
Abstract
By using PacBio HiFi technology, we produced over 700 Gb of long-read sequencing (LRS) raw data; and by using Illumina paired-end whole-genome shotgun (WGS) sequencing technology, we generated more than 70 Gb of short-read sequencing (SRS) data. With LRS data, we assembled one genome and then generate a set of annotation data for an early-matured Geng/japonica glutinous rice mega variety genome, Longgeng 57 (LG57), which carries multiple elite traits including good grain quality and wide adaptability. Together with the SRS data from three parents of LG57, pedigree genome variations were called for three representative types of genes. These data sets can be used for deep variation mining, aid in the discovery of new insights into genome structure, function, and evolution, and help to provide essential support to biological research in general.
Similar content being viewed by others


Background & Summary
In recent years, the planting area for rice (Oryza sativa L.) in Heilongjiang (HLJ) province of China has increased to around 4 million ha1. For this global largest planting region for early Geng/japonica rice, which is about 2.6 times larger than the rice planting area of Japan2, determining how to transfer its advantages in agriculture to other branches of the economy remains a significant challenge for agriculture researchers.
Early-matured Geng/japonica varieties provide the base for food security3, and supply critical agro-industrial materials, especially glutinous varieties. Glutinous rice, also called sticky rice, is becoming increasingly popular because of growing public awareness of health issues4. Glutinous rice has health benefits in managing diabetes, inhibiting chronic diseases, enhancing digestion, and reducing inflammation5. In addition to being an elite cooking material for a low gluten diet and ‘good food’6, glutinous rice also provides raw materials for environment-friendly industry7,8,9. Longgeng 57 (LG57), a glutinous early variety, has favorable quality and stable-yield behavior in the early Geng/japonica planting region; therefore, it is now planted over more than 120,000 ha per year on average.
Grain quality traits of rice are largely controlled by major genes, such as Waxy for the amylose content and OsNramp5 for the mineral nutritional quality10,11,12. Thus, further improvement of grain quality of glutinous rice, e.g., LG57, also requires more genome information.
Currently, joint analysis has become a trend in biotechnology-based rice breeding in HLJ. For example, the Rice Molecular Breeding (RMB) laboratory from the Institute of Crop Science (ICS), Chinese Academy of Agricultural Sciences (CAAS), has set up a genome-based breeding scheme with the aid of both core germplasms of 3K-RG13, and the Rice Functional Genomics Breeding (RFGB) information platform14. It also widely cooperates with local research institutes from HLJ, including Jiamusi Rice Research Institute (JMS-RRI) and Suihua RRI (SH-RRI)3. Herein, we present a dataset from a collaboration between the RMB laboratory and JMS-RRI for early-matured Geng/japonica including LG57. Information based on this dataset for certain target genes, such as Waxy and OsNramp5, were also included as examples for data validation. This dataset comprises more than 770 Gb of pedigree genome data that will be useful for researches in general.
Methods
Plant material and library construction
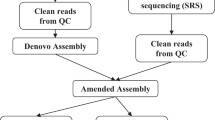
The early-matured Geng/japonica variety Longgeng57 (LG57) was developed by our own and licensed to be released in 2017 and is now one mega variety with multiple elite traits and widely planted (more than 120,000 hectare per year) in Heilongjiang province in Northeast of China. High-molecular-weight genomic DNA was extracted from 10-day-old leaves of LG57 pedigree members (multiple seeds) with modified CTAB method followed by 0.5x bead purification for twice. The DNA sample through the qualification processes by both 0.75% agarose gel assay and Nanodrop was quantified with Qubit. Then the sample of LG57 met the standard was submitted to the constructions of PacBio HiFi library for long-read sequencing (LRS). Samples of three parents (Longnuo 2 (LN2), Punian 8 (PN8), and Longgeng 29 (LG29)) were submitted to construct Illumina libraries short-read sequencing (SRS) (Fig. 1).

Outlines of the workflow used to generate and analyze the pedigree genome data for Longgeng 57 (LG57).
Genomic data were generated for all pedigree members, as listed in Table 1. Among them, PacBio (Menlo Park, CA, USA) protocols were adopted for long-read sequencing of LG57 and Illumina (San Diego, CA, USA) protocols were used for short-read sequencing. The details are as follows.
DNA sample testing
DNA extraction from samples was carried out using a routine method that met the quality standard required for sequencing according to a previous study3. Sample purity and quantity were detected using a Nano Photometer® (IMPLEN, Westlake Village, CA, USA) and a Qubit® 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), respectively, in combination with Agarose electrophoresis (concentration 1%, voltage 120 V for 45 min).
Library construction and Inventory inspection
Covaris® g-TUBE15 was used to break the genomic DNA into suitable large pieces. Magnetic beads were then used for enrichment and purification. SageELF (Sage, Newcastle upon Tyne, UK) was adopted to screen and purify the DNA fragments. An Annoroad® Universal DNA Library Prep Kit V2.0 (Annoroad Gene Technology, Beijing, China) was used for sample preparation, including end repair and ligation addition.
To ensure the quality of the library, a three-step quality check procedure was adopted as follows. After the library was constructed, the Qubit 3.0 was used for preliminary quantification. Then, the library was diluted to 1 ng/μL and the insert size was checked using an Agilent 2100 instrument (Agilent, Santa Clara, CA, USA). The effective concentration of the library was accurately quantified using quantitative real-time reverse transcription PCR (qRT-PCR) in a Bio-Rad CFX96 PCR instrument with a Bio-Rad IQ SYBR GRN Kit (both Bio-Rad, Hercules, CA, USA).
Sequencing
The single-molecule real-time (SMRT) method was adopted for the long-read sequencing (LRS) according to standard method (PacBio). Short-read sequencing (SRS) was carried out on the NovaSeq 6000 S4 platform (Illumina) to obtain a 250 bp double-ended sequencing reads.
Genome assembly, validation and annotation
For the LRS data obtained by HiFi library sequencing, the raw data (subreads) from the PacBio sequencing was filtered by using SMRT link v9.0.0.92188 (https://www.pacb.com/support/software-downloads/) with default parameters to obtain high-quality circular consensus sequences (CCS) data. For the assembly, hifiasm16 with default parameters were employed based on the CCS data. Merqury17 was adopted for the quality check of LG57 assembly. Also, BUSCO (Benchmarking Universal Single-Copy Orthologs)18 was used for genome assembly quality assessment. BUSCO analysis with default parameters was carried out using a single-copy gene set of several large evolutionary branches based on the OrthoDB (http://cegg.unige.ch/orthodb). The gene set was compared with the assembled genome using embryophyta_odb10, and the accuracy and completeness were assessed based on the proportions and completeness of the alignment.
Based on the LG57 assembly, two strategies were adopted for genome annotation. The first was a homologous strategy. RepeatMasker with default parameters19 based on RepBase20 was used to annotate repeats. For gene structures, BLAST21 with evalue = 1e-5 and GeMoMa22 with default parameters were used. Prediction of rRNAs, snRNAs, and miRNAs was carried out by aligning the assembly with known non-coding RNA libraries, e.g., Rfam23.The second was a de novo strategy. For repeat analysis, RepeatModeler (https://www.repeatmasker.org/RepeatModeler/) with -engine ncbi was adopted. For protein-coding gene prediction, Augustus24 with–genemodel = partial, SNAP(https://github.com/KorfLab/SNAP), and GeneMark25 with default parameters were adopted. Based on the above predictions, EVidence Modeler (EVM)26 with default parameters was used to integrate the gene sets predicted by various strategies into a non-redundant gene set. The resulting predictive gene set was compared with various functional databases using UniProt27, NCBI (https://www.ncbi.nlm.nih.gov/nucleotide/), PFAM28, eggNOG29, GO (gene ontology)30, and KEGG (Kyoto Encyclopedia of Genes and Genomes)31. For tRNA sequence prediction, we used tRNAscan-SE32 with parameters of -X 20 and –z 8.
The SRS data were aligned to the reference genome and variations were called using a pipeline comprising BWA33, SAMtools34, and GATK35 with default parameters, with Nipponbare IRGSP 1.036 as the reference genome.
Data Records
The assembly of LG57 is accessible at NCBI through GenBank37 or the following accession ID of JAXQPT00000000037. Additionally, the raw read data for LG57 in the bam format are also available with accession number of SRR2537649638. Other sequencing pedigree genomic data for parents of LG57, including PN8 (SRR24688636)39, LN2 (SRR24688637)40, and LG29 (SRR24688635)41. Annotation data for LG57 are accessible through figshare42. All above data except for the bam files are also accessible in RFGB website (https://rfgb.rmbreeding.cn/download/publicDataDownload/download?dataset=3).
Technical Validation
A total 1,671,418 of reads were obtained. The averaged read-length is 16,831.42 bp and N50 value is more than 17 Kb. The distribution of these reads was shown in Fig. 2. A rough assembly for LG57 was carried out. A quality checking for the assembly of Longgeng 57 was also carried out by using Merqury and BUSCO. Based on the output of Merqury, the completeness of assembly was 99.5% and the QV was 62.0 (Table 2). As shown in Table 3, N50 of contig has arrived at more than 27 Mb, which is over 10 times of our previous work with SJ183. As shown in Table 4, a total of 1614 groups were searched by BUSCO, the complete groups accounted for about 98.8%. Functional genes predicted in LG57 comparing with those from databases were shown in Table 5. Identified by RepeatMasker, the total length of the repeat sequences is approximately 170MB, accounting for 43.13% of the whole LG57 genome (Table 6). Prediction results of different types of non-coding RNA including miRNA, tRNA, rRNA, and snRNA were listed in Table 7. These RNAs together accounting for 81.3% of the LG57 genome. We also compared the parameters of LG57 to the other assemblies. Averaged gene length of LG57 is longer than those of the others (Table 8).

Distribution of lengths of circular consensus sequences (CCS) reads for Longgeng 57 (LG57).
For the SRS data of the three parents (LN2, PN8, and LG29), we firstly aligned them against reference genome IRGSPv1.0 to gain the genome variations. Then we adopted sequences of three representative types of major genes from IRGSPv1.0 as queries and BLAST against LG57 assembly to get target sequences.
More details about data validation cases from three key genes for LG57 breeding works based on the pedigree genome data especially the assembly data of LG57 and the alignment data of its three parents were listed in Table 9. The maturing time of Geng/japonica is largely affected by Hd1 gene43, which commonly harbors highly-diverse variation panels in rice genome44. In this region, LG57 and its three early Geng/japonica parents show extremely high consistency. The grain quality of glutinous rice is mainly controlled by Waxy gene45. LG57 possess better grain quality than other glutinous early Geng/japonica varieties, such as PN2 and LN2. There are three differences in the Waxy genes found between PN8 and LN2. Although a common variation in the 5th exon of Waxy was found in PN8, LN2, and their progeny, LG57, there is a unique 23 bp deletion in the 1st exon that is shared by LG57 and its non-glutinous parent, LG29. Variations in major gene OsNramp5 affects the mineral concentrations in rice10. It’s notable that LG57 has variations that are different from all three parents, which is supposed to be caused by spontaneous mutations in breeding process46,47. Three types of variations in three representative genes validated the genome data and indicated the possible applications with this dataset. In a word, the quality of the pedigree genome data of LG57 was sufficient for public reuse in the future.
Code availability
No custom code was used during this study for the curation and/or validation of the dataset.
References
NBSC. National Data, https://data.stats.gov.cn/english/ (2022).
FAO. FAOSTAT-Crops and livestock products, https://www.fao.org/faostat/en/ (2022).
Nie, S. J. et al. Assembly of an early-matured japonica (Geng) rice genome, Suijing18, based on PacBio and Illumina sequencing. Sci Data 4, 170195, https://doi.org/10.1038/sdata.2017.195 (2017).
EMR. Global Glutinous Rice Market Outlook. (2022).
Terashima, Y., Nagai, Y., Kato, H., Ohta, A. & Tanaka, Y. Eating glutinous brown rice for one day improves glycemic control in Japanese patients with type 2 diabetes assessed by continuous glucose monitoring. Asia Pac J Clin Nutr 26, 421–426, https://doi.org/10.6133/apjcn.042016.07 (2017).
Cadogan, M. Sticky rice & mango, https://www.bbcgoodfoodme.com/recipes/sticky-rice-and-mango/ (2022).
Ling, H. Y. et al. Amylopectin from Glutinous Rice as a Sustainable Binder for High-Performance Silicon Anodes. ENERGY & ENVIRONMENTAL MATERIALS 4, 263–268, https://doi.org/10.1002/eem2.12143 (2021).
GAREFU. What Is Glutinous Rice Glue?, https://www.garefutech-paste.com/news/what-is-glutinous-rice-glue-60117817.html (2022).
Yao, L. et al. Glutinous rice-derived carbon material for high-performance zinc-ion hybrid supercapacitors. Journal of Energy Storage 58, 106378, https://doi.org/10.1016/j.est.2022.106378 (2023).
Zhao, F. J. & Chang, J. D. A weak allele of OsNRAMP5 for safer rice. J Exp Bot 73, 6009–6012, https://doi.org/10.1093/jxb/erac323 (2022).
Liu, C. L. et al. Characterization of a major QTL for manganese accumulation in rice grain. Scientific Reports 7, 17704, https://doi.org/10.1038/s41598-017-18090-7 (2017).
Luo, J. S. et al. A defensin-like protein drives cadmium efflux and allocation in rice. Nature Communications 9, 645, https://doi.org/10.1038/s41467-018-03088-0 (2018).
Wang, W. et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557, 43–49, https://doi.org/10.1038/s41586-018-0063-9 (2018).
Wang, C. C. et al. Towards a deeper haplotype mining of complex traits in rice with RFGB v2.0. Plant Biotechnology Journal 18, 14–16, https://doi.org/10.1111/pbi.13215 (2020).
Covaris. g-TUBE, https://www.covaris.com/products-services/consumables/g-tube (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32, W20–25, https://doi.org/10.1093/nar/gkh435 (2004).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol 1962, 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res 31, 439–441, https://doi.org/10.1093/nar/gkg006 (2003).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Borodovsky, M. & Lomsadze, A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr Protoc Bioinformatics Chapter 4, 4.6.1–4.6.10, https://doi.org/10.1002/0471250953.bi0406s35 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Consortium, T. U. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research 51, D523–D531, https://doi.org/10.1093/nar/gkac1052 (2022).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230, https://doi.org/10.1093/nar/gkt1223 (2014).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314, https://doi.org/10.1093/nar/gky1085 (2018).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 6, 4, https://doi.org/10.1186/1939-8433-6-4 (2013).
Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, C. Oryza sativa Japonica Group cultivar Early Geng isolate Longgeng 57, whole genome shotgun sequencing project, NCBI GenBank, https://identifiers.org/ncbi/insdc:JAXQPT000000000 (2023).
Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, C. Genomic data of Long Geng 57 in bam format, NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR25376496 (2023).
Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, C. Genomic data for PN8 in Fastq format, NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR24688636 (2023).
Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, C. Genomic data for LN2 in Fastq format, NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR24688637 (2023).
Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, C. Genomic data for LG29 in Fastq format, NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR24688635 (2023).
Zheng, T.-Q. Annotation files for Longgeng 57. figshare https://doi.org/10.6084/m9.figshare.24799695 (2023).
Leng, Y. et al. Using Heading date 1 preponderant alleles from indica cultivars to breed high-yield, high-quality japonica rice varieties for cultivation in south China. Plant Biotechnology Journal 18, 119–128, https://doi.org/10.1111/pbi.13177 (2020).
Wu, C.-C. et al. Studies of rice Hd1 haplotypes worldwide reveal adaptation of flowering time to different environments. PLOS ONE 15, e0239028, https://doi.org/10.1371/journal.pone.0239028 (2020).
Zeng, D. et al. Rational design of high-yield and superior-quality rice. Nature Plants 3, 17031, https://doi.org/10.1038/nplants.2017.31 (2017).
Faruquee, M. et al. Dominant early heading without yield drag in a sister-line BC breeding progeny DEH_229 is controlled by multiple genetic factors with main-effect loci. The Crop Journal 9, 400–411, https://doi.org/10.1016/j.cj.2020.06.014 (2021).
Li, H. et al. A spontaneous thermo-sensitive female sterility mutation in rice enables fully mechanized hybrid breeding. Cell Res 32, 931–945, https://doi.org/10.1038/s41422-022-00711-0 (2022).
Acknowledgements
This work was mainly supported by the National Key Research and Development Program of China (2020YFE0202300, 2023ZD04076); the National Nature Science Fund of China (grant number 31871715); the Key Special Program (2022ZD0400404), Ministry of Science and Technology, China, International Science & Technology Innovation Program of Chinese Academy of Agricultural Sciences (grant numbers CAASTIPS, CAAS-ZDRW202109); the Guangxi Key Laboratory of Rice Genetics and Breeding (grant number 2022-36-Z01-KF10); the Hainan Yazhou Bay Seed Lab (B21HJ0216); and the Bill & Melinda Gates Foundation (grant number OPP1130530).
Author information
Authors and Affiliations
Contributions
T.Q.Z., Y.J.Z. and J.L.X. designed and conceived research; Y.B.L., L.Y.X., Z.Q.Z. and D.C.Y. prepared samples for sequencing; Y.B.L., J.L., P.Y.Q., M.F., Y.L.Z. and G.J.Q. performed data collection and analysis; X.Q.Z., B.L.L., W.S.W., F.Z. and Z.K.L. give valuable suggestions and contributed new reagents/analytic tools; T.Q.Z. and J.L.X. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, Y., Zhang, Y., Xu, L. et al. Pedigree genome data of an early-matured Geng/japonica glutinous rice mega variety Longgeng 57. Sci Data 11, 230 (2024). https://doi.org/10.1038/s41597-024-03057-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03057-x
- Springer Nature Limited