
Abstract
Elymus species, belonging to Triticeae tribe, is a tertiary gene pool for improvement of major cereal crops. Elymus sibiricus, a tetraploid with StH genome, is a typical species in the genus Elymus, which is widely utilized as a high-quality perennial forage grass in template regions. In this study, we report the construction of a chromosome-scale reference assembly of E. sibiricus line Gaomu No. 1 based on PacBio HiFi reads and chromosome conformation capture. Subgenome St and H were well phased by assisting with kmer and subgenome-specific repetitive sequence. The total assembly size was 6.929 Gb with a contig N50 of 49.518 Mb. In total, 89,800 protein-coding genes were predicted. The repetitive sequences accounted for 82.49% of the genome in E. sibiricus. Comparative genome analysis confirmed a major species-specific 4H/6H reciprocal translocation in E. sibiricus. The E. sibiricus assembly will be much helpful to exploit genetic resource of StH species in genus Elymus, and provides an important tool for E. sibiricus domestication.
Similar content being viewed by others


Background & Summary
The genus Elymus L. belongs to the grass tribe Triticeae, containing approximately 150 species1,2. The genus is entirely composed of polyploidy species with StH, StY, StHY, StPY, and StWY, including five basic genomes. The included basic genomes St, H, P, W are derived from Pseudoroegeneria (Neveski) Löve, Hordeum L., Agropyron Gaertn., and Australopyrum (Neveski) Love, respectively, although the origin of Y genome is still unknown3,4. Elymus species belong to the same tribe with staple food crops such as wheat (Triticum aestivum, 2n = 6x = 42; AABBDD genome), rye (Secale cereal, 2n = 2x = 14), and barley (Hordeum vulgare, 2n = 2x = 14), and which are important genetic resources with high diversity, constituting a tertiary gene pool for improvement of major cereal crops.
Elymus sibiricus L. (Siberian wild rye), a typical species of the genus Elymus, is a well-known perennial and caespitose grass. E. sibiricus is widely distributed in the northern hemisphere, with particular preponderance in Sweden, northern Asia, Japan, and North America5, which is mostly utilized as perennial forages in template regions6,7. E. sibiricus is an allotetraploid with a genome constitution of StStHH (2n = 4x = 28)2. Chromosomal polymorphisms and major rearrangements of E. sibiricus have been revealed by Florescence in situ hybridization (FISH) in different accessions8,9. Genomic SSR markers were exploited by screening enriched microsatellite DNA library for genetic diversity evaluation10. Transcriptome of E. sibiricus was profiled to reveal candidate genes connected to seed shattering11. Genome sequencing was carried out by Illumina HiSeq X-ten platform, and a draft genome of 4.34 Gb was assembled, and which was used for SSR markers development12.
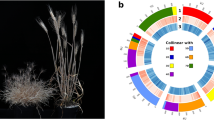
In this study, an E. sibiricus chromosome-scale reference genome by integrating PacBio HiFi reads and chromatin conformation capture data was assembled. The high-quality E. sibiricus assembly obtained in this study provides a reference for the StH genome of the genus Elymus in the Triticeae tribe (Fig. 1). It will be much helpful to facilitate genetic resource evaluation of StH species in genus Elymus. Furthermore, it can be served as important tool to directly domesticate E. sibiricus as a forage crop or even a cereal crop.

Overview of the assembled E. sibiricus genome. (a–g) are as followers: collinearity between the chromosomes (The color red is used to highlight the associations between different homologous), chromosomes, gene counts, GC content, Simple repeat density, DNA transposons density, LTR elements density (The window used for calculating the density of the above elements is 100,000 bp).
Methods
Plant materials and genome sequencing
The inbreed line Gaomu No.1 of E. sibiricus required for sequencing was self-crossed exactly 6 generations. Fresh young leaf tissue of it was collected, frozen in liquid nitrogen, The extraction of DNA samples follows the CATB method13. The DNA library preparation and sequencing were carried out according to the protocol provided in the SMRTbell® prep kit 3.0 instruction manual and sequencing was performed on the PacBio Revio platform. DNA required for Hi-C sequencing was purified using the QIAamp DNA Mini Kit (CAT#51306, Qiagen) following the manufacturer’s protocol, while for Next-Generation Sequencing (NGS) whole genome sequencing, libraries were constructed using the MGIEasy Universal DNA Library Prep Kit V1.0 (CAT#1000005250, MGI) following the standard protocol. The Hi-C library was sequenced on the DNBSEQ-T7 platform, while NGS for whole-genome sequencing was conducted on the MGISEQ-2000 platform. Fastp v0.23.414 with default parameters was used to obtain NGS clean reads. All genome sequencing and Hi-C sequencing data were derived from a single plant. The data obtained from each platform is shown in Table 1.
Raw reads from full-length transcriptome sequencing were processed into circular consensus (CCS) reads based on the adapter. Subsequently, full-length, non-chimeric (FLNC) transcripts were identified by detecting the poly A tail signal and 5′ and 3′ cDNA primers in CCS. Clustering was performed on full-length sequences from the same transcript, grouping similar full-length sequences into clusters, and obtaining a consensus sequence for each cluster. These consensus sequences were then corrected to obtain high-quality sequences for further analysis. High-quality FL transcripts from Iso-Seq were used to remove redundancy using cd-hit v4.8.115 (identity >0.99).
Genome assembly and chromosome construction
The genome of E. sibiricus at the contig level was assembled using the hifiasm v0.19.616, supplemented by Hi-C data and Pacbio HiFi data. Conserved homologous probes17 across A, B, D genome of common wheat (Triticum aestivum L.)18, and H genome of barley (Hordeum vulgar L.)19 were developed using CHORUS2 v2.0.120. BWA v0.7.1721 is utilized to align Hi-C data to the draft genome reference. Subsequently, contigs and Hi-C alignment were classified based on these homologous probes. Classified contigs were subjected to chromosome construction through the polyploid workflow of ALLHiC22. Juicebox v1.11.0823 was used to further manually correct the chromatin contact matrix and built the Hi-C interaction heatmap. SubPhase v1.2.624 (kmer = 15) with default parameters was used to distinguish between two subgenomes of E. sibiricus. An H genome specific transposable element (Gypsy-96_TAe-LTR) was obtained by a pipeline procedure of RepeatExplorer25,26 using low coverage NGS sequencing data of both H genome donor species Hordeum bogdanii and St genome donor species Pseudoroegneria stipifolia. The content of the Gypsy-96_TAe-LTR was estimated hundreds times more in H genome than St genome. We used this element to further confirm which set of subgenomes is H and which set is St (Table 2). Benchmarking Universal Single-Copy Orthologs27 (BUSCO v5.2.2) and LTR Assembly Index28 (LAI) were employed to evaluate the completeness and contiguity of genome assemblies. Finally the assembly resulted in a genome size of 6.929 Gb with an contig N50 of 49.518 Mb (Table 3). Using SubPhaser and subgenome-specific repetitive sequence, we were able to successfully separate the two sets of subgenomes (Fig. 2).

Principal component analysis (PCA) based on subgenome-specific kmers (kmer = 15).
Annotation of repetitive sequences and function gene
LTRfinder v1.0729 (-w 2 -C -D 15000 -d 1000 -L 7000 -l 100 -p 20 -M 0.85) and LTRHarvest v1.6.530 (-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes) were used to initially predict Long Terminal Repeat (LTR) sequences. Subsequently, LTR_retriever v2.9.531 was used to merge the results and obtain the final LTR predictions. A De Novo repeat sequence database for E. sibiricus was constructed using RepeatModeler v2.0.332 with default parameters. The final repeat sequence predictions were conducted using RepeatMasker v4.1.233 pipeline.
The BRAKER3 v3.0.334 pipeline was used for structural annotation of E. sibiricus genome. This comprehensive pipeline incorporated three sources of extrinsic evidence: short-read RNA-seq data obtained from the public NCBI Illumina dataset (SRP101478)35, full-length transcriptome sequencing from the current experiment, and protein sequences of Eukaryota sourced from OrthoDB36. BRAKER3 utilizes the GeneMark-ETP v1.0237 pipeline for gene prediction. This involves assembling transcript sequences with StringTie v2.2.138. Short RNA-Seq reads were aligned to the genome by HISAT2 v2.2.139. GeneMarkS-T analyzes the assembled transcripts to predict protein-coding genes, which are then searched against a protein database. ProtHint maps homologous proteins back to the genome, generating hints for another round of gene structure prediction. AUGUSTUS v3.4.040 is trained on the high-confidence gene set and predicts a second genome-wide gene set with hint support. The predictions from these components were integrated using TSEBRA41.
This study found that repetitive sequences accounted for 82.49% of the genome in E. sibiricus (Table 4). A total of 89,800 protein-coding genes were annotated, with an average gene length of 2,315 bp and an average CDS length of 1,075 bp (Table 5). Among these annotated genes, 85,250 genes were annotated in the NR42 database, 49,637 in the Swiss-Prot43 database, 63,623 in the Pfam44 database, 24,763 in the GO45 database, and 18,856 in the KEGG46 database. Additionally, 85,274 genes are annotated in at least one of these databases (Fig. 3).

The venn picture of the function genes of E. sibiricus by using different database.
Phylogenetic tree construction
We have selected the Coding DNA Sequences(CDS) of the following genomes for phylogenetic analysis: Oryza sativa47, Brachypodium distachyon48, Triticum aestivum (subgenomes A, B, and D), Secale cereale49, Thinopyrum intermedium (subgenomes St, Jr, and Jvs) (https://phytozome-next.jgi.doe.gov/info/Tintermedium_v3_1), Dasypyrum villosum50, Hordeum vulgare along with E. sibiricus (subgenomes H and St). Orthofinder v2.5.551 with the search engine Blast v2.14.152 was employed to identify orthologous genes. From the selected genomes, a total of 2,082 lineal homologous genes were obtained. MUSCLE v5.153 was used for multiple sequence alignment. The phylogenetic tree was constructed using RAxML v8.2.1254 with the maximum likelihood method. Divergence times were estimated with mcmctree v4.10.755 using the calibrated times (O. sativa - B. distachyon: 41.5–62.0 MYA) from the Time Tree56 website (Fig. 4).

Estimation of divergence time between E. sibiricus and related species. Divergence times (unit: MYA) are indicated at each node. Green represent Triticeae species; red represent Brachypodieae species; blue represent Oryzeae species.
synteny analysis
One Step MCScanX in TBtools-II57 was used for synteny analysis. First,coding protein sequences between subgenomes were aligned using blastp v2.15.0 + (−evalue 1e-5 -num_alignments 5), MCScanX v2022.11.0158 with default parameters was employed to identify collinear blocks.
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center59, China National Center for Bioinformation/Beijing Institute of Genomics60, Chinese Academy of Sciences (GSA: CRA014200)61. The final chromosome assembly of E. sibiricus was deposited at GenBank under the accession number JBDKXM00000000062. Genome assembly and annotation, conserved homologues probes and subgenome-specific repetitive sequnce were uploaded to figshare63.
Technical Validation
The genome-wide Hi-C interaction heatmap was generated using Juicerbox. The coordinates in the heatmap represent all bins on individual chromosomes, where the color of each point indicates the logarithmic value of the corresponding bin pair interaction strength in the genome (Fig. 5). The interaction strength intensifies from white to red, with darker colors indicating higher interaction strength. Notably, regions with higher interaction strength exhibit deeper colors, and the depth of colors along the diagonal is significantly higher than at the two ends. The anti-diagonals are typical for Triticeae genomes and correspond the Rabl configuration of Triticeae chromosomes64,65. Following manual adjustments, the current assembly of the E. sibiricus genome adheres to the distance-dependent interaction decay. From the global heatmap perspective, the overall assembly results appear satisfactory, with no apparent clustering errors between chromosomes.

Contact map after the integration of the Hi-C data and manual correction. blue boxes represent pseudo molecules; green boxes represent contigs.
The ultimate calculated LTR Assembly Index (LAI) value is 12.61, with a corresponding raw LAI of 18.02. In accordance with the criteria proposed by the authors of the LTR_retriever methodology, the assembly quality of the E. sibiricus is categorized at the reference level.
The BUSCO analysis of the entire genome indicates a high level of completeness and contiguity in the assembly of the E. sibiricus genome. Among the 4895 single-copy gene set, only 38 single-copy genes were found to be either missing or fragmented. We also conducted BUSCO analysis by extracting the longest transcript of each gene. The results indicate a relatively complete annotation, with the majority of genes on subgenomes being identified as single-copy (Table 6).
Phylogenetic analysis with the assembled CDS showed close relationships between St genome in E. sibiricus and St in Th. Intermidum, and those between H genome in E. sibiricus and H. vulgare, which is accordant with the recognized genome constitution of E. sibiricus.
The synteny analysis revealed an apparent collinearity distort in 4H and 6H chromosome (Fig. 1), which was confirmed by a species-specific 4H/6H reciprocal translocation detected by chromosomal Florescence in situ hybridization with single-gene probes in E. sibiricus8.
Code availability
All software and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and parameters of software have been described in Methods.
References
Löve, Á. Conspectus of the Triticeae. Feddes Repert. 95, 425–521 (1984).
Dewey, D. R. The Genomic System of Classification as a Guide to Intergeneric Hybridization with the Perennial Triticeae. in Gene Manipulation in Plant Improvement: 16th Stadler Genetics Symposium (ed. Gustafson, J. P.) 209–279 (Springer US, Boston, MA, 1984). https://doi.org/10.1007/978-1-4613-2429-4_9.
Wang, R. R. C. & Lu, B. Biosystematics and evolutionary relationships of perennial Triticeae species revealed by genomic analyses. J. Syst. Evol. 52, 697–705 (2014).
Wang, R., Jensen, K. & Jaussi, C. Proceedings of the 2nd International Triticeae Symposium. Triticeae 2nd International Triticeae Symposium (1994).
Baum, B. R., Edwards, T., Ponomareva, E. & Johnson, D. A. Are the Great Plains wildrye (Elymus canadensis) and the Siberian wildrye (Elymus sibiricus) conspecific? A study based on the nuclear 5S rDNA sequences. Botany 90, 407–421 (2012).
Klebesadel, L. J. Siberian Wildrye (Elymus sibiricus L.): Agronomic Characteristics of a Potentially Valuable Forage and Conservation Grass for the North1. Agron. J. 61, 855–859 (1969).
Pei-sheng, Ma. O., Jian-guo, Ha. N. & Xi-cai, W. Effects of Harvest Time on Seed Yield of Siberian Wildrye. Acta Agrestia Sin. 11, 33 (2003).
Liu, B. et al. Single-gene FISH maps and major chromosomal rearrangements in Elymus sibiricus and E. nutans. BMC Plant Biol. 23, 98 (2023).
Xie, J., Zhao, Y., Yu, L., Liu, R. & Dou, Q. Molecular karyotyping of Siberian wild rye (Elymus sibiricus L.) with oligonucleotide fluorescence in situ hybridization (FISH) probes. PLOS ONE 15, e0227208 (2020).
Lei, Y., Zhao, Y., Yu, F., Li, Y. & Dou, Q. Development and characterization of 53 polymorphic genomic-SSR markers in Siberian wildrye (Elymus sibiricus L.). Conserv. Genet. Resour. 6, 861–864 (2014).
Xie, W., Zhang, J., Zhao, X., Zhang, Z. & Wang, Y. Transcriptome profiling of Elymus sibiricus, an important forage grass in Qinghai-Tibet plateau, reveals novel insights into candidate genes that potentially connected to seed shattering. BMC Plant Biol. 17, 78 (2017).
Xiong, Y. et al. Genomic survey sequencing, development and characterization of single- and multi-locus genomic SSR markers of Elymus sibiricus L. BMC Plant Biol. 21, 3 (2021).
A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull.
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinforma. Oxf. Engl. 22, 1658–1659 (2006).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Liu, B. Chromosomal structure rearrangements and associated genes expression in Elymus nutants. (Northwest Institute of Plateau Biology, Chinese Academy of Sciences, 2023).
The International Wheat Genome Sequencing Consortium (Iwgsc). et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018).
Jayakodi, M. et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289 (2020).
Zhang, T., Liu, G., Zhao, H., Braz, G. T. & Jiang, J. Chorus2: design of genome-scale oligonucleotide-based probes for fluorescence in situ hybridization. Plant Biotechnol. J. 19, 1967–1978 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25, 1754–1760 (2009).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Jia, K.-H. et al. SubPhaser: a robust allopolyploid subgenome phasing method based on subgenome-specific k-mers. New Phytol. 235, 801–809 (2022).
Novák, P., Neumann, P., Pech, J., Steinhaisl, J. & Macas, J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinforma. Oxf. Engl. 29, 792–793 (2013).
Novák, P., Neumann, P. & Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11, 378 (2010).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. Clifton NJ 1962, 227–245 (2019).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. Chapter 4, 4.10.1–4.10.14 (2009).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-Seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. BioRxiv Prepr. Serv. Biol. 2023.06.10.544449 https://doi.org/10.1101/2023.06.10.544449 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP101478 (2017).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res. 51, D445–D451 (2023).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistency with Extrinsic Data. BioRxiv Prepr. Serv. Biol. 2023.01.13.524024 https://doi.org/10.1101/2023.01.13.524024 (2023).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Nachtweide, S. & Stanke, M. Multi-Genome Annotation with AUGUSTUS. Methods Mol. Biol. Clifton NJ 1962, 139–160 (2019).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 566 (2021).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26 (2022).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Ouyang, S. et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 35, D883–D887 (2007).
Vogel, J. P. et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010).
Rabanus-Wallace, M. T. et al. Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential. Nat. Genet. 53, 564–573 (2021).
Zhang, X. et al. A chromosome-scale genome assembly of Dasypyrum villosum provides insights into its application as a broad-spectrum disease resistance resource for wheat improvement. Mol. Plant 16, 432–451 (2023).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Edgar, R. C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 13, 6968 (2022).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma. Oxf. Engl. 30, 1312–1313 (2014).
Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Kumar, S. et al. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 39, msac174 (2022).
Chen, C. et al. TBtools-II: A ‘one for all, all for one’ bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics 19, 578–583 (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2021. Nucleic Acids Res. 49, D18–D28 (2021).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA014200 (2024).
NCBI GenBank https://identifiers.org/nucleotide:JBDKXM000000000 (2024).
Shen, W., Liu, B., Guo, J., Yang, Y. & Dou, Q. Chromosome-scale assembly of the wild cereal relative Elymus sibiricus. figshare https://doi.org/10.6084/m9.figshare.24964659 (2024).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Tiang, C.-L., He, Y. & Pawlowski, W. P. Chromosome Organization and Dynamics during Interphase, Mitosis, and Meiosis in Plants. Plant Physiol. 158, 26–34 (2012).
Acknowledgements
This work was supported by the Chinese Academy of Sciences strategic leading science and technology project (XDA24030502), and the team project of the Natural Science Foundation of Qinghai Province (Grant No. 2022-ZJ-902).
Author information
Authors and Affiliations
Contributions
Q.D. designed the research project and participated in the writing. J.G., Y.Y. and X.L. cultivated plants and collected the sample for sequencing. B.L. and J.C. developed homologous probes and the subgenome-specific repetitive sequence. W.S. assemble the genome, performed the bioinformatics analyses and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, W., Liu, B., Guo, J. et al. Chromosome-scale assembly of the wild cereal relative Elymus sibiricus. Sci Data 11, 823 (2024). https://doi.org/10.1038/s41597-024-03622-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03622-4
- Springer Nature Limited