
Abstract
The northeastern Indian Ocean exhibits distinct hydrographic characteristics influenced by various local and remote forces. Variations in these driving factors may alter the physiochemical properties of seawater, such as dissolved oxygen levels, and affect the diversity and function of microbial communities. How the microbial communities change across water depths spanning a dissolved oxygen gradient has not been well understood. Here we employed both 16S rDNA amplicon and metagenomic sequencing approaches to study the microbial communities collected from different water depths along the E87 transect in the northeastern Indian Ocean. Samples were collected from the surface, Deep Chlorophyll Maximum (DCM), Oxygen Minimum Zone (OMZ), and bathypelagic layers. Proteobacteria were prevalent throughout the water columns, while Thermoproteota were found to be abundant in the aphotic layers. A total of 675 non-redundant metagenome-assembled genomes (MAGs) were constructed, spanning 21 bacterial and 5 archaeal phyla. The community structure and genomic information provided by this dataset offer valuable resources for the analysis of microbial biogeography and metabolism in the northeastern Indian Ocean.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background & Summary
The Indian Ocean is bordered by the Southern Ocean to the south and enclosed by continental shelf and land masses on other sides, covering approximately 20% of the global surface ocean. The hydrographic characteristics of the Indian Ocean are influenced by a multitude of geological and physicochemical processes, including tectonic activities1, oceanic circulation patterns2, boundary currents3, climate modes4, and land-ocean interactions5, etc. Three distinct biomes have been proposed based on the biogeochemical characteristics of the Indian Ocean6, including the oligotrophic subtropical southern Indian Ocean, the iron-deficient low-productivity equatorial region, and the nutrient-rich high-productivity northern Indian Ocean6,7,8. The northern Indian Ocean, particularly the Bay of Bengal (BoB), also receives significant freshwater discharge9,10 and atmospheric deposition11, which increase surface productivity and strengthen stratification. In conjunction with the limited oxygen supply from deep overturning circulation and lateral advection in the northern Indian Ocean2, the mid-depth waters ranging approximately from 200 to 1000 m are oxygen deficient to a vast extent, forming two large oxygen minimum zones (OMZs) in the Arabian Sea and the Bay of Bengal12. Collectively, the areas covered by OMZs in the Indian Ocean account for more than half of the global OMZs (59%)13, with the Bay of Bengal standing as the world’s largest hypoxic bay14. Despite significant seasonal variations in monsoon winds and biological productivity, dissolved oxygen concentrations in these regions exhibit relatively minor fluctuations15.
Marine microorganisms play a central role in driving various elemental cycles within the global ocean due to their high abundance, immense diversity, and versatile metabolic capacity16,17. The Indian Ocean has a great influence on global biogeochemical cycles by contributing around 15% of oceanic net primary production18, with a particularly higher abundance of picocyanobacteria than most other oceanic basins19. Dissolved oxygen is one of the most important factors controlling microbial respiration and biogeochemical transformation in marine environments20. In oxygen-deficient waters, alternative electron acceptors such as nitrate were used or preferred by diverse marine organisms21,22. OMZs are characterized by significant REDOX gradients, and the nitrogen cycle dominates the biogeochemical processes23,24. The continuous expansion of marine OMZs will be accompanied by more widespread anammox and denitrification activities, which will have a profound influence on nitrogen bioavailability in marine environments25. To better understand the role of biological communities within OMZs, it is important to study their diversity, metabolic function, and ecological relationships26,27.
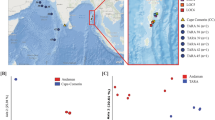
In this study, we conducted a comprehensive sampling expedition from April 15th to June 20th, 2020, along the E87 transect in the Northeast Indian Ocean, spanning from 10°S off the East India coast to 15°N in the BoB. A total of 25 water samples were collected from various depths, including the surface (5 m, n = 7), DCM (n = 7), OMZ (n = 6), bathypelagic (Bathy) layers (2000 m, n = 5), for studying microbial diversity and metabolic potentials (Fig. 1). Detailed sample metadata including geographic locations and environmental factors can be found in Table S1. Flow cytometry analysis showed that the abundance of Prochlorococcus and picoeukaryotes reached their maxima in the DCM layer. In contrast, a higher abundance of Synechococcus was observed near the surface (Table S1). The 16S rDNA amplicon data revealed that Proteobacteria constituted the dominant phylum, accounting for 49.31% of all reads. Within Proteobacteria, Alphaproteobacteria accounted for 59.72%, while Gammaproteobacteria represented 29.44%. Notably, Gammaproteobacteria dominated in both the OMZ and Bathy waters. Cyanobacteria, on the other hand, were primarily distributed in the DCM and higher layers, accounting for 12.46% of all reads. Thermoproteota (Marine Group I archaea, MGI) emerged as a significant component of the OMZ layer, accounting for 8.77% of all reads. MGII (Marine Group II archaea) was predominantly found in the DCM, and although the relative abundance of MGIII (Marine Group III archaea) was relatively low across the water column, it was significantly higher in the OMZ layer compared to other layers (Fig. 2 and Table S2).

Sampling sites and layers along the E87 transect in the northeastern Indian Ocean. Surface, the surface layer at 5 m. DCM, the Deep Chlorophyll Maximum layer. OMZ, the oxygen minimum zone layer. Bathy, the bathypelagic layer at 2000 m. Detailed sample metadata can be found in Table S1.

The relative abundance of different taxa across depths based on 16S rDNA amplicon sequencing in the northeastern Indian Ocean. Amplicon sequences were denoised and grouped into Amplicon Sequence Variants (ASVs) to calculate microbial relative abundance in each sample. Detailed 16S rDNA taxonomy assignment can be found in Table S2.
After metagenomic binning and refinement, a total of 675 non-redundant metagenome-assembled genomes (MAGs) with completeness ≥50% and contamination ≤10% were recovered, covering 21 bacterial and 5 archaeal phyla (Figs. 3, 4). Based on the MIMAG (Minimum Information about a Metagenome-Assembled Genome) standards28, 164 of these MAGs were classified as high-quality (completeness >90% and contamination <5%), accounting for 24.3% of the total MAGs. Compared with MAGs of OceanDNA29 and Tara Oceans30 at the identity threshold of 95%, we found that 62.45% of the MAGs reported here were not covered by either dataset, suggesting the uniqueness of MAGs recovered from the northeastern Indian Ocean. These MAGs were taxonomically classified into 104 archaeal and 571 bacterial genomes based on Genome Taxonomy Database (GTDB) release r20731. Bacterial phyla with >10 MAGs include Proteobacteria (n = 251), Bacteroidota (n = 50), Actinobacteriota (n = 45), Marinisomatota (n = 40), Planctomycetota (n = 40), Verrucomicrobiota (n = 40), Chloroflexota (n = 23), SAR324 (n = 15), Cyanobacteria (n = 13), and Acidobacteriota (n = 11) (Fig. 3 and Table S2). Archaeal phyla include Thermoplasmatota (n = 93), Thermoproteota (n = 7), Nanoarchaeota (n = 2), Asgardarchaeota (n = 1), and Micrarchaeota (n = 1) (Fig. 4 and Table S2). MAGs of MGIII archaea formed two distinct phylogenetic clusters with divergent GC contents, as previously reported32 (Fig. 4 and Table S2).

The phylogenomic tree of 571 bacterial MAGs reconstructed from the northeastern Indian Ocean. The universally conserved 160 single-copy marker genes were used to build this maximum-likelihood phylogenomic tree with 1000 bootstraps. Detailed MAG taxonomy assignment, associated with completeness and contamination information can be found in Table S2.

The phylogenomic tree of 104 archaeal MAGs reconstructed from the northeastern Indian Ocean. The universally conserved 49 single-copy marker genes were used to build this maximum-likelihood phylogenomic tree with 1000 bootstraps. Detailed MAG taxonomy assignment, associated with completeness and contamination information can be found in Table S2.
Complementary to the MAG-based analysis, genes were called on the contig level to construct a community-level gene catalog. After gene calling and deduplication, a total of 9,908,058 unique genes were recovered and function annotated with KEGG Orthology (KO) groups. The relative abundance of each unique gene in each sample was calculated in RPKM values. Gene sequences and a table of gene abundance across samples with functional annotations were provided (see the “Data records” section).
Materials and Methods
Sample collection and preparation
Samples were collected from the Northeast Indian Ocean, spanning latitude 10°S to 15°N along longitude 87°E, during the R/V “Shiyan3” cruise from April 15 to June 20, 2020 (Fig. 1). A total of 25 seawater samples were collected from 9 distant sites, covering both surface waters and deeper ocean regions. Fifteen liters of seawater were pre-filtered using a 20 μm nylon mesh (Sefar Nitex, Sweden), followed by subsequent filtration through a 0.22 μm pore size polycarbonate filter (Millipore, MA, USA). The filters were frozen in liquid nitrogen onboard and kept at −20 °C until DNA extraction. For microbial abundance estimation, 2 mL seawater samples were first filtered through a 20 μm nylon mesh, then fixed with 1% (vol/vol) glutaraldehyde, incubated in the dark for 15 minutes, and promptly frozen in liquid nitrogen and preserved at −20 °C for subsequent analysis. In-situ measurements of water temperature, salinity, dissolved oxygen (DO), and fluorescence were conducted using conductivity-temperature-depth (CTD) oceanic profilers (SBE-911 Plus). Other chemical parameters, including nitrite nitrogen, nitrate nitrogen, phosphate, and silicate concentrations were assessed using the Technicon AA3 Auto-Analyzer (Bran-Luebbe, Germany)33. Samples were named following the pattern of “station_name-water_depth”. For instance, the sample name “S10-1-5” indicates this sample was taken at station “S10-1” at a depth of “5” meters.
DNA extraction and sequencing
The phenol-chloroform-isoamyl alcohol method was applied to extract microbial DNA, as described previously34. The quality and concentrations of DNA were quantified using 1% agarose gel electrophoresis and Invitrogen Qubit 2.0 Fluorimeter (ThermoFisher Scientific), respectively. The V4-V5 hypervariable regions of the 16S rRNA gene sequences were amplified using a universal primer pair, 515Y (5′-GTGYCAGCMGCCGCGGTAA-3′) and 926 R (5′-CCGYCAATTYMTTTRAGTTT-3′)35. The amplified fragments were sequenced on the Illumina HiSeq 2500 platform using paired-end 2 × 250 bp chemistries as described previously36. To ensure data quality, raw reads of 16S rDNA gene sequencing were subjected to adapter trimming and quality control using the cutadapt v4.0 and the fastqc v0.12.1 plugins wrapped in the QIIME2 toolkit suite (version 2022.2)37. Amplicon sequence variants (ASVs) and a feature table were generated using the deblur v1.1.1 plugin in QIIME238. The taxonomy of representative ASV sequences was then assigned using the QIIME2 feature-classifier plugin with the pre-trained 99% clustered SILVA database (release 138) as the employed sklearn classifier (Fig. S1).
Qualified DNA samples were fragmented using the Covaris Ultrasonicator M220 (Covaris, USA) with a fragment size of ~500 bp. The resulting DNA fragments were subsequently used in the library preparation and sequencing on an Illumina HiSeq 2500 platform using paired-end 2 × 150 bp chemistries for metagenomic sequencing. All the sequencing jobs were carried out at MAGIGENE (Magigene Biotech, Guangzhou, China).
Metagenomic assembly, gene annotation and abundance quantification
Raw reads were trimmed and quality filtered using fastp v0.23.139 wrapped in the metaWRAP v1.3 pipeline40. Clean reads were assembled using MEGAHIT v1.2.941 with default parameters set by the metaWRAP pipeline. Gene-coding sequences of the assembled contigs were predicted using Prodigal v2.6.3 in “meta” mode42. To generate a gene catalog of non-redundant sequences, all the coding sequences were clustered into representative sequences at 95% identity using CD-HT v4.8.143 with parameters: -c 0.95 -d 400 -T 20 -M 20000 -n 5. For each sample, quality-controlled reads were mapped to the non-redundant gene database using bwa v2.2.144, and RPKM (reads per kilobase per million) values were calculated to determine the relative abundance of contigs using coverM v0.3.1 (https://github.com/wwood/CoverM) with parameters: contig mode, --trim-min 0.10 --trim-max 0.90 --min-read-percent-identity 0.95--min-read-aligned-percent 0.75 -m rpkm. Functions of the non-redundant genes were predicted by KofamScan45 using the prokaryotic, eukaryotic and viral KEGG gene database (Release 108.1) with default settings (Fig. S1).
Metagenomic binning
Contigs longer than 1000 bp were grouped into bins using the metaWRAP binning module with three binners: MaxBin2 v2.2.7, MetaBAT2 v2.12.1, and CONCOCT v1.1.046,47,48. The resulting bins from individual binners were further refined using the bin_refinement module of metaWRAP with >50% completeness and <10% contamination thresholds37. In addition, samples were compared using sourmash v4.8.449, and those ones with close community composition were co-assembled and further binned using BASALT v1.0.050 (via MaxBin2 v2.2.7, MetaBAT2 v2.12.1, and CONCOCT v1.1.0 with more-sensitivity parameter)46,47,48 (Fig. S1).
MAGs refinement and quality assessment
Bins meeting the criteria of ≥50% completeness and ≤10% contamination were subsequently clustered using dRep v3.4.251 at the 95% average nucleotide identity (ANI) threshold (-sa 0.95 -comp 50 -con 10), resulting in a total of 732 species-level bins. The refined bins were further quality checked using CheckM2 v1.0.252 to remove low quality bins, and the remaining 675 bins were classified into high-, medium-quality MAGs according to MIMAG criteria28. Taxonomy of each MAG was assigned using GTDB-Tk v2.3.253 based on the Genome Taxonomy Database (GTDB) version r20731. In addition, MAGs were functionally annotated using Prokka v1.14.554.
Phylogenomic tree construction
The 160 and 49 conserved bacterial and archaeal single-copy genes were extracted from these MAGs using GTDB-Tk v2.3.253, respectively. Only marker genes found in ≥30 MAGs were eventually selected to construct the bacterial and archaeal phylogenomic trees. MUSCLE v555 was used to align marker gene sequences extracted from MAGs, and then BMGE56 was used to prune the alignments. Phylogenomic trees were constructed using IQTree v2.0.357 with the optimal models (Bacteria: -m Q.pfam + F + I -B 1000, Archaea: -m LG + F + R5 -B 1000) estimated by ModelFinder58. The confidence of the maximum-likelihood tree was estimated using 1000 bootstraps.
Data Records
All sequencing products associated with this project can be found under National Center for Biotechnology Information (NCBI) BioProject ID PRJNA103156859. Clean reads of 16S rDNA amplicon and metagenomic sequencing have been deposited at NCBI with the Sequence Read Archive (SRA) project number SRP46822260. NCBI SRA accession numbers for each sample and sequencing type were also provided in Table S1 (Metagenomic Information sheet). Metagenomic assemblies have been deposited at NCBI GenBank database under the same BioProject, and accession numbers can be found in Table S1 (Metagenomic Information sheet). All reads uploaded to the NCBI SRA database were quality-controlled using the software as documented in the “Materials and methods” section. MAGs, Prokka annotations, and function-annotated non-redundant genes with abundance information have been deposited at Figshare61. The MAG names were identical to those of the genome bins in Table S2 (MAG information sheet).
Technical Validation
All raw data processing steps, software, and parameters used in this study were described in the “Materials and methods” section. The assessment of quality scores for the raw reads of the 25 16S rDNA amplicon was performed using FastQC v0.12.1. The results showed that >92.55% of the deduplicated percentage and GC content ≤54%. The assessment of quality scores for the raw reads of the 25 metagenomes was performed using fastp v0.23.139. The results showed that ~95.71% and ~90.14% of the bases have quality scores of ≥20 and ≥30, and GC content <56%, respectively, indicating that sequencing was performed adequately (Table S1). MAGs recovered here were compared with OceanDNA and Tara Oceans using dRep v3.4.251 at the 95% average nucleotide identity (ANI) threshold (-sa 0.95 -comp 50 -con 10) to show the novelty of our MAGs.
Code availability
All versions of third-party software and scripts used in this study are described and referenced accordingly in the “Materials and methods” section for ease of access and reproducibility.
References
Eittreim, S. L. & Ewing, J. Mid-plate tectonics in the Indian Ocean. J. Geophys. Res. 77, 6413–6421 (1972).
Phillips, H. E. et al. Progress in understanding of Indian Ocean circulation, variability, air–sea exchange, and impacts on biogeochemistry. Ocean Sci. 17, 1677–1751 (2021).
Hood, R. R., Beckley, L. E. & Wiggert, J. D. Biogeochemical and ecological impacts of boundary currents in the Indian Ocean. Progress in Oceanography. 156, 290–325 (2017).
Saji, N. H. & Yamagata, T. Possible impacts of Indian Ocean Dipole mode events on global climate. Climate Research. 25, 151–169 (2003).
Zinke, J. et al. Western Indian Ocean marine and terrestrial records of climate variability: a review and new concepts on land–ocean interactions since AD 1660. Int J Earth Sci. 98, 115–133 (2009).
Larkin, A. A. et al. Subtle biogeochemical regimes in the Indian Ocean revealed by spatial and diel frequency of Prochlorococcus haplotypes. Limnol Oceanogr. 65, S220–S232 (2019).
Grand, M. M. et al. Dust deposition in the eastern Indian Ocean: The ocean perspective from Antarctica to the Bay of Bengal. Global Biogeochem Cy. 29, 357–374 (2015).
Liao, J. et al. Microdiversity of the vaginal microbiome is associated with preterm birth. Nat Commun. 14, 4997 (2023).
Howden, S. D. & Murtugudde, R. Effects of river inputs into the Bay of Bengal. J. Geophys. Res. 106, 19825–19843 (2001).
Kumar, M. D. et al. A sink for atmospheric carbon dioxide in the northeast Indian Ocean. J. Geophys. Res. 101, 18121–18125 (1996).
Bikkina, S. & Sarin, M. M. Atmospheric deposition of phosphorus to the Northern Indian Ocean. Curr. Sci. 108, 1300–1305 (2015).
Rixen, T. et al. Reviews and syntheses: Present, past, and future of the oxygen minimum zone in the northern Indian Ocean. Biogeosciences. 17, 6051–6080 (2020).
Fernandes, G. L., Shenoy, B. D. & Damare, S. R. Diversity of Bacterial Community in the Oxygen Minimum Zones of Arabian Sea and Bay of Bengal as Deduced by Illumina Sequencing. Front Microbiol. 10, 3153 (2019).
Cui, W. et al. Statistical characteristics and thermohaline properties of mesoscale eddies in the Bay of Bengal. Acta Oceanol. Sin. 40, 10–22 (2021).
Mandar, S. P. et al. Microbial diversity of the Arabian Sea in the Oxygen minimum zones by metagenomics approach. Curr Sci. 118, 1042–1051 (2020).
Dlugosch, L. et al. Significance of gene variants for the functional biogeography of the near-surface Atlantic Ocean microbiome. Nat Commun. 13, 456 (2022).
Kirchman, D. L. Growth Rates of Microbes in the Oceans. Annu Rev Mar Sci. 8, 285–309 (2016).
Behrenfeld, M. J. & Falkowski, P. G. Photosynthetic rates derived from satellite-based chlorophyll concentration. Limnol. Oceanogr. 42, 1–20 (1997).
Flombaum, P. et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 110, 9824–9829 (2013).
Long, M. C., Deutsch, C. & Ito, T. Finding forced trends in oceanic oxygen. Global Biogeochem Cy. 30, 381–397 (2016).
Glock, N. et al. Metabolic preference of nitrate over oxygen as an electron acceptor in foraminifera from the Peruvian oxygen minimum zone. Proc. Natl. Acad. Sci. USA 116, 2860–2865 (2019).
Canfield, D. E. & Kraft, B. The ‘oxygen’ in oxygen minimum zones. Environ Microbiol. 24, 5332–5344 (2022).
Galloway, J. N. et al. Nitrogen Cycles: Past, Present, and Future. Biogeochemistry. 70, 153–226 (2004).
Codispoti, L. A. et al. The oceanic fixed nitrogen and nitrous oxide budgets: Moving targets as we enter the anthropocene? Sci Mar. 65, 85–105 (2007).
De Brabandere, L. et al. Vertical partitioning of nitrogen-loss processes across the oxic-anoxic interface of an oceanic oxygen minimum zone. Environ Microbiol. 16, 3041–3054 (2014).
Ding, C. et al. The Composition and Primary Metabolic Potential of Microbial Communities Inhabiting the Surface Water in the Equatorial Eastern Indian Ocean. Biology (Basel). 10, (2021).
Ding, C. et al. Comparison of Diazotrophic Composition and Distribution in the South China Sea and the Western Pacific Ocean. Biology (Basel). 10, 248 (2021).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 35, 725–731 (2017).
Nishimura, Y. et al. The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments. Sci Data. 9, 305 (2022).
Paoli, L. et al. Biosynthetic potential of the global ocean microbiome. Nature. 607, 111–118 (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Haro-Moreno, J. et al. New insights into marine group III Euryarchaeota, from dark to light. ISME J. 11, 1102–1117 (2017).
Dafner, E. V. Segmented continuous-flow analyses of nutrient in seawater: intralaboratory comparison of Technicon AutoAnalyzer II and Bran+Luebbe Continuous Flow AutoAnalyzer III. Limnol. Oceanogr. Methods. 13, 511–520 (2015).
Xia, X. et al. Rare bacteria in seawater are dominant in the bacterial assemblage associated with the Bloom-forming dinoflagellate Noctiluca scintillans. Sci Total Environ. 711, 135107 (2020).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 18, 1403–14 (2016).
Milke, F. et al. Selection, drift and community interactions shape microbial biogeographic patterns in the Pacific Ocean. ISME J. (2022).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 37, 852–857 (2019).
Amir, A. et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems. 2, e00191–16 (2017).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890 (2018).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 6, 158 (2018).
Li, D. et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31, 1674–6 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Fu, L. et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 28(23), 3150–3152 (2012).
Li, H. et al. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 26, 589–595 (2010).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 36, 2251–2252 (2019).
Wu, Y.-W. et al. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2, 26 (2014).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 7, e7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 11, 1144–6 (2014).
Ondov, B. D. et al. sourmash: a library for MinHash sketching of DNA. Journal of Open Source Software. (2016).
Yu, K. et al. Recovery of high-qualitied Genomes from a deep-inland Salt Lake Using BASALT. Preprint at https://www.biorxiv.org/content/10.1101/2021.03.05.434042v2 (2021).
Olm, M. R. et al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Chklovski, A. et al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods. 20, 1203–1212 (2023).
Chaumeil, P. A. et al. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics. 38, 5315–5316 (2022).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30(14), 2068–9 (2014).
Edgar, R. C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun. 13, 6968 (2022).
Criscuolo, A. & Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evolutionary Biology. 10, 210 (2010).
Nguyen, L. T. et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32, 268–74 (2015).
Kalyaanamoorthy, S. et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14, 587–589 (2017).
NCBI BioProject. https://identifiers.org/ncbi/bioproject:PRJNA1031568 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP468222 (2023).
Wang, X. M. Northeast Indian Ocean metagenomic dataset. Figshare https://doi.org/10.6084/m9.figshare.24314026.v2 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (42188102, 92251306, 42222604, and 42276163). S. Hou was supported by the MEL Visiting Fellowship of Xiamen University (MELRS2210) and by Shenzhen Science, Technology and Innovation Commission Programme (JCYJ20220530115401003). Data and samples were collected on board of R/V Shiyan3 during the open research cruise NORC2021-10 supported by NSFC Shiptime Sharing Project (Project number: 42049910).
Author information
Authors and Affiliations
Contributions
S.H. and Q.Z. conceived this study. X.W. conducted field sampling, DNA extraction, amplicon and metagenomic data analysis. X.W. produced all figures and wrote the first draft under the supervision of S.H. and Q.Z. M.A., S.H. and Q.Z. revised the draft. All authors reviewed and contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Zain Ul Arifeen, M., Hou, S. et al. Depth-dependent microbial metagenomes sampled in the northeastern Indian Ocean. Sci Data 11, 88 (2024). https://doi.org/10.1038/s41597-024-02939-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02939-4
- Springer Nature Limited