
Abstract
Recent advancements in plant regeneration and synthetic polyploid creation have been documented in Gossypium arboreum ZB-1. These developments make ZB-1 a potential model within the Gossypium genus for investigating gene function and polyploidy. This work generated the sequence and annotation of the ZB-1 genome. The contig-level genome was constructed using the PacBio high-fidelity reads, encompassing 81 contigs with an N50 length of 112.12 Mb. The Hi-C data assisted the construction of the chromosome-level genome, which consists of 13 pseudo-chromosomes and 39 un-anchored contigs, with a total length of about 1.67 Gb. Repetitive sequences accounted for about 69.7% of the genome in length. Based on ab initio and evidence-based prediction, we have identified 48,021 protein-coding genes in the ZB-1 genome. Comparative genomics analysis revealed conserved gene content and arrangement between ZB-1 and G. arboreum SXY1. The single nucleotide polymorphism occurrence rate between ZB-1 and SXY1 was about 0.54 per 1,000 nucleotides. This study enriched the genomic resources for further exploration into cotton regeneration and polyploidy mechanisms.
Similar content being viewed by others


Background & Summary
Cotton (Gossypium spp.) is one of the most crucial fiber and oilseed crops worldwide. The Gossypium genus comprises about 50 species, which include 45 diploids categorized into eight genome groups (A-G and K) and seven tetraploids denoted as AD1-AD71,2. The tetraploids originated from a single hybridization event between the A- and D-genome progenitors, followed by polyploidization3. Historically, four of these species have undergone independent domestication, i.e., the diploids G. arboreum (A2) and G. herbaceum (A1)4, as well as the tetraploids G. hirsutum (AD1) and G. barbadence (AD2)5. Notably, AD1 has emerged as the predominant species in contemporary cotton cultivation.
A2 is critical for investigating the mechanisms underlying natural polyploidy in the genus Gossypium. For an extended period, one of the major concerns in cotton biology is the origin of the A-genome in the natural tetraploids, with the controversy over whether it is from A1 or A2. In the last decade, the genome sequences of A1, A2, and several tetraploids have been successively generated6. With the insights gained from comparative genomics, researchers are now inclined to conclude that the ancestor (termed A0) of A1 and A2 is the A-genome donor7,8,9. However, as A0 no longer exists, A1 and A2 are still necessary substitutes for assessing the evolution of the natural cotton polyploids upon genome merge and doubling. Due to its broader distribution and, more significantly, the more profound understanding of its biology in comparison to A1, researchers often opt for A2 as the substitute progenitor when studying the evolution of natural tetraploids.
Besides, A2 has also frequently been used to generate synthetic cotton polyploids to investigate issues such as gene expression modulation in the early stage of polyploidy. For example, hybridization between A2 and G. thurberi (D1), G. raimondii (D5), and G. bickii (G1) gave rise to several hybrids and synthetic tetraploids10,11. Gene expression analysis of these synthetic hybrids or polyploids revealed that the subgenome expression pattern was rapidly established in the early generations of the progenies, which would then be reconciled during long-term evolution. Likewise, Ke et al. generated a novel cotton tetraploid between A2 (accession ZB-1) and G. stocksii (E1) by somatic hybridization12. As meiosis does not occur during somatic hybridization, the novel polyploid 2(A2E1) might be an ideal model for further exploring the transcriptional and epigenomic alteration upon genome doubling.
In addition, A2-derived new cotton polyploids provide valuable germplasm resources for improving agronomically important traits in cotton cultivars. For example, A2 and G1-derived tetraploids have glandular vegetative tissue and glandless seeds (without toxic gossypol)13, which provide novel germplasm to breed cultivars with the seeds could be more widely utilized in industrial production14. Likewise, by hybridizing A2 with E1 and following genome doubling, Nie and colleagues generated novel tetraploids 2(A2E1), of which the high generations produce high-strength fiber15. Moreover, Chen et al. generated a novel cotton hexaploid by hybridization between A2 (SXY1 or Shixiya 1) and AD1 (TM-1), which could be further utilized to develop the G. arboreum-introgressed lines to transfer the drought and Verticillium resistance to cultivars16.
In addition to serving as the progenitor for synthetic cotton polyploids in polyploidy research and breeding, A2 is also a potential model organism for gene function exploration in cotton species. Currently, the creation of transgenic cotton plants predominantly relies on AD16,17. Nevertheless, ascribed to the doubled genomes in AD1, each allele is associated with four haplotypes that are similar in sequence. The complexity of the genetic background poses a challenge in gene manipulation in AD1, such as the risk of off-target effects in CRISPR/Cas9-mediated genome editing. Furthermore, the gene duplication resulting from polyploidy also adds to the complexity of evaluating gene functions in AD1. For instance, several studies have highlighted that subfunctionalization and neofunctionalization of duplicated genes frequently occur after cotton polyploidization11,18,19. Consequently, diploids such as A2 are better models for characterizing gene functions in cotton species.
However, the method of regenerating plants by somatic embryogenesis has yet to be established for most cotton species, which significantly hindered the transgenic study in cotton cells. Recently, we have achieved plant regeneration using A2 (ZB-1) with alternative solid-liquid culture method20. The callus was able to be induced from different explants (hypocotyl, root, and cotyledon), and the regenerated plants could be regularly grown to maturity in soil or be grafted onto A2 seedlings. Upon further optimization, ZB-1 will become a readily available tool for the functional analysis of cotton genes.
Taken together, the accession ZB-1 with A2 genome is an essential material for the studies on cotton polyploidy and gene function exploration (Fig. 1). At the same time, we have noticed that although some genome assemblies of A2 are available now and tens of A2 accessions have been resequenced, the chromosome-level A2 genomes were almost exclusively generated from the accession SXY17,21,22,23,24. Therefore, a high-quality genome assembly of ZB-1 is necessary to provide additional genomic information for studies on polyploidy, gene manipulation, and the mechanisms underlying the regeneration of cotton plants.

Gossypium arboreum ZB-1 and its utilization in cotton biology study. a-f indicate the morphological characteristics of ZB-1: (a) whole plant, (b) leaf, (c) flower, (d) boll, (e) opening boll, and (f) fiber. (g) indicates the potential utilization of ZB-1. G. arboreum accessions including ZB-1 could hybrid with many other cotton diploids and generate synthetic polyploids. In addition, ZB-1 could produce regenerated cotton plants via somatic embryogenesis, which is essential for further manipulating the genes and construct the transgenic plants. ZB-1 derived novel polyploid and transgenic plants will help the future study on cotton polyploidy, gene function, and breeding.
In this work, we constructed a chromosome-level genome assembly of G. arboreum ZB-1. The contig-level genome assembly was generated using PacBio HiFi reads. The Hi-C data was used to scaffold the contigs into pseudo-chromosomes. Four transcriptomes of ZB-1 (boll, flower, sepal, and stem) were generated to assist the gene identification. The final gene set was generated by integrating the results from ab initio prediction, homologs-based gene annotation, and transcripts-derived gene identification using the PASA pipeline. The completeness of the genome assembly was evaluated by BUSCO assessment and the mapping back of the short reads, which showed the high quality of the provided genome assembly of ZB-1. Moreover, a comparative analysis was carried out between the assemblies of ZB-1 and SXY1, revealing a large number of single nucleotide polymorphisms (SNPs), suggesting this genome assembly is necessary for further studies using ZB-1 as a test material.
Methods
Plant material collection
G. arboreum ZB-1 was planted in the experimental field on the campus of Zhejiang Sci-Tech University in Hangzhou, Zhejiang Province, China, in May 2022. The tender leaves (the third to fifth), bolls (10 days post anthesis), flowers (bloom day, including petals, stamens and pistils), sepals, and stems were harvested from at least five adult plants and pooled. All the tissues were snap-frozen in liquid nitrogen and stored at −80 °C until use.
DNA library construction and genome sequencing
The genomic DNA (gDNA) was extracted from the tender leaves using the CTAB (cetyltrimethylammonium bromide) method. After chilling in liquid nitrogen, 20 mg leaves were ground with steel balls in a 2 mL polypropylene tube. 600 μL pre-heated (65 °C) CTAB buffer (2% w/v CTAB, 100 mM Tris-HCl, 20 mM EDTA, 1.5 M NaCl, pH 8) was added into the tube and thoroughly vortexed. The tube was placed in a 65 °C water bath for 1 h. The homogenate was then centrifuged for 10 min at 12,000 rpm. The supernatant was transferred to a new tube, and 5 μL of RNase A solution was added. After incubating at 37 °C for 20 min, an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) was added and mixed. The sample was centrifuged for 10 min at 12,000 rpm. The upper aqueous phase was transferred to a new tube, and repeat this extraction once. Cold isopropanol was added with a volume of about two-thirds of the upper phase. The sample was gently mixed and incubated at −20 °C for 30 min. Next, the sample was centrifuged for 10 min at 12,000 rpm at room temperature, and the pellet was washed using 500 μL ice-cold 70% ethanol. Decant the ethanol and naturally dry the pellet to remove the ethanol residual. Finally, about 20 uL TE buffer (10 mM Tris, pH 8, 1 mM EDTA) was added to dissolve the DNA pellet.
Illumina sequencing
For Illumina short-read sequencing, the purified gDNA was randomly sheared into fragments by a Covaris M220 Ultrasonicator (Covaris, USA). Subsequently, a library with an average length of about 350 bp for the inserted fragments was constructed according to the recommended protocols of the Illumina TruSeq DNA Sample Prep Kits. Qubit 2.0 Fluorometer (ThermoFisher Scientific, USA) was used to quantify the dsDNA of the library, which was then diluted to 1 ng/μL. Agilent 2100 Bioanalyzer (Agilent, USA) was used to assess the integrity and size of the inserted fragments. A final concentration of the library was determined with QuantStudio 3 Real-Time PCR Instrument (Thermo Fisher Scientific, USA) (>2 nM). The library was sequenced using the Novaseq 6000 platform (Illumina, USA) and a program of pair-end 150 bp. Quality control of the raw data was performed using Fastp (v0.21.0)25. The read pairs were removed if the ambiguous base (N) exceeds 10% or the low-quality bases (Q < 5) exceed 20% of a single read in length. The generated raw data contained about 101.03 Gb paired short reads, with 99.91% retained after quality control (Table 1).
Pacbio sequencing
A HiFi library was constructed using the SMRTbell Template Prep Kit (Pacific Biosciences, USA). The high molecular weight DNA fragments (ca. 20 Kb) were isolated and purified from the g-TUBE (Covaris, USA) sheared gDNA sample using the AMPure beads (Beckman Coulter, USA), followed by damage repair, end-repair/A-tailing and hairpin adapter ligation according to the manufacturer’s instructions. The SMRTbell library was sequenced on the Pacbio Sequel II platform with the Circular Consensus Sequencing (CCS) model. The HiFi reads were generated using SMRT Link software (v12.0) with default settings. About 4.01 M HiFi reads were finally acquired, with a total length and average length of about 71 Gb and 17.7 kb, respectively (Table 1).
Hi-C library construction and sequencing
The Hi-C library was constructed according to the method previously described by Belton et al. with certain modifications26. Tender leaves of ZB-1 were ground with liquid nitrogen and crosslinked by 4% formaldehyde at room temperature for 30 mins. 2. 5 M glycine was added to quench the crosslinking reaction. The pellet was centrifuged at 2,500 rpm at 4 °C and resuspended with the lysis buffer (1 M Tris-HCl, pH 8, 1 M NaCl, 10% CA-630, and 13 units protease inhibitor). The supernatant was centrifuged at 5,000 rpm at room temperature. The pellet was washed twice using the ice-cold NEB buffer and centrifuged again. Then, the nuclei were resuspended by the NEB buffer, solubilized with dilute sodium dodecyl sulphate (SDS), and incubated at 65 °C for 10 mins. Triton X-100 quenched SDS, and the crosslinked chromatin was digested overnight by the restriction enzyme DPN II (400 units, 37 °C). The 5’ overhangs were filled in with biotinylated residues (biotin-14-dCTP), and the proximate blunted ends were ligated. Biotin was removed from the un-ligated ends using T4 DNA polymerase. The ligated DNA was then sheared with the Covaris instrument, and the fragments with an average size of 350 bp were purified via agarose gel electrophoresis and gel elution. After end repairing by the mixture of T4 DNA polymerase, T4 polynucleotide kinase, and Klenow DNA polymerase, the biotin-labelled DNA fragments were pulled down using the streptavidin C1 magnetic beads. The library was then constructed with a standard protocol and sequenced on the Illumina Novaseq 6000 platform with 2 × 150 bp paired-end reads (PE150).
About 3.80 Gb short reads were generated from Hi-C library sequencing, with 3.78 Gb (99.6%) retained after removing the low-quality reads (Table 1). Further quality control was mainly carried out with HICUP27. The truncated sequences were identified and mapped with hicup_truncater and hicup_mapper. The re-ligated and same circularised sequences were removed by hicup_filter, and the PCR-raised duplication was filtered using hicup_deduplicator. As a result, 2,494,732 unique di-Tags were obtained, with the effect rate of Hi-C sequencing data about 23.32%.
RNA library construction and sequencing
Total RNA was isolated from the tissue samples (boll, flower, sepal, and stem) using the RNAprep Pure Plant Plus Kit (DP441, TIANGEN) and purified with RNase-free DNase I. The sequencing libraries were constructed using NEB Next Ultra RNA Library Prep Kit for Illumina (New England Biolabs, USA). The mRNAs were enriched using the oligo(dT) attached beads, and the cDNAs were synthesized using Superscript II reverse transcriptase. Fragmented cDNAs with a length of around 350 bp were used for sequencing library construction with standard protocol. The constructed libraries were sequenced on Novaseq 6000 with the PE150 strategy.
About 6.75 Gb, 6.16 Gb, 6.82 Gb, and 6.36 Gb raw data were generated from sequencing of the libraries of the boll, flower, sepal, and stem, respectively. After quality control, about 6.1 to 6.7 Gb clean data were retained for further analysis, with their effective ratio ranging from 98.21% to 98.93% (Table 1).
Genome assembly
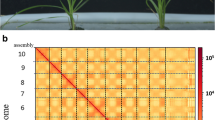
The Pacbio sequencing generated HiFi reads were used to construct the contig-level genome assembly with Hifiasm (v0.13.0-R307)28. 81 contigs were generated with a total length of 1,667.63 Mb. The length of the contigs ranged from 3.8 kb to 149.26 Mb, with the N50 and N90 lengths of 112.12 Mb and 41.97 Mb, respectively. The Hi-C data was then used to construct the chromosome-level assembly with the ALLHiC pipeline29. The tool juicebox (v2.20.00) was used for a manual check and accuracy of the generated assembly (Fig. 2)30. 52 scaffolds were finally acquired, with the N50 and N90 lengths of 137.59 Mb and 102.94 Mb, respectively (Table 2). The total length of 13 pseudo-chromosomes was 1,663.66 Mb, accounting for 99.77% of the entire assembly in length (Table 3).

Heat map of Hi-C interactions in Gossypium arboreum ZB-1. The color indicates the number of Hi-C reads supporting the interactions.
Genome annotation and gene function prediction
Annotation of repetitive sequences
A hybrid strategy was used to identify the repetitive sequences in the ZB-1 genome. The tandem repeats were predicted using TRF (v4.09) (Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, MaxPeriod = 2000)31. The homology-based prediction was carried out using the Repbase database (v202101) and the software RepeatMasker (v4.1.0) (-a -nolow -no_is -norna) and RepeatProteinmask (v4.1.0) (-noLowSimple -pvalue 0.0001 -engine ncbi) (http://www.repeatmasker.org/)32. The de novo repetitive elements database was built with LTR_FINDER (v1.06) (-C -w 2)33, RepeatScout (v1.0.5)34, and RepeatModeler (v2.0.1) (-engine ncbi), then all repeat sequences with lengths greater than 100 bp and gap ‘N’ less than 5% constituted the raw transposable element (TE) library. This TE library was combined with the results from the homology search against Repbase to construct the custom library using Uclust (-id 80)35, and supplied to RepeatMasker for DNA-level repeat identification.
A total of 1,161.62 Mb of repetitive sequences were identified in the ZB-1 genome, representing 69.7% of the entire assembly in length. Apart from the tandem repeats (61.4 Mb, 3.68%), the other repetitive sequences were mainly classified into long terminal repeats (LTR), DNA, and long interspersed nuclear elements (LINE) (Table 4).
Annotation of coding genes
The coding genes were identified by integrating the results from ab initio prediction, homology-based prediction, and cDNA-assisted prediction. Augustus (v3.2.3) (--genemodel = complete,--noInFrameStop = true)36, GlimmerHMM (v3.0.4)37, SNAP (v2013.11.29)38, Geneid (v1.4), and Genscan (v1.0) were used for ab initio prediction39. The predicted proteomes of closely related species, i.e., A2 (‘SXY1’ genome WHU-updated v1)7, E1 (ZSTU_v1)40, D5 (JGI_v2_a2.1)41, AD1 (‘TM-1’ genome NBI_v1.1)42, and Theobroma cacao43, were downloaded from cottongen (https://www.cottongen.org/) and Ensembl Genomes (http://ensemblgenomes.org/) and used for homology-based gene prediction. These protein sequences were aligned to the ZB-1 genome assembly using TBLASTN (v2.2.26) (-F T -e 1e-5), and the matching ones were further aligned to the genome with GeneWise (v2.4.1) for an accurate prediction of the gene structures44,45.
In addition, RNA-seq data of the ZB-1 tissues (leaf, boll, flower, sepal, and stem) were used to assist the gene prediction. Notably, the RNA-seq data of ZB-1 seedling leaves were generated and submitted to the public database by our previous work40, including SRR13933598, SRR13933601, and SRR13933602. Trinity (v2.1.1) (--normalize_reads --full_cleanup –min_glue 2 –min_kmer_cov 2 --KMER_SIZE 25) was used to generate de novo transcripts46. In addition, the short reads from RNA-seq were aligned to the ZB-1 genome using Hisat2 (v2.2.1), and the resulting bam files were used for re-constructing the gene models using Stringtie (v1.3.3)47,48.
The non-redundant gene annotation was then generated by merging the results derived from three methods with EvidenceModeler (EVM, v1.1.1) (--segmentSize 200000 --overlapSize 20000 --min_intron_length 20) and was improved by using PASA pipeline and manual check49.
This study finally identified 50,058 loci of coding genes, including 48,236 genes of high confidence and 1,822 pseudogenes (Table 5). The majority of coding genes (48,021) were found within the constructed chromosomes, in contrast to a small number (215) of genes in the unanchored contigs. The high confidential genes spanned an average of 2,620 bp in the genome, with an average exon number of 4.3 and an average coding sequence (CDS) length of 1,047 bp.
Functional annotation
Functional annotation of the coding genes was performed by homology search against the non-redundant protein database (NR) in NCBI using BLASTP (cutoff evalue 1e-4 and identity 25%). The domain architectures and Gene Ontology annotations of the predicted proteins were characterized by Interproscan (v5.35–74.0) (-appl ProDom, SMART, ProSiteProfiles, PRINTS, Pfam, Panther -iprlookup -dp -goterms) and online service of eggNOG (http://eggnog-mapper.embl.de/)(--evalue 0.001 --score 60 --pident 35)50,51. The combined analysis assigned tentative functions for about 99% of the predicted coding genes. 17,700 genes were mapped to the KEGG pathways, and a similar number of genes (17,774) were assigned GO terms.
Annotation of noncoding RNAs
The tRNAs in the ZB-1 genome were identified using tRNAscan-SE (v2.0.12)52, and the rRNA genes were identified via homology search with BLASTN (v2.2.26) (-e 1e-10). In addition, Rfam (v14.1) and INFERNAL (v1.1.3) (http://infernal.janelia.org/) were used to predict the miRNAs and snRNAs53,54. 274 miRNA genes and 1,369 tRNA genes were identified, as well as 14,383 rRNA and 7,467 snRNA loci (Table 6).
Comparative genomics analyses
Genomic synteny
The predicted proteomes of ZB-1 and SXY1 (WHU-updated v1) were compared using BLASTP (-evalue 1e-5, cutoff identity 50%, coverage 50%). Based on the homology analysis, MCScanX (-s 10) was then used to identify the collinear blocks55, with the result visualized using the R package RIdeogram (0.2.2)56. Extensive genomic synteny was observed between the genomes of these two A2 accessions (Fig. 3), i.e., 99.1% of the coding genes (including pseudogenes) were encoded by the collinear blocks.

Genomic synteny between Gossypium arboreum accessions ZB-1 and SXY1.The collinear regions were determined based on the similarity of coding genes, with each collinear block contained at least ten genes.
SNP calling
Discovery of the SNPs between ZB-1 and SXY1 was carried out using the MUMmer tools (v4.0.0)57. The script nucmer was used to align the two genome sequences, and the script delta-filter was subsequently used to remove the redundancy in the alignments (−1 -q -r). The structural variations were then called using the script show-snaps (-C), and the result was displayed using the R package of CMplot (v4.5.0)58. The overall rate of the SNP occurrence was about 0.53 in every 1,000 nucleotides (Fig. 4). Specifically, 19,017 SNPs were identified in the coding regions (cSNPs), with the occurrence rate of about 0.38 in 1,000 nucleotides.

A graphic view of the single nucleotide polymorphisms between Gossypium arboreum SXY1 and ZB-1. Single nucleotide polymorphisms (SNPs) were identified through genome alignment between the SXY1 and ZB-1 strains, and their distribution across the chromosomes of ZB-1 is presented here. The count of SNPs was determined within a window size of 1 Mb. The color (green to red) indicates the density (low to high) of SNP in the chromosomes.
Data Records
All the high-throughput sequencing data generated in this work were available through NCBI with the project PRJNA935667. The clean data generated by sequencing the genome libraries were deposited in the Sequence Read Archive (SRA) under the accession numbers SRR27009933 (Illumina PE150) and SRR27009931 (Pacbio CCS)59,60. The data generated from sequencing of the Hi-C library was deposited under SRR2700993261. The RNA-seq data of the boll, flower, sepal, and stem could be retrieved from the SRA with the accession numbers SRR27009934-SRR2700993762,63,64,65.
The assembled genome sequence of G. arboreum ZB-1, together with the information on the coding gene structures, has been deposited in GenBank under the accession number JARKNE00000000066. The annotation of noncoding genes and repeat sequence, as well as the gene function prediction, were available in the Figshare database (https://doi.org/10.6084/m9.figshare.24736338)67.
Technical Validation
Evaluation of the completeness and quality of the genome assembly
Mapping the short reads to the genome
The short reads generated from Illumina sequencing of the genome library were aligned to the assembly using BWA (v0.7.17)68, which showed that 99.22% of the reads were mapped to 99.95% of the genome sequence. The average depth of short reads mapping was about 50, with 99.73% of the genomic regions covered by at least four reads, suggesting high concordance and completeness of the ZB-1 genome assembly (Table 7).
Based on the result of short reads alignment, SNP calling was performed using samtools (v1.7)69, which showed an extremely low rate of heterozygosis SNP (0.000563%) and rare homology SNP (<10). In addition, the k-mer based method Merqury was also used to estimate the quality of the genome assembly70, which showed high base accuracy of the genome assembly (Q-value about 50).
Mapping of RNA-seq data
The RNA-seq data generated by this work, i.e., for the ZB-1 samples of flower, sepal, stem, and boll, were aligned to the genome assembly using HiSat2 (v2.2.1) with default settings71. According to the statistics of the alignment, 94.5% (boll), 96.2% (flower), 91.9% (sepal), and 94.4% (stem) of the properly paired short reads from RNA-seq could be mapped to the pseudo-chromosomes.
BUSCO assessment
The BUSCO (Benchmarking Universal Single-Copy Orthologs) method was also used to evaluate the completeness of the genome assembly72,73. The ZB-1 genome assembly captured 99.4% of the BUSCO 1614 reference gene set (embryophyta_odb10), indicating the completeness of the ZB-1 genome assembly (Table 8).
Evaluation of the genome annotation
The genomic features of the coding genes were compared between G. arboreum ZB-1 and closely related species within the genus Gossypium, including G. arboreum SXY1, G. hirsutum, G. raimondii, and G. stocksii, with the versions of their genome assemblies as aforementioned. According to the distribution of the exon number and the length of CDS, exon, gene, and intron, the gene features of ZB-1 were highly consistent with other cotton species (Fig. 5), suggesting the overall high accuracy of the gene annotation. The predicted proteome of ZB-1 was also subjected to a search against the BUSCO 1614 reference gene set (embryophyta_odb10) using HMMER (v3.4) (http://hmmer.org/). With the recommended cutoff by BUSCO, this assessment showed that 98.4% of the reference genes have been annotated in this study.

Comparison of the gene features among Gossypium species. a-e indicate the distribution of the (a) CDS length, (b) exon length, (c) exon number, (d) gene length, and (e) intron length. The untranslated regions were not included when calculating the exon length and number. The gene length was estimated by the length of the genomic region from the translation initiation site to the translation termination site.
Evaluation of the SNP calling
About 95.1 Gb of short reads from sequencing the genome library of SXY1 were downloaded from SRA with the accession number SRR1306194374. About 97.5% of the reads were retained after quality control using Trimmomatic (v0.33) (SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50)75. 97.1% of the clean data were aligned to the ZB-1 genome sequence using BWA (v0.7.17) with default settings. GATK (v4.0.5.1) tools MarkDuplicates and HaplotypeCaller were used to remove the redundancy raised by PCR amplification and identify the structure variations (default parameters), respectively76. According to the result, 88.1% (16,752/19,017) of the cSNPs identified from genome sequence alignment (by MUMMER) were validated by the short reads, suggesting high accuracy of the estimation of the SNP occurrence rate between SXY1 and ZB-1 and also high quality of the ZB-1 genome assembly.
Code availability
All the bioinformatics tools were used according to the users’ manuals. Otherwise specified in the context, default settings were used during data processing. No specific custom code has been developed in this study.
References
Huang, G., Huang, J. Q., Chen, X. Y. & Zhu, Y. X. Recent Advances and Future Perspectives in Cotton Research. Annu Rev Plant Biol 72, 437–462 (2021).
Wendel, J. F., Brubaker, C. L. & Seelanan, T. The Origin and Evolution of Gossypium. in Physiology of Cotton (eds. Stewart, J. M., Oosterhuis, D. M., Heitholt, J. J. & Mauney, J. R.) 1-18 (Springer Netherlands, Dordrecht, 2010).
Chen, Z. J. et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat Genet 52, 525–533 (2020).
Grover, C. E. et al. Dual Domestication, Diversity, and Differential Introgression in Old World Cotton Diploids. Genome Biology and Evolution 14, evac170 (2022).
Yuan, D. et al. Parallel and Intertwining Threads of Domestication in Allopolyploid Cotton. Adv Sci (Weinh) 8, 2003634 (2021).
Wen, X. et al. A comprehensive overview of cotton genomics, biotechnology and molecular biological studies. Sci China Life Sci 66, 2214–2256 (2023).
Huang, G. et al. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat Genet 52, 516–524 (2020).
Feng, Y. L. et al. Assembly and phylogenomic analysis of cotton mitochondrial genomes provide insights into the history of cotton evolution. Crop Journal 11, 1782–1792 (2023).
Wu, Y. et al. An insight into the gene expression evolution in Gossypium species based on the leaf transcriptomes. BMC Genomics 25, 179 (2024).
Flagel, L. E. & Wendel, J. F. Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol 186, 184–193 (2010).
Rapp, R. A., Udall, J. A. & Wendel, J. F. Genomic expression dominance in allopolyploids. BMC Biol 7, 18 (2009).
Ke, L. et al. Differential transcript profiling alters regulatory gene expression during the development of Gossypium arboreum, G.stocksii and somatic hybrids. Sci Rep 7, 3120 (2017).
Li, B., Zhu, S., Wang, H. & Zhang, B. Bred and studied of a new allotetraploid cotton germplasm with glandless seeds/glanded plant trait. Acta Gossypii Sinica 3, 27–32 (1991).
Gao, W. et al. Development of the engineered “glanded plant and glandless seed” cotton. Food Chem (Oxf) 5, 100130 (2022).
Nie, Y. & Liu, J. The botanical and agronomic characters of new allotetraploid germplasm of Gossypium arboreum x G. stocksii. Journal of Huazhong Agricultural University 14, 333–337 (1995).
Chen, Y. et al. A new synthetic amphiploid (AADDAA) between Gossypium hirsutum and G. arboreum lays the foundation for transferring resistances to Verticillium and drought. PLoS One 10, e0128981 (2015).
Khan, Z. et al. Genome editing in cotton: challenges and opportunities. Journal of Cotton Research 6 (2023).
Dong, Y. et al. Parental legacy versus regulatory innovation in salt stress responsiveness of allopolyploid cotton (Gossypium) species. Plant J 111, 872–887 (2022).
Peng, Z. et al. Expression patterns and functional divergence of homologous genes accompanied by polyploidization in cotton (Gossypium hirsutum L.). Sci China Life Sci 63, 1565–1579 (2020).
Ke, L., Jiang, Q., Wang, R., Yu, D. & Sun, Y. Plant regeneration via somatic embryogenesis in diploid cultivated cotton (Gossypium arboreum L.). Plant Cell, Tissue and Organ Culture (PCTOC) 148, 177–188 (2022).
Li, F. et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46, 567–572 (2014).
Du, X. et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat Genet 50, 796–802 (2018).
Wang, M. et al. Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium. Nat Genet 54, 1959–1971 (2022).
Wang, M. et al. Comparative Genome Analyses Highlight Transposon-Mediated Genome Expansion and the Evolutionary Architecture of 3D Genomic Folding in Cotton. Mol Biol Evol 38, 3621–3636 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, 265–268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), 351–358 (2005).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2), 215–225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Parra, G., Blanco, E. & Guigo, R. GeneID in Drosophila. Genome Res 10, 511–515 (2000).
Yu, D. et al. Multi-omics assisted identification of the key and species-specific regulatory components of drought-tolerant mechanisms in Gossypium stocksii. Plant Biotechnol J 19, 1690–1692 (2021).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Zhang, T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33, 531–537 (2015).
Argout, X. et al. The genome of Theobroma cacao. Nat Genet 43, 101–108 (2011).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278 (2019).
Haas, B. J., Zeng, Q., Pearson, M. D., Cuomo, C. A. & Wortman, J. R. Approaches to Fungal Genome Annotation. Mycology 2, 118–141 (2011).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res 51, D418–D427 (2023).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci 6, e251 (2020).
Marcais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol 14, e1005944 (2018).
Yin, L. et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genomics Proteomics Bioinformatics 19, 619–628 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009933 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009931 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009932 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009934 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009935 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009936 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27009937 (2023).
Yu, D. ENA https://identifiers.org/insdc.gca:GCA_036320975.1 (2024).
Sun, R. et al. Chromosome-level genome assembly and annotation of a potential model organism Gossypium arboreum ZB-1. Figshare https://doi.org/10.6084/m9.figshare.24736338 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–60 (2015).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654 (2021).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr Protoc 1, e323 (2021).
NCBI Sequence Read Archive https://identifers.org/ncbi/insdc.sra:SRR13061943 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20 (2014).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11.10.1–11.10.33 (2013).
Acknowledgements
The authors would like to thank Dr. Wang Tingzhang and Dr. Hua Xuejun for their assistance during the gene annotation and manuscript writing processes. This work was supported by the National the National Natural Science Foundation of China (32170623 and U190320) and the Fundamental Research Funds of Zhejiang Sci-Tech University (19042398-Y).
Author information
Authors and Affiliations
Contributions
D.Y. and Y.S. conceived and designed the study; R.S., Y.W. and D.Y. analysed the sequencing data; X.Z. and M.L. collected the samples; R.S. and D.Y. wrote the draft manuscript; Y.S. and D.Y. modified the manuscript. All authors have read and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, R., Wu, Y., Zhang, X. et al. Chromosome-level genome assembly and annotation of a potential model organism Gossypium arboreum ZB-1. Sci Data 11, 620 (2024). https://doi.org/10.1038/s41597-024-03481-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03481-z
- Springer Nature Limited