Variant filtration was performed by applying the Variant Quality Score Recalibration (VQSR) approach19 in GATK (v 3.8-0) using about one million validated SNPs20 as a training and true set, and over 20 M known chicken SNPs from the Ensembl database as known variants. During the VQSR step the following annotations or context statistics were considered: read depth (DP), variant quality by depth (QD), root mean square of mapping quality (MQ), mapping quality rank sum test statistic (MQRankSum), read position rank sum test statistic (ReadPosRankSum), and strand bias statistics (FS and SOR). A tranche sensitivity threshold of 99% was applied for filtering variants. The “Code availability” section below shows the specific codes for each mapping and variant-calling step. As the final quality control of the called variants, any SNPs with a missing genotype rate of more than 20% across the samples were filtered out using VCFtools (option – max-missing 0.8).
Data Records
The raw full-length sequencing data (in FASTQ format) have been submitted to the European Nucleotide Archive (ENA) under the accession number PRJEB3927513. The VCF file of ~15 M SNPs detected from this dataset has been deposited in the European Variation Archive (EVA) with the accession number for Project: PRJEB4649421 and Analyses: ERZ2899764.
Technical Validation
Quality control of sequencing data
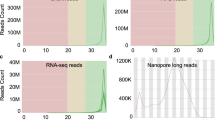
For each sample, 41 Gb to 148 Gb sequencing yield (number of bases generated) was obtained, of which 74–83% of the bases (average 79%) had a minimum Phred scaled quality score of 30, indicating expected base calling accuracy of 99.9% (Fig. 2). The average estimated coverage for the samples varied from 38X to 139X (average across all samples 57X) (Fig. 2). Figure 3 shows selected features from FASTQC reports regarding sequencing quality (consolidated for all samples by the MultiQC package). This confirmed overall high quality sequencing data. Although Fig. 3b shows “Fails” signal for many reads, this should not be a matter of concern. All these “Fails” signals are associated with Read2 of the paired reads. Typically, Read2 often has a lower average quality than Read122. A gradual drop in sequencing quality towards the end of the reads is also typical and expected of Illumina sequencing. It is important to note that Fig. 3d confirms a high average quality score for all reads. The mapping success rates of the sequence reads against the chicken reference genome were very high – 98.2% to 99.5% - which further confirmed the high quality of the sequencing data.

Boxplots showing the distributions of sequencing yield, yield Q30 and estimated coverage for Ethiopian chicken samples (n = 234).

Quality control metrics from FastQC analysis of sequencing data. The metrics from all sequence FASTQ files (total 540) are combined using the MultiQC package.
Quality control of SNP data
Joint genotyping of all samples originally identified about 25 M SNPs. To ensure variant quality and minimize false positives, VQSR filtration was applied. By using machine learning algorithms, the VQSR method clusters the called variants based on annotation profiles of a set of known true positive SNPs (training set) in the detected set and calculates, for each variant, a new score called VQSLOD (https://gatk.broadinstitute.org/hc/en-us/articles/360035531612-Variant-Quality-Score-Recalibration-VQSR-). For filtration of the variants, we applied a VQSLOD threshold that retained 99% of the training variants. This filtration retained about 19 M SNPs. Further filtration based on missing genotypes (removed any SNPs with missing rate >20%) retained ~15 M good quality SNPs. About 86% of these variants have already been reported in the public databases. This provides extra confidence in the validity of the detected SNPs.
Transition and transversion ratio (Ti/Tv) is used as a quality control metric for SNP calling. For whole genome sequencing data, the typical value is ~223. A higher ratio generally indicates better SNP calling unless the ratio is too high (>4)24. We obtained a Ti/Tv ratio of 2.38 for 19 M SNPs after VQSR filtration and a ratio of 2.5 for the 15 M final set.
Table 2 and the heat maps of SNP density across different chromosomes in Fig. 4 show a good representation of most chromosomes and regions except some microchromosomes (e.g., chr16, 22, 25, 30–33) and the sex chromosomes (Fig. 4). Chromosome 16 is known to have a high repeat content25 whereas most microchromosomes have higher GC contents26; both causing difficulty in sequencing and mapping. The detected SNPs also had a good representation of different annotation categories in relation to their positions within or outside genes (Table 3).

Chromosome-wise SNP distribution heat map across the Ethiopian indigenous chicken genomes based on 15 M SNPs. X-axis denotes the chromosome size in base pairs (bp) and Y-axis the chromosome number. The SNP count was calculated for 10 kb non-overlapping windows.
Code availability
Most of the data analyses were completed by standard bioinformatic tools running on the Linux system. The version and code/parameters of the main software tools are described below.
(1) BWA-mem (v0.7.15); code for mapping reads:
bwa mem -t 1 -M -R
“@RG\tID:${SAMPLE}\tSM:${SAMPLE}\tPL:Illumina\tLB:${SAMPLE}\tPU:unkn-0.0” ${REF} ${READS_1} ${READS_2} > ${SAMPLE}.sam
(2) Picard (2.9.2): code for sorting sam file and converting to bam:
java -jar picard.jar SortSam I = ${SAMPLE}.sam
O = ${SAMPLE}_sorted.bam SORT_ORDER = coordinate
TMP_DIR = tmp_${SAMPLE}
(3) Picard (2.9.2): code for marking duplicate reads:
java -jar picard.jar MarkDuplicates
I = ${SAMPLE}_sorted.bam O = ${SAMPLE}_mdup.bam CREATE_INDEX = true M = ${SAMPLE}_mdup_metrics.txt
TMP_DIR = tmp_${SAMPLE}
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP = 4000
OPTICAL_DUPLICATE_PIXEL_DISTANCE = 2500
(4) GATK (3.8-0): codes for BQSR steps
# Analyse patterns of covariation in the sequence dataset
java -jar $gatk -T BaseRecalibrator -R ${REF} -I
${SAMPLE}_mdup.bam -knownSites ${KNOWNVAR} -o
${SAMPLE}_recal_data.table
# Analyse covariation post-recalibration
java -jar $gatk -T BaseRecalibrator -R ${REF} -I
${SAMPLE}_mdup.bam -knownSites ${KNOWNVAR} -BQSR
${SAMPLE}_recal_data.table -o ${SAMPLE}_post_recal_data.table
# Generate before/after plots
# Requires R packages gsalib, reshape and ggplot2 installed
java -jar $gatk -T AnalyzeCovariates -R ${REF} -before ${SAMPLE}_recal_data.table -after
${SAMPLE}_post_recal_data.table
-plots ${SAMPLE}_recalibration_plots.pdf
# Apply the recalibration to your sequence data
java -jar $gatk -T PrintReads -R ${REF} -I ${SAMPLE}_mdup.bam -
BQSR ${SAMPLE}_recal_data.table -o ${SAMPLE}_recal.bam
(5) GATK (3.8-0) Variant calling in GVCF mode by HaplotypeCaller
java -jar $gatk -T HaplotypeCaller -R ${REF} -I ${SAMPLE}_recal.bam
-o ${SAMPLE}.g.vcf.gz -ERC GVCF
(6) GATK (3.8-0) Joint genotyping of a cohort of samples
# used the --variant option as many times as needed to specify the gvcf files to be used for joint genotyping (the code below shows three samples only as example).
java -Xmx4g -jar $gatk -T GenotypeGVCFs -R ${REF} --variant
SAMPLE1.g.vcf.gz --variant SAMPLE2.g.vcf.gz --variant
SAMPLE3.g.vcf.gz -o ${COHORT}.vcf.gz -D ${KNOWNVAR}
(7) GATK (3.8-0) VQSR steps
# Variant recalibration step
java -Xmx4g -jar $gatk -T VariantRecalibrator -R ${REF}
-input ${COHORT}.vcf.gz
-resource:GRCg6a_dbsnp,known = true,training = false,truth = false,prior = 2.0 ${KNOWNVAR}
-resource:GRCg6a_validated_snp,known = false,training = true,truth = true,prior = 12 ${TRUEVAR}
-an DP -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR -mode SNP -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 -recalFile ${COHORT}.SNPs.recal.gz
-tranchesFile ${COHORT}.SNPs.tranches -rscriptFile ${COHORT}_recalSNPS.plots.R
# Apply Recalibration
java -Xmx4g -jar $gatk -T ApplyRecalibration -R ${REF} -input
${COHORT}.vcf.gz -mode SNP --ts_filter_level 99.0 -recalFile
${COHORT}.SNPs.recal.gz -tranchesFile ${COHORT}.SNPs.tranches -o
${COHORT}_recalSNPs_rawIndel.vcf.gz
References
Shapiro, B. I. et al. Ethiopia Livestock Sector Analysis. ILRI Project Report (Nairobi, Kenya: International Livestock Research Institute, 2017).
Bettridge, J. M. et al. The role of local adaptation in sustainable village chicken production. Nat. Sustain. 1, 574–582 (2018).
Woldekiros, H. S. & D’Andrea, A. C. Early evidence for domestic chickens (Gallus gallus domesticus) in the Horn of Africa. Int. J. of Osteoarchaeol. 27, 329–341 (2017).
Mwacharo, J. M., Bjornstad, G., Han, J. L. & Hanotte, O. The history of African village chickens: an archaeological and molecular perspective. Afr. Archaeol. Rev. 30, 97–114 (2013).
Getu, A. Review on Ethiopian poultry origin, domestication, classification and characterization of its production systems. Middle-East. J. of Sci. Res. 22(7), 1025–1032 (2014).
Padhi, M. K. Importance of indigenous breeds of bhicken for rural economy and their improvements for higher production performance. Scientifica 2016, Article ID 2604685 (2016).
Fazzini, M., Bixci, C. & Billi, P. in Landscapes and Landforms of Ethiopia, World Geomorphological Landscapes (ed. Billi, P.) Ch. 3 (Springer, Dordrecht 2015).
Gheyas, A. A. et al. Integrated environmental and genomic analysis reveals the drivers of local adaptation in African indigenous chickens. Mol. Biol. Evol. 38, 4268–4285 (2021).
Wang, M. S. et al. 863 genomes reveal the origin and domestication of chicken. Cell Res. 30, 693–701 (2020).
Gheyas, A. A. et al. Functional classification of 15 million SNPs detected from diverse chicken populations. DNA Res. 22, 205–217 (2015).
Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Luo, W. et al. Genome diversity of Chinese indigenous chicken and the selective signatures in Chinese gamecock chicken. Sci. Rep.-Uk 10, Article number: 14532 (2020).
European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB39275 (2021).
Illumina. bcl2fastq conversion user guide. Illumina http://www.bea.ki.se/documents/bcl2fastq.pdf (2013).
Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997v2 (2013).
Broad Institute. Best practices for variant calling with the GATK. Broad Institute https://www.broadinstitute.org/partnerships/education/broade/best-practices-variant-calling-gatk-1 (2015).
GATK. Variant Quality Score Recalibration. Broad Institute https://gatk.broadinstitute.org/hc/en-us/articles/360035531612-Variant-Quality-Score-Recalibration-VQSR (2017).
Kranis, A. et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 14, Article number: 59 (2013).
Eurpean Variation Archive. https://www.ebi.ac.uk/eva/?eva-study=PRJEB46494 (2021).
Tan, G., Opitz, L., Schlapbach, R. & Rehrauer, H. Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci.Rep.-Uk 9, Article number: 2856 (2019).
Wang, G. T., Peng, B. & Leal, S. M. Variant association tools for quality control and analysis of large-scale sequence and genotyping array data. Am. J. Hum. Genet. 94, 770–783 (2014).
Wang, J., Raskin, L., Samuels, D. C., Shyr, Y. & Guo, Y. Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 31, 318–323 (2015).
Guizard, S., Piegu, B., Arensburger, P., Guillou, F. & Bigot, Y. Deep landscape update of dispersed and tandem repeats in the genome model of the red jungle fowl, Gallus gallus, using a series of de novo investigating tools. BMC Genomics 17, 659, https://doi.org/10.1186/s12864-016-3015-5 (2016).
Burt, D. W. Chicken genome: current status and future opportunities. Genome Res. 15, 1692–1698 (2005).
Acknowledgements
The authors would like to acknowledge the Edinburgh Genomics Facility (Edinburgh, UK) for generation of the sequence data. This study was funded by the Bill and Melinda Gates Foundation (BMGF) and with UK aid from the UK Government’s Department for International Development (Grant Agreement OPP1127286) under the auspices of the Centre for Tropical Livestock Genetics and Health (CTLGH), established jointly by the University of Edinburgh, SRUC (Scotland’s Rural College), and the International Livestock Research Institute. The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the BMGF nor the UK Government. Adriana Vallejo is a Ph.D. student funded by a Vice-Chancellor Scholarship for Research Excellence International at the University of Nottingham and Administrative Department of Science, Technology and Innovation (Colciencias) – Colombian Government (Call 2015 N°728). We thank the CGIAR livestock program (CRP) for supporting the sampling component of the research. We also thank Prof. Nick Sparks (CTLGH, SRUC) for his valuable support in conducting this research.
Author information
Authors and Affiliations
Contributions
J.S., A.G. and O.H. conceived the research project. A.K., T.D. and O.H. led the collection of samples and population metadata. A.G., A.V. and A.K. performed the bioinformatic analyses. A.G. led the writing of the manuscript. All authors contributed critically to the drafts.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Gheyas, A., Vallejo-Trujillo, A., Kebede, A. et al. Whole genome sequences of 234 indigenous African chickens from Ethiopia. Sci Data 9, 53 (2022). https://doi.org/10.1038/s41597-022-01129-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01129-4
- Springer Nature Limited