
Abstract
In Japan, wasabi (Eutrema japonicum) is an important traditional condiment, and is recognized as an endemic species. In the present study, we generated a chromosome-level and haplotype-resolved reference genome for E. japonicum using PacBio CLR (continuous long reads), Illumina, and Hi-C sequencing data. The genome consists of 28 chromosomes that contain 1,512.1 Mb of sequence data, with a scaffold N50 length of 55.67 Mb. We also reported the subgenome and haplotype assignment of the 28 chromosomes by read-mapping and phylogenic analysis. Three validation methods (Benchmarking Universal Single-Copy Orthologs, Merqury, and Inspector) indicated that our obtained genome sequences were a high-quality and high-completeness genome assembly. Comparison of genome assemblies from previously published genomes showed that our obtained genome was of higher quality. Therefore, our genome will serve as a valuable genetic resource for both chemical ecology and evolution research of the genera Eutrema and Brassicaceae, as well as for wasabi breeding.
Similar content being viewed by others


Background & Summary
Eutrema japonicum (Miq.) Koidz, wasabi, is endemic to Japan1. It serves as a traditional and crucial condiment in the consumption of ‘sushi’ or ‘soba’ in Japan. The major wasabi variety “Mazuma” boasts both a giant rhizome and a potent pungency, and is currently the most valuable cultivar. Wasabi is a member of the Brassicaceae family, which includes numerous scientifically and commercially significant species1. Currently, the National Center for Biotechnology Information (NCBI) genome database has sequenced eighty-seven species of the Brassicaceae family. Within the genus Eutrema, whole-genome sequences have been released for the wild species Eutrema heterophyllum (W. W. Smith) H. Hara, and Eutrema yunnanense Franch2. However, in Japan, only the chloroplast genomes of Eutrema species, including wasabi, have been sequenced3. The Brassicaceae family is well known for its high frequency of hybridization4 and polyploidy, or whole-genome duplication (WGD) events, with over 43% of species exhibiting such characteristics5. These features may have played a crucial role in the diversification and adaptive survival of the species, particularly during the Quaternary period6. The chromosome number of wasabi was 287, and it is a tetraploid, similar to that of E. yunnanense (2n = 4x = 28)7. While wasabi polyploidy has been posited, it is yet to be confirmed in Japan, and its genome constitution has not been reported (i.e. allo- or auto-polyploid).
Additionally, the phylogenetic relationships between E. yunnanense, a close relative to wasabi, and Eutrema species in Japan are yet to be investigated using nuclear DNA polymorphisms.
Recent advancements in long-read sequencing have enabled the acquisition of uninterrupted genomic sequence data from highly heterozygous plant species8. In this study, we provide the first report of whole-genome sequences of wasabi, and investigate its chromosome structure using chromosome-level de novo genome sequencing. We also directly compared the genomes of other Eutrema species. We anticipate that our data will provide the genetic basis for understanding the variable pungent components and defense systems, as well as the evolutionary history of wasabi as an endemic plant in Japan. Our data will serve as a valuable genetic resource for both chemical ecology and evolutionary research of the genus Eutrema, and family Brassicaceae, as well as for wasabi breeding.
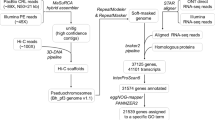
Methods
Sample collection and sequencing
The variety “Mazuma” No. 3 (wasabi, Fig. 1) from MIYOSHI AGRI-TECH CO., LTD. was selected for the whole genome sequencing of the wasabi. Individual used for genome sequencing is stored in the Laboratory of Plant Genetics and Breeding, Faculty of Applied Biological Sciences, Gifu University. Whole-genome shotgun sequencing was performed using the PacBio and Illumina sequencing platforms. Genomic DNA from Eutrema japonicum was isolated using a NucleoBond® HMW DNA kit (Macherey-Nagel, Germany) and sheared into fragment sizes ranging from 30 kb to 100 kb with a g-tube device (Covaris Inc., MA, USA). A Continuous Long Read (CLR) SMRTbell library was prepared using a SMRTbell Express Template Prep Kit 2.0 (Pacific Bioscience, CA, USA) according to the manufacturer’s instructions. The CLR library was size-selected using the BluePippin system (Saga Science, MA, USA) with a lower cutoff of 30 kb. One SMRT Cell 8 M was sequenced on the PacBio Sequel II system with Binding Kit 2.0 and Sequencing Kit 2.0 (20 h collection times). As a result of PacBio sequencing, 175.73 Gb of CLR reads were obtained (Table 1). In addition, genomic DNA was fragmented to an average size of 600 bp using a M220 Focused-ultrasonicator (Covaris Inc., MA, USA). A paired-end library with insert sizes from to 550–650 bp was constructed using a TruSeq DNA PCR-Free Library Prep kit (Illumina, CA, USA) and was size-selected on an agarose gel with a Zymoclean Large Fragment DNA Recovery Kit (Zymo Research, CA. USA). The final library was sequenced using a 2 × 250 bp paired-end protocol for the HiSeq. 2500 system (Illumina, San Diego, CA, USA). As a result of Illumina sequencing, we obtained 73.47 Gb of Illumina paired-end reads (Table 1). The Arima Hi-C library was constructed using an Arima-HiC + Kit (Arima Genomics, CA, USA) according to the manufacturer’s instructions for Plant Tissues (A160135 v01) and library preparation was done using the KAPA Hyper Prep Kit (A160139 v00). In brief, 2.0 grams of the frozen leaves were crosslinked with 37% formaldehyde solution under a vacuum for 25 min and stopped in 125 mM glycine. Crosslinked leaves were ground into powder in liquid nitrogen, and 1.8 g of the powder was suspended in 6.0 mL nuclei isolation buffer. Nuclear extraction was performed using a CelLytic PN Isolation/Extraction Kit (Sigma-Aldrich, MO, USA) according to the manufacturer’s recommendations for cell lysis and semi-pure preparation of nuclei. The cross-linked DNA was digested with two restriction enzymes (^GATC and G^ANTC). After incorporating biotinylated nucleotides into the digested DNA ends, both ends were ligated to the spatially proximal ends. To reverse formaldehyde cross-linking, the crosslinked DNA was incubated as follows: 55 °C for 30 min, 68 °C for 90 min, and 25 °C for 10 min. The ligated DNA was mechanically sheared into average sizes of 400–500 bp using the M220 focused ultrasonicator, and the ligation junctions were enriched with streptavidin magnetic beads. The sequencing library was prepared from enriched DNA fragments with a KAPA Hyper Prep Kit (Roche Molecular Systems, CA, USA) and amplified for six PCR cycles. The concentration and quality of the libraries were evaluated using a Qubit 4 Fluorometer (Thermo Fisher Scientific, MA, USA), 2100 Bioanalyzer system (Agilent Technologies, CA, USA), and 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific, MA, USA). The final Hi-C libraries were run on the Illumina NovaSeq. 6000 system (Illumina, San Diego, CA, USA) with 2 × 150 bp read length, and 160.84 Gb of Arima Hi-C reads were generated (Table 1). The Omni-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics, Scotts Valley, CA, USA) according to the manufacturer’s protocol. Approximately 300 mg of leaf tissue was ground in liquid nitrogen with a mortar and pestle. The pulverized material was processed into a proximity ligation library using the Omni-C Proximity Ligation Assay Non-Mammalian Sample Protocol v1.2 of the Omni-C Kit (Dovetail Genomics, Scotts Valley, CA, USA). The final library was sequenced on a NovaSeq 6000 instrument with 2 × 100 bp read length, and 82.53 Gb of Omni-C reads were generated (Table 1).

The variety ‘Mazuma’ No.3 of Wasabi, Eutrema japonicum.
Genome size and heterozygosity estimation
The genome size of E. japonicum was estimated from Illumina sequencing data using the k-mer-based method9. Illumina sequenced reads were filtered using platanus_trim v1.0.7 (http://platanus.bio.titech.ac.jp/pltanus_trim) with default parameters. To remove highly copied reads derived from the chloroplast genome, filtered reads were mapped to the E. japonicum chloroplast genome10, and unmapped reads were extracted. Using the unmapped reads, Jellyfish v2.2.1011 was first applied to extract and count canonical k-mers at k = 32. Subsequently, GenomeScope 2.09 was used to estimate haploid genome size and heterozygosity from k-mer count data with parameters of “-k 32 -p 4” (Fig. 2). As a result, we estimated a haploid genome size of 397.3 Mb with a heterozygosity of 5.9% (aaab: 2.14%, aabb: 3.12%, aabc: 0.01%, abcd: 0.658%, respectively); therefore, the homozygosity rate was estimated to be 94.1% (Fig. 2).

K-mer analysis of E. japonicum genome.
Chloroplast genome assembly
Illumina sequenced reads were filtered using platanus_trim v1.0.7 (http://platanus.bio.titech.ac.jp/pltanus_trim) with the following parameters “-q 0.” The trimmed reads were assembled using NOVOPlasty v4.3.112 with the following parameters: “Type = chloro, K-mer = 36, Read Length = 250, Insert size = 600, Platform = illumina, Single/Paired = PE, Insert size auto = yes”. The chloroplast genome of E. japonicum was used as the reference10, and psbA chloroplast gene sequences were used as seeds to assemble the plastome. As a result, a chloroplast genome of 153,794 bp was obtained13,14.
Mitochondrial genome assembly
Firstly, we performed a de novo assembly of the trimmed reads using NOVOPlasty v 4.3.112 with the following parameters: “Type = mito_plant, K-mer = 36, Read Length = 250, Insert size = 600, Platform = illumina, Single/Paired = PE, Insert size auto = yes”. Then, the cox115 and nad615 genes were used for the seed sequence in an independent assay. Preliminary draft mitochondrial contigs were obtained. We used mitochondrial contigs as bait files to extract PacBio reads from the sequenced mitochondrial genome. Then, the extracted PacBio reads were assembled using Flye v2.9-b176816 and polished using Racon v1.4.2017. PacBio-based assemblies were used as the new bait file to extract Illumina reads from the sequenced mitochondrial genome. Finally, using both the extracted Illumina and PacBio reads, we applied Unicycler v0.4.818 to perform a hybrid assembly. We obtained two independent contigs, totaling 309,141 bp. The length of the longer contig was 201,312 bp14,19, while that of the shorter contig was 107,829 bp14,20.
De novo genome assembly
PacBio sequenced reads were used for genome assembly by the Canu v2.1.121 with parameters of “genomeSize = 600 m corOutCoverage = 200 “batOptions = -dg 3 -db 3 -dr 1 -ca 500 -cp 50” correctedErrorRate = 0.035 -pacbio-raw”. The draft assembly contigs were polished with two rounds of Arrow (Pacific Biosciences) and three rounds of HapoG v1.3.422 and then the Arrow-identified variants were filtered via Merfin23 using Illumina sequenced reads. Organelle contigs were identified by alignment against the already generated mitochondrial and chloroplast genomes using nucmer v4.0.0beta224. After removing organelle contigs, redundant contigs, short contigs (<15,000 bp), and contigs of aberrant GC content (<1% or >99%), we obtained 616 contigs with a total size of 1,529.3 Mb, which was approximately four times the estimated haploid genome size (397.3 Mb). This result suggested that not only the subgenome but also each of the haplotype genomes that make up the tetraploid chromosomes are constructed into contigs in a separate form.
Chromosome assembly using Hi-C data
To obtain a haplotype-resolved chromosome assembly of E. japonicum, we performed Hi-C scaffolding using two different Hi-C datasets (Arima Hi-C and Omni-C). The Arima Hi-C reads were mapped to the cleaned contigs and processed to generate Hi-C contacts by Juicer v1.625 with the following parameter settings: “-s Arima.” Bridge sequences were trimmed from the Omni-C reads using cutadapt v4.126. The cleaned Omni-C reads were then processed using the Juicer pipeline with default parameters. Two distinct Hi-C contacts files (merged_nodups.txt) were combined and used for scaffolding by the 3D-DNA v18092227 with the following parameter settings: “--rounds 0 --editor-coarse-resolution 2500000 --editor-coarse-region 12500000 --editor-coarse-stringency 1 --polisher-coarse-resolution 2500000-- polisher-coarse-region 15000000 --polisher-coarse-stringency 1 --splitter-coarse-resolution 2500000 --splitter-coarse-region 15000000 --splitter-coarse-stringency 1,” so as to avoid unnecessary fragmentation. We visualized the Hi-C contact map and performed extensive manual curation using Juicebox v1.11.0828 to fix mis-assemblies and mis-scaffoldings. After manual curation, overlapping contig ends (identity ≥99%) of flanking contigs were merged using an in-house script (https://github.com/th2ch-g/Canu-Contig-Overlap-Merge). As a result, Hi-C data helped to anchor 477 contigs of 1,512.1 Mb sequence to 28 chromosomes (Fig. 3). The scaffold N50 size was 55.67 Mb (Table 2).

Genome-wide Hi-C heatmap of E. japonicum. The blue squares represent chromosomes.
Chromosome, sub-genome and haplotype assignment
A comparison of sequence similarity with Eutrema salsugineum enabled the identification of homoeologous sets of E. japonicum chromosomes, with each of the four chromosomes showing clear sequence similarity to each E. salsugineum chromosome as a set (Fig. 4). To assign chromosomes within each homoeologous set to the A or B sub-genome, we mapped and visualized the location of sequencing reads from diploid E. yunnanense2,29,30,31 and E. japonicum32,33. Fourteen chromosomes showed a clear overabundance of mapped diploid E. yunnanense reads (Fig. 5) and were assigned to subgenome A. The remaining 14 chromosomes were assigned to the B sub-genome. Subsequently, seven chromosomes belonging to the A sub-genome and seven chromosomes belonging to the B sub-genome showed a clear overabundance of mapped E. japonicum reads (Fig. 5) and were assigned to haplotype A1 and haplotype B1, respectively. The remaining chromosomes belonging to the A sub-genome and 7 chromosomes belonging to the B sub-genome were assigned to haplotype A2 and haplotype B2, respectively.

Comparison of sequence similarity with E. salsugineum. Dot plot analysis comparing E. japonicum homoeologous chromosomes and E. salsugineum chromosomes.

Mapping analysis of other Eutrema sequence reads against chromosomes of E. japonicum. The normalized sequencing depth of the diploid E. yunnanense reads (a) and previously published E. japonicum reads (b) are shown. Coverage corresponding to haploid is set to 1. Colored by identity of mapped read (Red: 99%≥ identity, Orange: ≥97.5% identity, Green: ≥95% identity, Blue: ≥90% identity).
Intra-chromosomal comparative genome analysis
Within each of the seven homoeologous chromosome groups, the chromosome-level sequences of the four haplotypes were aligned to each other using nucmer v4.0.0beta224 with default parameters (Fig. 6) and visualized using a mummerplot and gnuplot. High collinearity was detected between all homoeologous chromosomes, but several large genomic inversions (>5 Mbp) were detected between chromosomes within the subgenome, for example, chr1A1 vs. chr1A2 and chr1B1 vs. chr1B2 (Fig. 6). To validate the predicted inversion, we selected six candidate inversion loci and performed PCR validation, which resulted in missed assemblies that were not observed. A high-confidence alignment subset (filtered with delta-filter -q -r) was used to calculate the average genome sequence similarity. The average genome sequence similarity between the chromosomes within the subgenome is 97.6–98.0%, while inter-subgenome homoeologous chromosomes are 95.7–96.3%.

Self Dot-plot analysis of four haplotype-resolved E. japonicum chromosomes. X-axis: From left to right: Chromosomes A1, A2, B1, and B2. Y-axis: From bottom to top: Chromosomes A1, A2, B1, and B2.
Genome sequence feature (repetitive sequence analysis)
To identify the repetitive elements in the E. japonicum chromosome assembly, RepeatModeler v2.034 was used to construct a repetitive library, followed by RepeatMasker v4.0.9 (http://www.repeatmasker.org). For RepeatModeler, in addition to the default parameters, the parameters, the following parameter “-LTRStruct” was used. For RepeatMasker, the additional parameters were “a” and “gff.” Repeats accounted for 66.9% of the genome, with LTR-retrotransposons representing the most abundant repeat class (41.0%). Other repeats, including LINEs, SINEs, and DNA elements, made up only minor genome proportions (Table 3). The 28 chromosomes were all characterized by a high density of repetitive sequences and transposable elements in the pericentromeric and centromeric regions (Fig. 7).

Characterization of the E. japonicum genome. From the outer to the inner layers: chromosomes with scales (a), links between intra-subgenomes syntenic blocks (b), repeat element abundance (c), LTR element abundance (d), GC rate (e), and links between inter-subgenome syntenic blocks (f) are shown. The links between intra-subgenome syntenic blocks (b) are indicated by green lines for subgenome A and by cyan lines for subgenome B, and the links between inter-subgenome syntenic blocks (f) are indicated by gray lines. The links between inverted syntenic blocks are indicated by red lines (b and f).
Comparative genomic analysis
To investigate the phylogenetic relationships of the four haplotype-resolved sets of E. japonicum chromosomes with related species, comparative genomic analysis was conducted based on orthologous gene information. For comparison, we selected the following plant species as targets. The genome sequences of Thlaspi arvense35, E. salsugineum36, E. heterophyllum37, and E. yunnanense38 were downloaded from NCBI GenBank. Then, Benchmarking Universal Single-Copy Orthologs (BUSCO)39 analysis with the brassicales_odb10 database against each genome sequence was conducted. Based on the BUSCO gene id information predicted by this analysis, 2,524 ortholog groups were extracted with a one-to-one gene relationship across all eight genomes (T. arvense, E. salsugineum, E. heterophyllum, E. yunnanense, E. japonicum haplotype-A1, E. japonicum haplotype-A2, E. japonicum haplotype-B1, and E. japonicum haplotype-B2). For each ortholog group, multiple alignments were performed with MAFFT v7.40740,41, and sites containing gaps (“−”) or ambiguous characters (“X”) were excluded. All alignments were concatenated and used for phylogenetic analysis. A phylogenetic tree was constructed using RAxML v8.2.1242. We applied the JTT substitution matrix using a gamma model of rate heterogeneity (-m PROTGAMMAJTT). Phylogenetic analysis performed with each chromosome dataset showed a similar topology (Fig. 8). Four E. japonicum haplotypes were placed into two separate clades (subgenome A and subgenome B), and diploid E. yunnanense was included in the subgenome A clade (Fig. 8).

The phylogenetic analysis of E. japonicum and closely related species. As ortholog groups, 515, 415, 333, 439, 534, 448, and 443 groups were used for chromosomes 1, 2, 3, 4, 5, 6, and 7, respectively. T. arvense was used as an outgroup.
Data Records
The genomic sequencing data (Illumina, PacBio, Hi-C) are available in the NCBI SRA database under BioProject ID PRJDB15095. The accession number of Illumina sequencing data is DRR43837043. The accession number of PacBio sequencing data is DRR43310944. The accession number of Hi-C sequencing data are DRR43936545 and DRR43936646. The final chromosome assemblies are available in the NCBI GenBank database under BioProject ID PRJDB15185 and PRJDB15186. The accession number of chromosome assembly haplotype-1 (principal haplotype; subgenome A1 and subgenome B1) is BSQW01000001-BSQW0100001447. The accession number of chromosome assembly haplotype-2 (alternate haplotype; subgenome A2 and subgenome B2) is BSQX01000001- BSQX0100001448. The organelle genome assemblies are available in the NCBI GenBank database and FigShare database14. The accession number of chloroplast genome is LC770997.113. The accession number of mitochondrial genomes are LC770998.119 and LC770999.120. Other data, such as structure annotation of BUSCO genes, predicted CDS and protein sequences of BUSCO genes, and annotation of TEs, are available at FigShare database14.
Technical Validation
DNA quality
Agarose gel electrophoresis and Pippin Pulse (Saga Science, MA, USA) were used to confirm the absence of total RNA and the fragment size of the purified DNA molecules. The concentration was measured using the Qubit 4 Fluorometer (Thermo Fisher Scientific, MA, USA). The main bands of genomic DNA fragments were over 40 kb, and the Nanodrop ND-1000 DNA spectrophotometer (LabTech, Corinth, MS, USA) ratio (260/280) was 1.82.
Assembly evaluation
The quality and completeness of chromosome assembly were evaluated using three independent approaches. First, the QV value and completeness were estimated using Merqury v1.349 by comparing k-mers in the assembly to those found in the Illumina sequence reads. The results revealed that the QV value for chromosome assembly was 51.33, and the completeness value was 97.82%. Secondly, the completeness of the chromosome assembly was also assessed using BUSCO v5.4.339 with 4,596 single-copy orthologs from the brassica_odb10 database. BUSCO analysis identified 99.24% (4, 561) complete BUSCOs (0.61% single-copy and 98.63% duplicated BUSCOs) and 0.04% (2) fragmented BUSCOs in the genome of E. japonicum. Thirdly, we further evaluated the assembly quality using Inspector50 by aligning PacBio sequence reads to the assembled contigs to generate read-to-contig alignment and performed downstream assembly evaluation. The read-to-contig mapping rate and QV value were 91.98% and 43.95, respectively. All these indicators suggest a high-quality and high-completeness genome assembly for further genetic research of E. japonicum.
Comparison of genome assembly and gene models with the previous study
A comparison was performed between the E. japonicum genome and the previously published genom32. The scaffold N50 size of our E. japonicum genome was 55.67 Mb, which was significantly longer than the previously published assembly (N50 = 356 kb). Regarding its quality, our assembly showed that the BUSCO evaluation achieved 99.24% completeness, whereas the previous result was extremely low at 67.35%. Therefore, these results indicate that our E. japonicum genome was of higher quality than the previously published genome.
Code availability
The code is available at Github (https://github.com/th2ch-g/Canu-Contig-Overlap-Merge) for merging overlapped contig ends. Other software and pipelines were executed according to the manual and protocols of the published bioinformatic tools. The version and code/parameters of software have been described in Methods.
References
Yamane, K. et al. Genetic differentiation, molecular phylogenetic analysis, and ethnobotanical study of Eutrema japonicum and E. tenue in Japan and E. yunnanense in China. Hort. J. 85, 46–54 (2016).
Guo, X. et al. The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 25, 307–315 (2018).
Haga, N. et al. Complete chloroplast genome sequence and phylogenetic analysis of wasabi (Eutrema japonicum) and its relatives. Scientific Reports 9, 14377 (2019).
Marhold, K. & Lihová, J. Polyploidy, hybridization and reticulate evolution: lessons from the Brassicaceae. Plant Syst. Evol. 259, 143–174 (2006).
Hohmann, N., Wolf, E. M., Lysak, M. A. & Koch, M. A. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell 27, 2770–2784 (2015).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Du, N. & Gu, Z. J. A comparative karyological study of the cultured Eutrema wasabi and its three related wild species. Acta Botanica Yunnanica 6, 645–650 (2004).
Michael, T. P. & VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 54, 26–33 (2020).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
NCBI GenBank https://identifiers.org/ncbi/insdc:LC500901 (2023).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45, e18 (2017).
NCBI GenBank https://identifiers.org/ncbi/insdc:LC770997 (2023).
Tanaka, H. et al. Dataset for “Haplotype-resolved, chromosomal-level assembly of wasabi (Eutrema japonicum) genome”. FigShare https://doi.org/10.6084/m9.figshare.22045403.v2 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:NC037304 (2023).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
NCBI GenBank https://identifiers.org/ncbi/insdc:LC770998 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:LC770999 (2023).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Aury, J. M. & Istace, B. Hapo-G, Haplotype-aware polishing of genome assemblies with accurate reads. NAR Genom. Bioinform. 3, lqab034 (2021).
Formenti, G. et al. Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation. Nat. Methods 19, 696–704 (2022).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Martin, M. Cutadapt Removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17, 10–12 (2011).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR6306016 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR6306020 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR6306023 (2018).
Liu, H. et al. The reference genome and organelle genomes of wasabi (Eutrema japoniacum). Front. Genet. 13, 1048264 (2022).
China National Center for Bioinformation https://ngdc.cncb.ac.cn/gsa/browse/CRA008347 (2022).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_911865555.2 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_016617915.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_002933915.1 (2018).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_002933935.1 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR438370 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR433109 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR439365 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:DRR439366 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:BSQW00000000 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:BSQX00000000 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biology 22, 312 (2021).
Acknowledgements
We would like to thank the MIYOSHI AGRI-TECH CO., LTD. for providing materials. Besides, we would like to thank the technical staff member at National Institute of Genetics and Itoh laboratory for library preparation, sequencing, and data management. This work was supported by JSPS KAKENHI, Grant Number 16H06279 (PAGS).
Author information
Authors and Affiliations
Contributions
Kyoko Yamane, Kentaro Yano, and Takehiko Itoh conceived the study. Kyoko Yamane collected the samples. Shohei Yamamoto and Atsushi Toyoda performed DNA extraction. Atsushi Toyoda performed sequencing. Hiroyuki Tanaka, Tatsuki Hori, and Takehiko Itoh performed the analyses. Hiroyuki Tanaka, Kyoko Yamane, and Takehiko Itoh wrote the manuscript. All authors have read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanaka, H., Hori, T., Yamamoto, S. et al. Haplotype-resolved chromosomal-level assembly of wasabi (Eutrema japonicum) genome. Sci Data 10, 441 (2023). https://doi.org/10.1038/s41597-023-02356-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02356-z
- Springer Nature Limited