
Abstract
Waterlily aphid, Rhopalosiphum nymphaeae (Linnaeus), is a host-alternating aphid known to feed on both terrestrial and aquatic hosts. It causes damage through direct herbivory and acting as a vector for plant viruses, impacting worldwide Prunus spp. fruits and aquatic plants. Interestingly, R. nymphaeae’s ability to thrive in both aquatic and terrestrial conditions sets it apart from other aphids, offering a unique perspective on adaptation. We present the first high-quality R. nymphaeae genome assembly with a size of 324.4 Mb using PacBio long-read sequencing. The resulting assembly is highly contiguous with a contig N50 reached 12.7 Mb. The BUSCO evaluation suggested a 97.5% completeness. The R. nymphaeae genome consists of 16.9% repetitive elements and 16,834 predicted protein-coding genes. Phylogenetic analysis positioned R. nymphaeae within the Aphidini tribe, showing close relations to R. maidis and R. padi. The high-quality reference genome R. nymphaeae provides a unique resource for understanding genome evolution in aphids and paves the foundation for understanding host plant adaptation mechanisms and developing pest control strategies.
Similar content being viewed by others
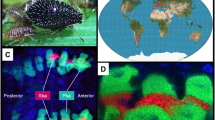
Background & Summary
Rhopalosiphum nymphaeae (Linnaeus), also known as the waterlily aphid, is a polyphagous host-alternating aphid that has been reported to feed on both terrestrial hosts plants like Prunus spp.1 and various aquatic hosts belonging to Nympahaeaceae, Araceae etc.2 (Fig. 1a). As R. nymphaeae is a devastating pest through direct herbivory and as plant virus vectors to some domesticated fruits and crops2, the comprehensive understanding of this insect is of great agricultural value. On the other hand, R. nymphaeae has a contrasting host range compared to its closely related species—likely the only aphid to live in both aquatic and terrestrial conditions2—making it a distinctive study model for revealing how insects adapt to diverse hosts.

Evolution of R. nymphaeae. (a) Pictures show R. nymphaeae alatae (up left) and apterae (down right) feeding on the great duckweed [Spirodela polyrhiza (L.) Schleid., Araceae]. Black scale bars indicate 1 mm. (b) The plot shows the Maximum-likelihood phylogenomic tree reconstructed based on the one-to-one orthologs of 12 aphids (Schlechtendalia chinensis, Aphis gossypii, Aphis glycines, Rhopalosiphum nymphaeae, Rhopalosiphum maidis, Rhopalosiphum padi, Pentalonia nigronervosa, Sitobion miscanthi, Acyrthosiphon pisum, Diuraphis noxia, Myzus cerasi, and Myzus perisicae) and the grape phylloxera (Daktulosphaira vitifoliae) as the outgroup. The blue dots on the internal nodes indicate 100% bootstrapping support. (c) Genome synteny analysis between R. nymphaeae and R. maidis genomes. The up panel bars show four assembled chromosomes of R. maidis with their names above, while the down panel shows 36 long contigs (with lengths greater than 1 Mb) of R. nymphaeae with names below.
Here, we report the first high-quality draft genome assembly of R. nymphaeae, generated using PacBio long-read sequencing (~31.7 Gb HiFi reads, with N50 = 19.3 kb). After assembling long reads into contigs, we removed bacterial comtaminations (298 contigs comprising 19.5 Mb; see Supplementary Fig. 1 and Supplementary Data 1). Among them, 145 contigs matched the well-studied aphids’ endosymbiotic bacterium, Buchnera aphidicola3,4 (Supplementary Data 1). The final monoploid genome assembly of R. nymphaeae consists of 91 contigs with a total size of 324.4 Mb (Table 1). The contig N50 reaches 12.7 Mb, and the longest contig is 47.9 Mb (Table 1). These data suggest the contiguity of the R. nymphaeae genome assembly is one of the highest compared to 13 previously published aphid genomes5,6,7,8,9,10,11,12 (Supplementary Data 2). We identified 54.8 Mb repetitive elements, which account for 16.9% of the R. nymphaeae assembly (Table 2). After soft-masking the R. nymphaeae genome, we predicted 16,834 protein-coding genes with an average length of 6,760 bp (Table 3) using the BRAKER pipeline13,14,15,16,17,18 that incorporated empirical evidence of transcripts assembled from short-reads sequencing (RNA-seq) data and full-length transcripts from long-read PacBio sequencing (Iso-seq) data, and extrinsic evidence based on the homology from other aphids (see methods).
We constructed a maximum likelihood phylogenetic tree based on the low-copy (often referred as single-copy) orthologs to determine the relationship of R. nymphaeae with the other 11 members from Aphidoidea (Fig. 1b). In accordance with the previously constructed phylogeny based on the mitochondrial sequences19, R. nymphaeae is positioned within the Aphidini tribe. It is closely related to R. maidis and R. padi (Fig. 1b).
We conducted a genome synteny analysis between R. nymphaeae and R. maidis20 (Fig. 1c). Despite observing several genomic rearrangements, there is a notable conservation between the two genomes. Among the 38 longest contigs (lengths greater than 1 Mb) from the R. nymphaeae genome, 36 exhibited synteny with four chromosomes of R. maidis. Most chromosomal regions from the R. maidis genome aligned with the R. nymphaeae genome assembly.
This study presents the first genome assembly for R. nymphaeae, providing a valuable dataset for understanding genome evolution in aphids. This genome assembly not only serves as a crucial resource for exploring potential pest control strategies, but also paves the way for subsequent comparative genomics and experimental evolution studies, aiming to decipher the adaptive mechanisms of this organism to a changing environment.
Methods
Sample preparation and sequencing
The aphid was collected in the summer of 2020 on a duckweed population growing near the University of Münster, Germany. A population derived from a single aphid individual was maintained in the lab on Spirodela polyrhiza plants. We extracted DNA from the aphids using the Monarch HMW DNA Extraction Kits. The DNA was sequenced on a Pacbio sequel II at Novogene, Beijing, China. To assist the protein-coding gene prediction, we generated both PacBio Iso-seq (27.2 Gb, N50 = 2,191 bp) and Illumina short-reads RNA-seq libraries (150 bp paired-end, 41.9 million reads) using total RNAs from the whole body of R. nymphaeae.
Genome assembly and contamination screening
We assembled the genome using Hifiasm (v.0.19.3-r572)21,22 with high-quality HiFi reads. We trimmed both ends of reads by 20 bp (with -z20 option). Next, the assembled genome was screened using two strategies to eliminate contamination from potential sequencing adaptors and foreign DNA: the NCBI Foreign Contamination Screen (FCS) tool suite23 and BlobTools (v 1.1.1)24. For the FCS-adaptor (v 0.5.0)23 screening, default settings were used, and no adapter sequence was found in the assembly. Both FCS-GX (v 0.5.0)23 and BlobTools (v 1.1.1)24 identified foreign DNA, which mostly originated from bacteria. Contaminated contigs identified by FCS-GX (v 0.5.0)23 or BlobTools (v 1.1.1)24 were removed from the assembly. In the case of screening using Blobtools24, assembly contigs longer than 1 Mb were split into smaller fragments of 1 Mb each (with a 1 Kb overlap between two consecutive fragments) to reduce the computational burden during alignment against the UniProt Reference Proteomes25 using Diamond (v2.1.8.16)26.
Repetitive element annotation
RepeatModeler (v2.0.2)27 was used to generate a de novo repeat library from the R. nymphaeae genome. The “-LTRStruct” flag was added in this step to also identify long terminal repeat structure. Next, based on the classified repeat library generated from the RepeatModeler, RepeatMasker (v4.1.5) was used to predict and soft mask the repeats in the R. nymphaeae genome.
Protein-coding gene annotation
We used the BRAKER13,14,15,16,17,18 pipeline for protein-coding gene prediction, which combines gene models predicted based on short-read RNA-seq transcriptome (BRAKER1 method15,28,29,30,31,32) and protein homologs from other aphids (BRAKER2 method17,28,29,33,34,35,36,37). We then used TSEBRA18 to compare these predictions with full-length transcripts derived from Iso-seq data, ultimately selecting the most optimal gene models.
For the BRAKER1 run, paired-end RNA-seq reads from R. nymphaeae were processed using Trimmomatic (v0.39)38 with parameters of “ILLUMINACLIP:TruSeq 3-PE-2.fa:2:30:10 SLIDINGWINDOW:4:15 MINLEN:36 HEADCROP:10”, which trimmed the first 10 bp and to filter the possible Illumina sequencing adaptor sequences. FastQC (v0.11.9, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to perform the quality control before and after the filtration. Next, the cleaned reads were aligned to the R. nymphaea genome using HISAT2 (v2.2.1)39 with default settings. After that, BRAKER1 was fed with the repeat soft-masked R. nymphaea genome and RNA-seq aligned BAM file. It automatically performed GeneMark-ET training using spliced alignment information from the RNA-seq and, based on which, predicted gene models using AUGUSTUS40. For the BRAKER2 run, similar automatic training of GeneMark-EP+37 was done to guide the AUGUSTUS’s gene prediction, but this time, BRAKER2 utilised protein-coding exon boundaries information, which was from the alignment of the protein sequences from Aphidoidea that were downloaded from UniProt41. For the Iso-seq data processing, the command-line tools from SMRT Link software from PacBio (https://www.pacb.com/) were used. In brief, consensus sequences generated from raw subread were filtered to remove primers, concatemers and poly(A) tails to get Full-Length Non-concatemer (FLNC) reads. These FLNC reads were then clustered, aligned to the R. nymphaea reference genome using minimap2 (v2.24)42 and collapsed using Cupcake (v0.1.4, https://github.com/Magdoll/cDNA_Cupcake) to get the full-length transcripts. GeneMarkS-T43 was then used to predict the protein-coding region for each full-length transcript. The gene models predicted independently from BRAKER1 and BRAKER2 were then merged and compared with full-length transcripts from Iso-seq data using TSEBRA with default options. Only the longest isoform was kept for each gene model.
After the best gene models were selected by TSEBRA, we adopted AGAT (v0.8.0, https://github.com/NBISweden/AGAT) for three rounds of filtration, including the removal of 1) genes with length less than 100 bp, 2) genes with coding sequences harbour repetitive elements higher than 20%, 3) genes have only one exon and don’t have a complete start or/and stop codon predicted.
For the functional annotation, proteins sequences translated from the gene annotation were aligned to the UniProtKB41 database using Blastp (BLAST + v2.12.0)29 with “-evalue 1e-6 -max_hsps 1 -max_target_seqs. 1 -outfmt 6” parameters and processed using InterProScan (v5.63–95.0)44 with “-goterms -iprlookup” options, respectively.
Phylogenetic tree reconstruction and genome synteny analyses
We identified 3,550 low-copy (often referred as single-copy) ortholog groups based on protein sequences (translated from the longest isoform of each gene) from 12 aphid genomes, including Schlechtendalia chinensis12, Aphis gossypii8, Aphis glycines10, R. nymphaeae, Rhopalosiphum maidis20, Rhopalosiphum padi45, Pentalonia nigronervosa9, Sitobion miscanthi7, Acyrthosiphon pisum11, Diuraphis noxia5, Myzus cerasi9, and Myzus perisicae11, and the grape phylloxera (Daktulosphaira vitifoliae)46 genome using OrthoFinder (v2.5.4)47,48. Those low-copy ortholog groups were concatenated and aligned automatically by OrthoFinder and generated a multiple sequence alignment file, which was used for phylogenetic analysis. For the phylogenetic tree reconstruction, ModelTest-NG (v0.2.0)49 was used first and found “JTT + I + G4” to be the best model, which was later used in the maximum likelihood phylogenetic tree reconstruction using RAxML-NG (v1.2.0)50. We used iTOL (v6)51 for tree visualization.
For the genome synteny analysis, the one-to-one orthologs between R. nymphaeae and R. maidis genomes were extracted from OrthoFinder’s result and fed to MCScanX_h52, which was used with “-b 2” option to get the inter-species collinearity between R. nymphaeae and R. maidis. SynVisio53 was used to visualize the genome synteny.
Evaluations of R. nymphaeae genome assembly and protein-coding gene annotation
Merqury (v1.3)54, an assembly evaluation software that compares the distribution of k-mers in sequencing reads and the final assembly, was used to estimate the base-level accuracy and completeness of the R. nymphaeae assembly, with an estimated optimal k-mer size of 19. Benchmarking Universal Single-Copy Orthologs (BUSCO, v5.4.3)55 was used to evaluate the genome assembly and protein-coding gene annotation of R. nymphaeae with “-m genome” and “-m proteins”, respectively. The “hemiptera_odb10” reference lineage database (2,510 BUSCOs) was chosen for both runs. In addition, DOGMA (v3.7)56,57 with “insects” reference core set was also used to assess the completeness of gene annotation in R. nymphaeae genome based on conserved protein domains. For the gene model structure visual checking, JBrowse 258 was used.
Data Records
The genomic PacBio sequencing, RNA-seq and Iso-seq data have been updated to the National Center for Biotechnology Information (NCBI) under the BioProject of PRJNA101528859. R. nymphaeae genome assembly and the gene annotation have been deposited in Genbank under the accession number JAZAQC00000000060 and Figshare61.
Technical Validation
We assessed the completeness and accuracy of R. nymphaeae genome assembly from five aspects. First, the summary statistics of the genome assembly revealed that the longest contig reaches 47.9 Mb, contig N50 reaches 12.7 Mb, and 38 contigs are longer than 1 Mb, constituting 98.45% of the assembly. All these data indicate that this assembly is one of the highest contiguous genome assemblies among 14 aphids that were in comparison (Supplementary Data 2). Second, the blob plots show that contaminant contigs, which were mainly from symbiotic bacteria, were completely removed from the assembly (Supplementary Figs. 1 and2). Third, using Merqury (v 1.3)54, we estimated the base-level accuracy and completeness of the R. nymphaeae assembly by comparing k-mers from the final assembly to those in the PacBio HiFi reads. Merqury reported a consensus quality (QV) of 69 and a completeness of 96.15% for the R. nymphaeae assembly, as visualized by the spectra-cn plot (Supplementary Fig. 3). In the spectra-cn plot, a homozygous peak was found at 90X coverage, suggesting a highly complete and accurate assembly. Fourth, the BUSCO evaluation indicated that 97.5% of gene orthologs (97% are single copy and 0.5% are duplicated) are present in the R. nymphaeae genome assembly (Supplementary Table 1). Lastly, the mapping rate of RNA-seq and Iso-seq reads are as high as 95.85% and 95.98%, respectively. These results, together, support our conclusion of a high-quality genome assembly.
Two methods were adopted to check the quality of protein-coding gene annotation in the R. nymphaeae genome assembly. First, BUSCO evaluation was used again, but this time under the “-m proteins” mode, and it suggested that the completeness of the annotation reached 95.3% (94.1% are single copy and 1.2% are duplicated, Supplementary Table 1). Second, DOGMA, a tool that assesses predicted proteins based on the conserved protein domains, indicated that 85.03% of conserved domains could be found in the R. nymphaeae gene annotation.
Code availability
All the data analysis procedures were done following published manuals or public protocols of the software described in the Methods. Parameters for each software were detailed. Codes used to run gene annotation pipelines were deposited in GitHub with the link: https://github.com/Xu-lab-Evolution/Waterlily_aphid_genome_project.
References
Blackman, R. L. & Eastop, V. F. Aphids on the world’s trees: an identification and information guide. (Cab International, 1994).
Ted D. Center, F. A. D. Jr., Greg P. Jubinsky, & Michael J. Grodowitz. Insects and other arthropods that feed on aquatic and wetland plants (United States Department of Agriculture) (Technical Bulletin, 1999).
Braendle, C. et al. Developmental origin and evolution of bacteriocytes in the aphid-Buchnera symbiosis. PLoS Biol. 1, E21 (2003).
Wilson, A. C. et al. Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect Mol. Biol. 19, 249–258 (2010).
Nicholson, S. J. et al. The genome of Diuraphis noxia, a global aphid pest of small grains. BMC Genomics 16, 429 (2015).
Thorpe, P., Escudero-Martinez, C. M., Cock, P. J. A., Eves-van den Akker, S. & Bos, J. I. B. Shared Transcriptional Control and Disparate Gain and Loss of Aphid Parasitism Genes. Genome Biol. Evol. 10, 2716–2733 (2018).
Jiang, X. et al. A chromosome-level draft genome of the grain aphid Sitobion miscanthi. Gigascience 8 (2019).
Quan, Q. M. et al. Draft genome of the cotton aphid Aphis gossypii. Insect Biochem. Mol. Biol. 105, 25–32 (2019).
Mathers, T. C., Mugford, S. T., Hogenhout, S. A. & Tripathi, L. Genome Sequence of the Banana Aphid, Pentalonia nigronervosa Coquerel (Hemiptera: Aphididae) and Its Symbionts. G3-Genes Genom. Genet. 10, 4315–4321 (2020).
Wenger, J. A. et al. Whole genome sequence of the soybean aphid, Aphis glycines. Insect Biochem. Mol. Biol. 123 (2020).
Mathers, T. C. et al. Chromosome-Scale Genome Assemblies of Aphids Reveal Extensively Rearranged Autosomes and Long-Term Conservation of the X Chromosome. Mol. Biol. Evol. 38, 856–875 (2021).
Wei, H. Y. et al. Chromosome-level genome assembly for the horned-gall aphid provides insights into interactions between gall-making insect and its host plant. Ecol. Evol. 12 (2022).
Stanke, M., Schoffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7 (2006).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-Genome Annotation with BRAKER. Gene Prediction: Methods and Protocols 1962, 65–95 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP plus and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3 (2021).
Gabriel, L., Hoff, K. J., Bruna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22 (2021).
Park, J., Kim, Y., Xi, H., Park, J. & Lee, W. The complete mitochondrial genome of Rhopalosiphum nymphaeae (Linnaeus, 1761) (Hemiptera: Aphididae). Mitochondrial DNA B Res. 5, 1613–1615 (2020).
Chen, W. B. et al. Genome sequence of the corn leaf aphid (Rhopalosiphum maidis Fitch). Gigascience 8 (2019).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. bioRxiv, 2023.2006. 2002.543519 (2023).
Laetsch, D. R. & Blaxter, M. L. BlobTools: Interrogation of genome assemblies. F1000Research 6, 1287 (2017).
UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Stromberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42 (2014).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Iwata, H. & Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 40 (2012).
Gotoh, O., Morita, M. & Nelson, D. R. Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC Bioinformatics 15 (2014).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP plus: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907-+ (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Bateman, A. et al. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Tang, S. Y. Y., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Morales-Hojas, R. et al. Population genetic structure and predominance of cyclical parthenogenesis in the bird cherry-oat aphid Rhopalosiphum padi in England. Evol. Appl. 13, 1009–1025 (2020).
Rispe, C. et al. The genome sequence of the grape phylloxera provides insights into the evolution, adaptation, and invasion routes of an iconic pest. BMC Biol. 18, 1–25 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16 (2015).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (2019).
Darriba, D. et al. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 37, 291–294 (2020).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (2012).
Bandi, V. & Gutwin, C. in Graphics Interface 2020.
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Gene Prediction: Methods and Protocols 1962, 227–245 (2019).
Dohmen, E., Kremer, L. P., Bornberg-Bauer, E. & Kemena, C. DOGMA: domain-based transcriptome and proteome quality assessment. Bioinformatics 32, 2577–2581 (2016).
Kemena, C., Dohmen, E. & Bornberg-Bauer, E. DOGMA: a web server for proteome and transcriptome quality assessment. Nucleic Acids Res. 47, W507–W510 (2019).
Diesh, C. et al. JBrowse 2: a modular genome browser with views of synteny and structural variation. Genome Biol. 24 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP459763 (2024).
Wang, Y. & Xu, S. Genome assembly of the waterlily aphid Rhopalosiphum nymphaeae. Genbank https://identifiers.org/ncbi/insdc:JAZAQC000000000 (2024).
Wang, Y. & Xu, S. Genome assembly and gene annotation of waterlily aphid (Rhopalosiphum nymphaeae L.). figshare https://doi.org/10.6084/m9.figshare.24118587.v3 (2024).
Acknowledgements
We thank Laura Bötnner for collecting the aphid sample in the field, and Marie Sarazova and Thoomke Brüning for keeping the aphid colony in the lab. We are grateful to Martine Ursula for isolating the HMW DNA from the aphids. This project is funded by the German Research Foundation (project number 438887884 to SX). Parts of this research were conducted using the supercomputer Mogon and/or advisory services offered by the Johannes Gutenberg University Mainz (hpc.uni-mainz.de), which is a member of the AHRP (Alliance for High-Performance Computing in Rhineland Palatinate) and the Gauss Alliance e.V.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
S.X. conceived and supervised the study. Y.W. and S.X. performed the data analyses. Y.W. and S.X. wrote the manuscript. All authors have read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Xu, S. A high-quality genome assembly of the waterlily aphid Rhopalosiphum nymphaeae. Sci Data 11, 194 (2024). https://doi.org/10.1038/s41597-024-03043-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03043-3
- Springer Nature Limited