
Abstract
Black scorch disease (BSD), caused by the fungal pathogen Thielaviopsis punctulata (Tp) DSM102798, poses a significant threat to date palm cultivation in the United Arab Emirates (UAE). In this study, Chicago and Hi-C libraries were prepared as input for the Dovetail HiRise pipeline to scaffold the genome of Tp DSM102798. We generated an assembly with a total length of 28.23 Mb comprising 1,256 scaffolds, and the assembly had a contig N50 of 18.56 kb, L50 of three, and a BUSCO completeness score of 98.6% for 758 orthologous genes. Annotation of this assembly produced 7,169 genes and 3,501 Gene Ontology (GO) terms. Compared to five other Thielaviopsis genomes, Tp DSM102798 exhibited the highest continuity with a cumulative size of 27.598 Mb for the first seven scaffolds, surpassing the assemblies of all examined strains. These findings offer a foundation for targeted strategies that enhance date palm resistance against BSD, and foster more sustainable and resilient agricultural systems.
Similar content being viewed by others
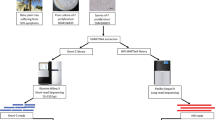


Background & Summary
Date palm (Phoenix dactylifera L.) is one of the oldest key fruit crop that is traditionally cultivated in arid regions of the Arabian Peninsula, Middle East and North Africa1, including the United Arab Emirates (UAE)2,3. More than 8.5 metric tons of dates are produced annually4, with an estimated 258,000 tons produced by 17,000 farmers in the UAE alone5,6. Many fungal diseases, however, wreak havoc on date palm farming and yield, resulting in significant losses in date production.
For example, Bayoud disease caused by the soil-borne fungal pathogen, Fusarium oxysporum f. sp. albedinis (Foa), specifically infects the roots and the vascular system of date palms, leading to widespread destruction of date palm plantations in North Africa7. Although Foa and Bayoud disease were not detected in the UAE, other Fusarium spp., such as F. oxysporum DSM106834, F. proliferatum DSM106835 and F. solani DSM106836, cause sudden decline syndrome (SDS) on date palm8,9. Black scorch disease (BSD, also known as Medjnoon) is a fungal disease that also affects date palms, leading to significant economic losses10. Disease symptoms, such as the formation of black charcoal-like lesions on leaves, inflorescence blight, and heart and bud rot, often appear on infected date palm trees11. Eventually, tissue necrosis, wilting, neck bending, and death of terminal buds and whole plant are associated with later stages of infection.
In 1932, Koltz first detected BSD on date palm trees, and identified Thielaviopsis paradoxa as the causative agent of the symptoms in the United States11. The same fungal pathogen was also diagnosed in Egypt on date palms in 200712. Recent reports, however, identified Thielaviopsis punctulata (Tp) on date palm trees showing symptoms of BSD in Spain13, Egypt14, Qatar15, and Saudi Arabia16. In the UAE, Tp DSM102798 was associated with BSD of date palm10. This soil-borne wound pathogen can produce two types of conidia: thick-walled, oval-shaped aleuroconidia (chlamydospores) and smooth-walled, cylindric phialoconidia (endoconidia)10. In general, aleuroconidia are larger than phialoconidia in all Thielaviopsis spp.14. Although aleuroconidia help Tp adapt to extreme desert conditions for prolonged periods, phialoconidia enable the fungus to grow fast under favourable conditions.
Even though chemical pesticides are extensively used in agriculture, they do not provide a sustainable long-term solution for managing plant diseases17,18,19. Whole-genome studies, including genomics and transcriptomics, offer valuable tools for understanding the genetic basis of resistance, susceptibility, and other factors related to plant diseases9,20,21. Therefore, we performed highly accurate de novo genome sequencing and assembly of Tp DSM102798 using high-throughput sequencing libraries along with Hi-C for chromosome-scale scaffolding22. We also corrected misjoins, scaffolding uncertainty and errors in contigs by comparing with other reference genomes. Finally, we assessed the quality of Chicago and Hi-C assemblies according to the contiguity of assembled sequences (N50), completeness of conserved protein-coding genes, and Gene ontology (GO) analysis. The assembled and annotated high-quality genome of Tp DSM102798 not only provides genetic resources for comparative genome studies among Thielaviopsis spp. but also addresses the potential application of genetic-based approaches to improve sustainable date palm production.
Methods
Sample collection and DNA extraction
Samples of entirely dried leaves and black scorched basal parts were collected from diseased date palms from the Al-Wagan area, Abu Dhabi, UAE (latitude 24.13; longitude 55.74). The rotting tissues were sectioned into smaller pieces and used as colony starter in potato dextrose agar (PDA; Sigma Aldrich) supplemented with penicillin-streptomycin to avoid bacterial contamination. The fungus was frequently sub-cultured from the initial plates every 10–14 days until pure cultures of Tp were obtained.
DNA extraction was carried out on pure cultures of Tp grown on PDA. High molecular weight (HMW) DNA was extracted by first scraping all visible fungal material from the Petri dish, which was then transferred to a 50 ml tube containing 2 ml H2O. This mixture was flash-frozen to create a pellet of ~500 mg that was then ground. In the ground sample, 10 ml of cetyltrimethylammonium bromide (CTAB) and 100 µl of β-mercaptoethanol (BME) were added and incubated at 68°C for 15 minutes. After incubation, 10 µl of protease and 1 µl of RNase were added to the sample and incubated at 60°C for 30 minutes. Phenol/chloroform/isoamyl-alcohol was used to extract DNA from the cell lysate, centrifuged into a pellet, and resuspended in 200 µl Tris-EDTA (TE) buffer.
Library preparation and sequencing
The isolated HMW DNA fragments were subjected to quality control (QC) check by measuring the concentration, the 260/280 and 260/230 ratios, and the average fragment size using pulsed-field gel electrophoresis (PFGE). After successfully passing the QC assessment, the fragments were employed in library preparation. First, Chicago libraries were prepared using ~500 ng of HMW DNA with mean fragment length = 100, which was reconstituted into chromatin in vitro and fixed with formaldehyde. Fixed chromatin was digested with DpnII, the 5′ overhangs filled in with biotinylated nucleotides, and then free blunt ends were ligated. After ligation, crosslinks were reversed and DNA was purified. The purified DNA was treated to remove biotin that was not internal to ligated fragments. The DNA was then sheared to ~350 bp mean length fragment size and sequencing libraries were generated using NEB Next Ultraenzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. For a 1 Gb genome, it is recommended to use one library and 200 million read pairs. The Chicago sequencing library was 2213.48 times larger than the 28.2 Mb genome size of Tp. The Chicago libraries were then subjected to QC by sequencing 1–2 M PE, 75 bp reads on the Illumina MiSeq instrument and the reads were mapped back to the draft assembly, GCA_000968615.123. The second library was constructed for Hi-C sequencing. It was prepared in manner similar to the Chicago library, with a coverage depth of 1904.26 times of the genome size. The same library preparation protocol was used, and QC was also applied. These libraries prepared by Dovetail Genomics (Scotts Valley, California, USA) were sequenced using an Illumina HiSeq X instrument.
Genome assembly and downstream analysis
The genome assembly was carried out in two steps. Initially, the Chicago assembly was generated using the Dovetail HiRise pipeline24, where the draft assembly (GCA_000968615.1) was used as a reference to map the Chicago reads. The Chicago assembly was then used as a reference to map the Hi-C reads to generate the final genome assembly, again using the Dovetail HiRise pipeline24. The assembled genome was also compared against the draft genome (GCA_000968615.1) to check for improvements in the overall quality of the assembly. The genome assembly was then annotated using FunAnnotate25, a fungal genome annotation pipeline that identifies protein-coding genes in a fungal genome assembly. First, repetitive contigs were cleaned from the genome for using minimap226. Next, the genome was masked for repeats using RepeatMasker27, and Repbase (v20170127)28 as the reference database for repetitive elements. FunAnnotate was first run in training mode to improve gene prediction using RNA-seq data from the closely related T. paradoxa (SRR15533162)29. Then, FunAnnotate was run in prediction mode using the transcriptome of T. paradoxa (SRR15533162) assembled with Trinity30, a list of Expressed Sequence Tags (ESTs) collected from the National Center for Biotechnology Information (NCBI) using Taxonomy ID: 6049631 via Entrez E-utilities32, and a list of related protein sequences retrieved from Uniprot33. The predicted gene models subjected to the FunAnnotate used InterProScan34, Eggnog-mapper35,36, and antiSMASH37 for functional annotation. In addition, FunAnnotate employed SignalP38 to predict the secretome, and HMMer39 to map protein models against dbCAN40 for predicting carbohydrate-active enzymes (CAZymes), and diamond41 blastp search of MEROPS42 database for peptidases prediction.
Assessment of completeness and continuity of genome assembly
For assembly continuity comparison, genome sequences along with annotations of five Thielaviopsis strains: T. ethacetica (BCFY00000000.1)43, T. populi (JADILG000000000.1)44, T. cerberus (JACYXV000000000.1)45, T. euricoi (BCHJ00000000.1)46, and T. musarum (LKBB00000000.1)47 were downloaded from the NCBI database. These strains were compared against the newly sequenced Tp DSM102798 genome using the sequence length of each assembly with the average scaffold length. The completeness analysis was performed by comparing the results of BUSCO analysis of each genome against fungi_odb10 lineage-specific profile48.
Data Records
All sequence data, including raw Chicago reads and Hi-C short reads, were deposited to the NCBI database under BioProject PRJNA1060910 with accessions SRR2742121649 and SRR2742121750, respectively. The genome assembly is available through NCBI GenBank with accession JAYKOR00000000051. The genome annotation information was deposited in the Figshare database52.
Technical Validation
Genome assembly
The Chicago library generated 208 M read pairs (2 × 150 bp) was used to create the primary Chicago assembly using the publicly available genome assembly of Tp GCA_000968615.1 as the reference. This produced a Dovetail HiRise assembly of 28.22 Mb with larger scaffolds than GCA_000968615.1 (Fig. 1a). During the assembly process, the HiRise pipeline made 55 breaks and 1,055 joins in GCA_000968615.1. The Chicago assembly then served as a reference to generate the Hi-C assembly against the Hi-C library of 179 M read pairs (2 × 150 bp), where the overall scaffold size was significantly improved due to 60 scaffolds being joined by the HiRise pipeline (Fig. 1b). At the basic level, the quality of the final Hi-C assembly was significantly better than GCA_000968615.1 assembly based on various factors such as scaffold length, N50, N90, and the total number of scaffolds (Table 1). Hi-C contact maps were created from the output of HiRise using Juicer53, and the contact map was configured to identify Topologically Associated Domains and A/B genome compartments. The configured contact map was visualised using Juicebox54, which revealed seven scaffolds, and made up the genome of Tp DSM102798 (Fig. 2).

Comparison of the contiguity of the input assembly and final HiRise assembly of Thielaviopsis punctulata DSM102798 genome. (a) Chicago assembly was first scaffolded with an estimated physical coverage (1–100 kb) that was 1,472.25X; and (b) Hi-C assembly which was further improved with estimated physical coverage (10–10,000 kb) of 77,155.15X. Each curve shows the fraction of the total length of the assembly present in scaffolds of a given size or smaller. The fraction of the assembly (scaffolds) is presented on the Y-axis, whereas the scaffold length (bp) is provided on the X-axis. The two dashed lines indicate N50 and N90 lengths of each assembly. Scaffolds less than 1 kb were excluded from the analysis.

Link density histogram mapped with Hi-C reads. X- and Y-axes represent the mapping positions of the first and second read in each read pair groups, respectively, into bins. For each square container, the color indicates the number of read pairs within the bin. White (vertical) and black (horizontal) lines are provided to show borders between scaffolds. Scaffolds less than 1 Mb were excluded from the analysis.
Genome annotation
The annotation of Hi-C genome assembly using FunAnnotate predicted 7,169 genes and 18,306 exon sequences; thus, providing important information about the function, structure, and location of genes and other biologically significant elements (Table 2; Fig. 3). GO analysis was carried out using Blast2GO55 and eggNOG, yielding 3,501 sequences with 33,829 annotations. There were 1,100 clusters of orthologous genes related to information storage and processing, 1,190 to cellular processes and signaling, and 1,473 to metabolism. GO terms were further categorized based on cellular components (Fig. 4a), biological processes (Fig. 4b), and molecular function (Fig. 4c). The orthologous group distribution revealed that out of 7,169 genes, 6,451 were predicted to be in Kingdom Fungi, 6,438 were specific to Division Ascomycota, and 6,154 belonged to Class Sordariomycetes which perfectly correspond to the taxonomy of Tp30.

Circos plot of Thielaviopsis punctulata DSM102798 genome assembly. (a) The seven longest scaffolds of the genome assembly, (b) gene density, (c) exon density, (d) mRNA density and (e) long terminal repeats (LTR) density of each scaffold.

Functional annotation and Gene Ontology (GO) for Thielaviopsis punctulata DSM102798. Distribution of sequences for (a) cellular components, (b) biological process, (c) molecular function, and (d) number of secondary metabolite biosynthesis gene clusters identified from the first 7 scaffolds of Tp DSM102798 genome.
Secondary metabolite biosynthesis gene clusters were identified from scaffolds 1–5 of Tp DSM102798 genome (Fig. 4d). Dimethylcoprogen has been identified as a siderophore produced by many pathogenic fungi to conquer the battle for iron acquisition56. In addition, The complex class of fungal metabolites, squalestatin S1 (zaragozic acid), which Is an inhibitor of squalene synthase that controls the use of cholesterol biosynthesis57 was also among the gene clusters of Tp.
In addition, 6811 protein families and domains were identified from the genome, including major facilitator superfamily, fungal transcription factor, and cytochrome P450 (Fig. 5a). These superfamily proteins play a significant role in various biological processes such as transporting small solutes across cell membranes and metabolism of drugs and synthesis of cholesterol, steroids, and other lipids. Notable protein domains, such as α/ß-hydrolases, kinase domains and S-adenosyl-L-methionine-dependent methyltransferases that were associated with specific biochemical activities includung enzyme catalysis, substrate binding, and molecular interactions were identified (Fig. 5b).

Prediction of protein-related genes identified in Thielaviopsis punctulata DSM102798 genome. Top 10 protein (a) families and (b) domains by sequence count. In (a & b), the top 10 identified protein families and domains with their respective sequence counts were presented.
Genome continuity and completeness analysis
Our analysis revealed that Tp DSM102798 exhibited the highest continuity among the five Thielaviopsis genomes. The cumulative size of the first seven scaffolds/contigs was 27.598 Mb, which surpassed the assemblies of all other Thielaviopsis strains, ranging from 0.360 Mb in T. cerberus to 18.391 Mb in T. euricoi (Fig. 6a). The same genomes were compared for their completeness using BUSCO, and Tp DSM102798 also achieved a completeness rate of 98.6% for the 758 orthologous genes in the Fungi_odb10 database (Fig. 6b).

Comparisons in genome assembly of Thielaviopsis punctulata DSM102798 and the genomes of other Thielaviopsis species. (a) Contiguity of the genomes of five Thielaviopsis spp. compared to Hi-C genome Tp DSM102798 based on the first 20 longest scaffolds from each genome. (b) Completeness of the genome assembly of Tp DSM102798 compared to that of five other Thielaviopsis genomes collected from NCBI database.
Code availability
This work did not utilise a custom script. Data processing was carried out using the protocols and manuals of the relevant bioinformatics software.
References
Hadrami, I.E., Hadrami, A.E. Breeding date palm. In: (eds. Jain, S. M., Priyadarshan, P. M.) Breeding Plantation Tree Crops: Tropical Species. Springer. https://doi.org/10.1007/978-0-387-71201-7_6 (2009).
Beech, M. & Shepherd, E. Archaeobotanical evidence for early date consumption on Dalma Island, United Arab Emirates. Antiquity 75, 83–89 (2001).
Tengberg, M. Beginnings and early history of date palm garden cultivation in the Middle East. Journal of Arid Environments 86, 139–147 (2012).
FAO. World Food and Agriculture – Statistical Yearbook 2021. https://doi.org/10.4060/cb4477en (FAO, 2021).
Agthia announces dates marketing season for Al Foah. TradeArabia https://www.tradearabia.com/news/MISC_399451.html (2022).
Date palm agriculture in UAE significantly developed in recent years: ADAFSA. WAM (Emirates News Agency) https://www.wam.ae/en/details/1395302969882 (2021).
El Hassni, M. et al. Biological control of bayoud disease in date palm: Selection of microorganisms inhibiting the causal agent and inducing defense reactions. Environmental and Experimental Botany 59, 224–234 (2007).
Alwahshi, K. J. et al. Molecular identification and disease management of date palm sudden decline syndrome in the United Arab Emirates. International Journal of Molecular Sciences 20, 923 (2019).
Purayil, G. P., Almarzooqi, A. Y., El-Tarabily, K. A., You, F. M. & AbuQamar, S. F. Fully resolved assembly of Fusarium proliferatum DSM106835 genome. Scientific Data 10, 705 (2023).
Saeed, E. E. et al. Chemical control of black scorch disease on date palm caused by the fungal pathogen Thielaviopsis punctulata in United Arab Emirates. Plant Disease 100, 2370–2376 (2016).
Klotz, L. Black scorch of the date palm caused by Thielaviopsis paradoxa. Journal of Agricultural Research 44, 155 (1932).
El-Deeb, H. M., Lashin, S. M. & Arab, Y. A. Distribution and pathogenesis of date palm fungi in Egypt. Acta Horticulturae 736, 421–429 (2007).
Abdullah, S. K. et al. Incidence of the two date palm pathogens, Thielaviopsis paradoxa and T. punctulata in soil from date palm plantations in Elx, south-east Spain. Journal of Plant Protection Research 49, 276–279 (2009).
Ammar, M. I. First report of Chalaropsis punctulata on date palm in Egypt, comparison with other Ceratocystis anamorphs and evaluation of its biological control. Phytoparasitica 39, 447–453 (2011).
Nishad, R. & Ahmed, T. A. Survey and identification of date palm pathogens and indigenous biocontrol agents. Plant Disease 104, 2498–2508 (2020).
Alhudaib, K. A., El-Ganainy, S. M., Almaghasla, M. I. & Sattar, M. N. Characterization and control of Thielaviopsis punctulata on date palm in Saudi Arabia. Plants 11, 250 (2022).
Saeed, E. E. et al. Streptomyces globosus UAE1, a potential effective biocontrol agent for black scorch disease in date palm plantations. Frontiers in Microbiology 8, 1455 (2017).
Alwahshi, K. J. et al. Molecular identification and disease management of date palm sudden decline syndrome in the United Arab Emirates. International Journal of Molecular Science 20, 923 (2019).
Alblooshi, A. A. et al. Biocontrol potential of endophytic actinobacteria against Fusarium solani, the causal agent of sudden decline syndrome on date palm in the UAE. Journal of Fungi 8, 8 (2022).
AbuQamar, S. F., Moustafa, K. & Tran, L.-S. P. ‘Omics’ and plant responses to Botrytis cinerea. Frontiers in Plant Science 7, 1658 (2016).
Mengiste, T., Laluk, K. & AbuQamar, S. Mechanisms of induced resistance against B. cinerea. In Post-Harvest Pathology, Vol. 2, Ch. 2 (eds. Prusky, D. & Gullino, M. L.) 13–30 (Springer Science + Business Media, 2010).
Kadota, M. et al. Multifaceted Hi-C benchmarking: what makes a difference in chromosome-scale genome scaffolding? GigaScience 9, 158 (2020).
Wingfield, B. D. et al. Draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagonosporopsis tanaceti, and Thielaviopsis punctulata. IMA Fungus 6, 233–248 (2015).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Research 26, 342–350 (2016).
Palmer, J. & Stajich, J. nextgenusfs/funannotate: funannotate v1.5.3. Zenodo https://doi.org/10.5281/zenodo.2604804 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2013-2015).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
NCBI Sequence Reads Archive https://identifiers.org/ncbi/insdc.sra:SRR15533162 (2022).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013).
Schoch, C. L. et al. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford) 2020, baaa062 (2020).
Kans, J. Entrez Direct: e-utilities on the unix command line. in Entrez Programming Utilities Help. National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/books/NBK179288/ (2010-2024).
The UniProt consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Research 51, D523–D531 (2023).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Research 51, D418–D427 (2023).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Blin, K. et al. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Research 49, W29–W35 (2021).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology 40, 1023–1025 (2022).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 39, W29–37 (2011).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Research 40, W445–51 (2012).
Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Research 46, D624–D632 (2018).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:BCFY00000000.1 (2018).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:JADILG000000000.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:JACYXV000000000.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:BCHJ00000000.1 (2018).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:LKBB00000000.1 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
NCBI Sequence Reads Archive https://identifiers.org/ncbi/insdc.sra:SRR27421216 (2024).
NCBI Sequence Reads Archive https://identifiers.org/ncbi/insdc.sra:SRR27421217 (2024).
NCBI GenBank https://identifiers.org/ncbi/nucleotide:JAYKOR000000000 (2024).
Purayil, G. P., Saeed, E. E., Mathai, A., El-Tarabily, K. A., & AbuQamar, S. F. A high-quality genome assembly and annotation of Thielaviopsis punctulata DSM102798., Figshare, https://doi.org/10.6084/m9.figshare.c.7012431.v1 (2024).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research 36, 3420–3435 (2008).
Jalal, M. A. F., Love, S. K. & van der Helm, D. N. Alpha-dimethylcoprogens. Three novel trihydroxamate siderophores from pathogenic fungi. Biology of Metals 1, 4–8 (1988).
Lebe, K. E. & Cox, R. J. Oxidative steps during the biosynthesis of squalestatin S1. Chemical Science 10, 1227–1231 (2019).
Acknowledgements
This work is supported by Khalifa Center for Genetic Engineering and Biotechnology-UAEU (Grant #: 12R028) to S. AbuQamar.
Author information
Authors and Affiliations
Contributions
G. Purayil: data curation, methodology, software, and writing – original draft; E. Saeed: Investigation; A. Mathai: methodology; K. El-Tarabily: resources, and supervision; S. AbuQamar: conceptualisation, data curation, writing – review, editing, and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Purayil, G.P., Saeed, E.E., Mathai, A.M. et al. A high-quality genome assembly and annotation of Thielaviopsis punctulata DSM102798. Sci Data 11, 745 (2024). https://doi.org/10.1038/s41597-024-03458-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03458-y
- Springer Nature Limited