
Abstract
Asian citrus psyllid (Diaphorina citri, D. citri) is the important vector of “Candidatus Liberibacter asiaticus” (CLas), associated with Huanglongbing, the most devastating citrus disease worldwide. CLas can affect endosymbiont abundance of D. citri. Here, we generated the high-quality gut endosymbiont metagenomes of Diaphorina citri on the condition of CLas infected and uninfected. The dataset comprised 6616.74 M and 6586.04 M raw reads, on overage, from CLas uninfected and infected psyllid strains, respectively. Taxonomic analysis revealed that a total of 1046 species were annotated with 10 Archaea, 733 Bacteria, 234 Eukaryota, and 69 Viruses. 80 unique genera in CLas infected D. citri were identified. DIAMOND software was used for complement function research against various functional databases, including Nr, KEGG, eggNOG, and CAZy, which annotated 84543 protein-coding genes. These datasets provided an avenue for further study of the interaction mechanism between CLas and D. citri.
Similar content being viewed by others


Background & Summary
Huanglongbing (HLB) is the most destructive citrus disease around the world. Asian citrus psyllid (ACP), Diaphorina citri Kuwayama, is the important carrier of Candidatus Liberibacter asiaticus (CLas), the causal agent of HLB. ACP has been found in many countries such as the Arabian Peninsula, Indian Subcontinent, South East and South West Asia, the USA, Central and South America, and the Indian Ocean islands of Mauritius and R´eunion1,2. CLas can infect many organs and tissues of ACP including the alimentary canal, hemolymph, hemocytes, muscle tissue, fat tissue, bacteriome, neural tissue, epidermis, reproductive organs, and salivary glands2, which affects not only the ACP’s physiological function, but also its endosymbiont communities3,4. However, it is little known about the relation between the change of endosymbiont communities and the physiological function alteration of ACP after being infected by CLas.

Flow chart used to process psyllid gut samples to get metagenomes.

Data preprocessing statistic map. Adapter: reads with adapters; Low Quality: low-quality reads; PolyA: number of reads containing polyA (%); N: single-ended reads containing more than 10% of N bases; Duplication: total length of the removed repeat reads sequence.
Analysis of the change of endosymbiont communities is the basis for understanding the interaction between CLas and ACP. Recent studies have shown that three predominant endosymbionts including Candidatus Carsonella ruddii, Candidatus Profftella, and Wolbachia were harbored in ACP5,6. These endosymbionts play different roles in participating in the ACP’s physiological activities. For instance, Carsonella has been considered to be a nutritional provider and Profftella has been verified to be a defender because of producing diaphorin6,7. Wolbachia titer has been demonstrated to have a positive correlation with CLas titer8. However, the key function of Wolbachia in ACP remains unclear. Data of endosymbiont communities in ACP have been reported based on 16 S rRNA sequencing9,10. Despite bacteria communities of genus level can be verified by conserved regions analysis in 16 S rRNA hypervariable regions, detailed composition on endosymbiont at species level comprised of bacteria, archaea, and Eukaryota and function characteristics are still needed. High-throughput metagenome sequencing technology can provide new views from a nearly complete composition of endosymbiont communities11.
Here, we characterized two endosymbiont metagenomes from the ACP strains before and after infection by CLas, which were named CLas-free and CLas-infected, respectively. Sample information was presented in Table 1. Raw data among 6175.81–7204.40 M were produced via metagenomic sequencing, in total. After filtration of the contaminated reads, clean data of six samples were between 6168.97 M and 7193.66 M. Comparatively, approximate 17.41–18.85 M data of microbiota from ACP were obtained using 16 s amplicon sequencing, suggesting that our data is more complete than the previous results10. Based on the metagenomes assembling, the number of scaffolds was separately distributed into 185390–196132 with 36.81–37.49% GC content in CLas-free and CLas-infected. The Q20 values and Q30 values were all above 96% and 90% respectively, and the N50 values were all above 1000 bp (Table 2). Table 3 generalized the overview of the composition of gut endosymbiont communities between CLas-free and CLas-infected and Table 4 summarized the gene number with function annotated after alignment in different functional databases. Figure 1 summarized the process of experiment.The endosymbiont orders with biomarkers were differed between the two groups, where Proteobacteria and Wolbachia pipientis were the most abundant in taxonomic phylum and species, respectively (Figs. 3, 4). KEGG orthology groups (KOs) relating to the category of metabolism were the most abundant in the intestinal microbiota of CLas-free and CLas-infected D. citri, such as biosynthesis and metabolism of amino acid, cofactors, vitamins, and glycan (Fig. 5). Five categories including the metabolism of other amino acids, cofactors, and vitamins were enriched in CLas-free D. citri samples and thirteen categories including immune and nervous systems were enriched in CLas-infected D. citri samples, indicating that two different gut endosymbionts of D. citri had a distinct contribution for host function compensation (Fig. 6). These KOs were unevenly found in the metagenomic of the taxa. For instance, KOs belonging to the category of metabolism were enriched in the bacteria of Firmicutes and Proteobacteria and the Eukaryota of Ascomycotab (Fig. 7).

The composition of microbiota at the phylum level. Taxonomic assignments of the 10 most abundant taxa are presented.

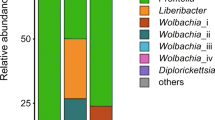
The composition of microbiota at species level. Taxonomic assignments of the 10 most abundant taxa are presented.

Hierarchically clustered heat map analysis of the highly represented KOs category of KEGG level 2.

LEfSe analysis of the category of KEGG level 2 that differ between HLB-free and HLB-infected D. citri. G1: HLB-free D. citri, G2: HLB-infected D. citri.

Heatmap of the distribution of KEGG. The horizontal axis represents 6 different kinds of KEGG, and the different colors of the vertical axis represent different endosymbiont taxonomic information.
In summary, these metagenomic data offer compelling evidence on the composition and function of intestinal endosymbionts from D. citri, which not only give further comprehension for gut-associated endosymbionts and host insect coevolution under distinguished ecological niches but also open a new avenue to pest management.
Methods
Psyllid sampling, tissue collection and DNA isolation
Adult D. citri newly emerged (five days old) was initially harvested from Citrus Tachibana uninfected CLas in 2020 at Guangxi province, China, and was successively reared on Murraya paniculata at Guangxi Special Crops Research Institute for more than 15 generations. All cages and experimental treatment were maintained on the conditions of 27 ± 1 °C, 60–70% RH, and at 14:10 h of photoperiod. It was confirmed by PCR detection that D. citri did not carry CLas.
A field population of D. citri newly emerged (five days old) infected CLas was collected from Guilin City, China, in 2022. Ninety adult psyllids from Citrus Tachibana infected CLas were randomly collected for calculating the infection rate. DNA extraction of one adult psyllid followed by Yu et al.12. with slight modifications. Briefly, genome DNA was extracted using the QIAGEN DNeasy Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Subsequently, HLB pathogen-specific primer set OI1/OI2c (forward primer 50-GCGCGTATGCAATACGAGCGGCA-30 and reverse primer 50-GCCTCGCGACTTCGCAACCCAT-30) were performed to amplify the target fragment. Afterward, the CLas infection rate was evaluated by agarose gel electrophoresis. When the CLas-infected proportion reached over 80%, D. citri was considered to be the CLas-infected population12.
To eliminate the host plant microbial endosymbionts contamination, a 6 h starvation treatment was necessary. For the metagenome sequencing, one hundred and fifty newly emerged (five days old) CLas-free and CLas-infected D. citri respectively were surface sterilized with 70% ethanol for 60 s and rinsed three times with sterile water13, with three biological replicates. Then all samples were dissected to obtain the intestinal tissue, which was homogenized with 200 ml sterile water, and frozen at - 80 °C before DNA extraction. The QIAGEN DNeasy Kit was used for microbial genomic DNA extraction. The DNA quality checking and concentrations quantification were performed by Agilent Bioanalyzer 2100 system. The high-quality DNA was sent to Novogene Company (Beijing, China) for metagenomics sequence analyses.
Metagenome sequence
NEBNext® UltraTM DNA Library Prep Kit for Illumina (NEB, USA) was used to construct the library. The genomic DNA was randomly sheared into short fragments which were end-repaired, A-tailed, and further ligated with an Illumina adapter. Then fragments with adapters were PCR amplified, size selected, and purified to complete library establishment. The library quality was checked with Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Massachusetts, USA). Library quantification and size distribution detected by real-time PCR and bioanalyzer. Quantified libraries were pooled and sequenced on Illumina PE150 platforms, according to effective library concentration and data amount required.
Readfq (V8, https://github.com/cjfields/readfq) was used for preprocessing raw data from the Illumina sequencing platform to obtain clean data for subsequent analysis14. Then MEGAHIT software (v1.0.4-beta) was used for assembly analysis of clean data, and scaftigs without N were obtained by breaking the resulted scaffolds from the N junction15,16. MetaGeneMark (V3.05, http://topaz.gatech.edu/GeneMark/) was used to perform ORF prediction for Scaftigs (> = 500 bp) of each sample14,17,18. For the ORF prediction results, CD-HIT software (V4.5.8, http://www.bioinformatics.org/cd-hit/) was used to eliminate redundancy and obtain the non-redundant initial gene catalogue19. Clean data of each sample were aligned with the initial gene catalog using Bowtie2 (Bowtie2.2.4, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) to calculate the number of reads of the genes on each sample alignment. The abundance of each gene in each sample was calculated based on the number of reads aligned and the length of gene20.
Metagenome annotation
DIAMOND21 (V0.9.9, https://github.com/bbuchfink/diamond/) was used for the alignment of unigenes sequences with those of bacteria, fungi, archaea, and viruses extracted from NCBI’s NR database (Version 2018-01-02, https://www.ncbi.nlm.nih.gov/), and LCA algorithm was adopted to determine the species annotation information of the sequence. Out of the results of the LCA annotation and gene abundance table, the abundance of each sample at each taxonomy and the corresponding gene abundance tables were acquired14,18,22. Based on the abundance tables at each taxonomy level, Krona analysis23, relative abundance overview, and abundance clustering heatmap were performed, combined with PCA24 (R ade4 package, Version 3.2.1) and NMDS25 (R vegan package, Version 2.15.3) analysis of dimension reduction. LEfSe analysis was used to search for species differences between groups26.
DIAMOND software was used to align unigenes with those in the functional database, including the KEGG database (Version 2018-01-01, http://www.kegg.jp/kegg/)27,28, eggNOG database (Version 4.5, http://eggnogdb.embl.de/#/app/home)29, and CAZy database (Version 201801, http://www.cazy.org/)30. According to the alignment results, the relative abundance at different functional levels was calculated. Based on the abundance table at each taxonomy level, annotated genes statistics, relative abundance overview, and abundance clustering heat map were carried out, combined with PCA and NMDS analysis of dimension reduction, metabolic pathway comparative analysis, as well as Metastatic and LEfSe analysis on inter-group functional differences.
Technical Validation
Contaminants and trim reads of the metagenomic sequencing data were removed and the clean reads accounted for more than 99% (Fig. 2), illustrating that the data had incredible quality. To ensure unbiased data production, Randomization principles were carried out and abided by sample collection, extraction, and CLas infection rate detection. Using several pieces of software, we confirmed the technical validation of the taxonomic assignments, completeness, and function annotations.
Code availability
No custom code was used to generate or process these data.
References
Bové, J. M. Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J. Plant Pathol. 88, 7–37 (2006).
Coates, L. C. et al. Development on Citrus medica infected with ‘Candidatus Liberibacter asiaticus’ has sex-specific and -nonspecific impacts on adult Diaphorina citri and its endosymbionts. PLoS One. 15(10), e0239771 (2020).
Liu, K. et al. Comparative microbiome analysis reveals bacterial communities associated with Candidatus Liberibacter asiaticus infection in the Huanglongbing insect vector Diaphorina citri. J. Asia Pac. Entomol. 25, 101884 (2022).
Kruse, A. et al. Combining ‘omics and microscopy to visualize interactions between the Asian citrus psyllid vector and the Huanglongbing pathogen Candidatus Liberibacter asiaticus in the insect gut. PloS One. 12(6), e0179531 (2017).
Nakabachi, A. et al. The 160-kilobase genome of the bacterial endosymbiont. Carsonella. Science. 314, 267 (2006).
Ramsey, J. S. et al. Protein interaction networks at the host–microbe interface in Diaphorina citri, the insect vector of the citrus greening pathogen. Royal Soc. Open Sci. 4, 160545 (2017).
Nakabachi, A. & Fujikami, M. Concentration and distribution of diaphorin, and expression of diaphorin synthesis genes during Asian citrus psyllid development. J. Insect Physiol. 118, 103931 (2019).
Fagen, J. R. et al. Characterization of the relative abundance of the citrus pathogen ca. Liberibacter asiaticus in the microbiome of its insect vector, Diaphorina citri, using high throughput 16S rRNA sequencing. Open Microbiol. J. 6, 29–33 (2012).
Meng, L. X., Xia, C. X., Jin, Z. X. & Zhang, H. Y. Investigation of gut bacterial communities of Asian Citrus Psyllid (Diaphorina citri) reared on different host plants. Insects. 13, 694 (2022).
Meng, L. X., Li, X. Y., Cheng, X. Q. & Zhang, H. Y. 16S rRNA gene sequencing reveals a shift in the microbiota of Diaphorina citri during the psyllid life cycle. Front. Microbiol. 10, 1948 (2019).
Lewis, R. W., Islam, A. A., Dilla-Ermita, C. J., Hulbert, S. H. & Sullivan, T. S. High-throughput siderophore screening from environmental samples: plant tissues, bulk soils, and rhizosphere soils. J Vis Exp. 144, e59137 (2019).
Yu, H. Z. et al. Transcriptome analyses of Diaphorina citri midgut sesponses to Candidatus Liberibacter Asiaticus infection. Insects. 11, 17 (2020).
Li, D. Y. et al. Fitness and evolution of insecticide resistance associated with gut symbionts in metaflumizone-resistant Plutella xylostella. Crop Prot. 124, 104869 (2019).
Karlsson, F. H. et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245 (2012).
Nielsen, H. B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32(8), 822–828 (2014).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 498(7452), 99–103 (2013).
Chen, K. & Pachter, L. Bioinformatics for whole-genome shotgun sequencing of microbial communities. PLoS Comput. Biol. 1(2), e24 (2005).
Qin, J. J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 464(7285), 59–65 (2010).
Qin, J. J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 490(7418), 55–60 (2012).
Oh, J. et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 514(7520), 59–64 (2014).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12, 59–60 (2015).
Zeller, G. et al. Potential of fecal microbiota for early - stage detection of colorectal cancer. Mol. Syst. Biol. 10(11), 766 (2014).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC bioinformatics. 12(1), 385 (2011).
Avershina, E., Trine, F. & Knutm, R. De novo semi-alignment of 16S rRNA gene sequences for deep phylogenetic characterization of next generation sequencing data. Microbes Environ. 28(2), 211–216 (2013).
Rivas, M. N. et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J. Allergy Clin. Immunol. 131(1), 201–212 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12(6), 1–18 (2011).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34, D354–7 (2006).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–205 (2014).
Powell, S. et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic acids Res. 42, D231–239 (2014).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37, D233–238 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454939 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454940 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454941 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454942 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454943 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23454944 (2023).
Acknowledgements
This work was supported by the National Key R&D Program of China 2021YFD1400800 and 2018YFD0201500.
Author information
Authors and Affiliations
Contributions
Qi Pan: Investigation, Data Curation, Data Analysis, Validation, Visualization, Writing- original draft, Writing-review & editing. Shi-Jiang Yu: Investigation, Data Analysis, Writing-original draft, Writing-review & editing. Shuang Lei: Investigation, Data Curation, Resources. Si-Chen Li: Investigation, Resources. Li-Li Ding: Validation, Resources. Lu-Yan Cheng: Data Curation. Ren Luo: Resources. Cui-Yun Lei: Resources. Bing-Hai Lou: Project administration. Lin Cong: Validation, Resources. Hao-Qiang Liu: Investigation, Resources. Xue-Feng Wang: Investigation, Resources. Chun Ran: Project administration, Conceptualization, Writing-original draft, Writing-review & editing, Supervision. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, Q., Yu, SJ., Lei, S. et al. Effects of Candidatus Liberibacter asiaticus infection on metagenome of Diaphorina citri gut endosymbiont. Sci Data 10, 478 (2023). https://doi.org/10.1038/s41597-023-02345-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02345-2
- Springer Nature Limited