
Abstract
Diomorus aiolomorphi Kamijo (Hymenoptera: Torymidae) is an inquiline of gall maker Aiolomorphus rhopaloides Walker (Hymenoptera: Eurytomidae). They are of significant economic significance and predominantly inhabit bamboo forest. So far, only four scaffold-level genomes have been published for the family Torymidae. In this study, we present a high-quality genome assembly of D. aiolomorphi at the chromosome level, achieved through the integration of Nanopore (ONT) long-read, Illumina pair-end DNA short-read, and High-through Chromosome Conformation Capture (Hi-C) sequencing methods. The final assembly was 1,084.56 Mb in genome size, with 1,083.41 Mb (99.89%) assigned to five pseudochromosomes. The scaffold N50 length reached 224.87 Mb, and the complete Benchmarking Universal Single-Copy Orthologs (BUSCO) score was 97.3%. The genome contained 762.12 Mb of repetitive elements, accounting for 70.27% of the total genome size. A total of 18,011 protein-coding genes were predicted, with 17,829 genes being functionally annotated. The high-quality genome assembly of D. aiolomorphi presented in this study will serve as a valuable genomic resource for future research on parasitoid wasps. The results of this study may also contribute to the development of biological control strategies for pest management in bamboo forests, enhancing ecological balance and economic sustainability.
Similar content being viewed by others

Background & Summary
Diomorus aiolomorphi Kamijo (Hymenoptera: Torymidae) is a parasitic inquiline associated with the gall maker Aiolomorphus rhopaloides Walker (Hymenoptera: Eurytomidae). D. aiolomorphi and A. rhopaloides are of significant economic significance and predominantly inhabit bamboo forest. Notably, these two species constitute approximately 90% of the insects within this group in such environments1.
The gall maker A. rhopaloides lays its eggs in the internode at the base of the new branch buds, stimulating the paraplegma tissue in these areas. This process inhibits the growth of bamboo plants, leading to a reduction in both the quantity and quality of bamboo shoots. It has been observed that bamboo galls are contagiously distributed across both the culms and branches in a bamboo stand2,3. Its harm makes it a significant factor hindering effective management and economic value of bamboo forests, with notable impacts on both society and the environment4. It not only leads to reduced bamboo yield, lower quality, and decreased market prices but also results in indirect losses such as control and restoration costs and ecological impacts2,3,4,5. Adults of D. aiolomorphi, known as inquilines, oviposit on these young bamboo galls. Unlike typical phytophagous insects, D. aiolomorphi cannot create its own galls but instead feeds on the gall tissues induced by other gall makers4,5. Understanding the attack pattern of D. aiolomorphi on bamboo galls is crucial for assessing and managing the population density of A. rhopaloides1. Despite the commonality of D. aiolomorphi among gall makers and its economic significance, it has received relatively little scientific attention6. Consequently, there is a substantial gap in our understanding of the genetic makeup underpinning the genome of D. aiolomorphi.
In this study, we have assembled the chromosome-level genome of D. aiolomorphi, representing the first chromosome-level sequenced genome of the family Torymidae. The genome size is 1,084.56 Mb, with 1,083.41 Mb (99.89%) assigned to five pseudochromosomes. The scaffold N50 of the genome is 224.87 Mb in length, and the complete Benchmarking Universal Single-Copy Orthologs (BUSCO) score reached 97.3%. A total of 762.12 Mb repetitive elements were identified, accounting for 70.27% of the total genome size. 18,011 protein-coding genes, with functional annotations available for 17,829 of these genes. The high-quality genome assembly of D. aiolomorphi provides a valuable repository for understanding the genomic traits of the Torymidae genomes.
Methods
Sampling
Galls were sampled from bamboo branches at Fuyang, Hangzhou, China (30°03′ N, 119°57′ E) before gall maker emergence, and a total of 1,467 galls were collected. An inquiline is an organism that lives within or on the structure of another organism. The inquiline, D. aiolomorphi, emerged from galls 15–20 days later than the gall maker A. rhopaloides. Before sequencing, both morphological examination7 and COI barcode information confirmed the identification of the species as D. aiolomorphi. The specimens were deposited at the Institute of Insect Sciences, Zhejiang University (ZJUH_20231101). They were preserved in 100% ethanol prior to DNA extraction to maintain the integrity of the genetic material, and subsequently kept in the scientific specimen repository.
Library preparation and genomic DNA sequencing
Genomic DNA was prepared by the sodium dodecyl sulfate (SDS) method followed by purification with QIAGEN® Genomic kit (Qiagen, Hilden, Germany) according to the manufacturer’s standard operating procedure for both long-read and short-read whole genome sequencing (https://www.qiagen.com/us/resources/resourcedetail?id = 566f1cb1-4ffe-4225-a6de-6bd3261dc920&lang = en). RNA extraction was conducted with the TRlzol reagent (Vazyme, Nanjing, China) (https://bio.vazyme.com/product/730.html). The quality of the extracted RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA Integrity Number (RIN) was determined for each sample, ensuring that only high-quality RNA (RIN > 7.0) was used for subsequent sequencing processes. The total data produced from RNA extraction amounted to 4.73 Gb, with a duplication rate of 66.07%. The Q20 (Quality scores > 20) bases totaled 3,607,313,837 (97.8713%), while the Q30 (Quality scores > 30) bases amounted to 3,455,480,380 (93.7518%). Long-read sequencing was performed on the Nanopore GridION X5/PromethION sequencer (Oxford Nanopore Technologies, UK) at Nextomics. Short-read and transcriptome sequencing were sequenced on the Illumina Novaseq/MGI-2000 platforms. The total data generated from the long-read sequencing was 81.21 Gb, while the output from the short-read sequencing totaled 28.37 Gb (Table 1).
Genome survey and assembly
K-mer analysis was performed using Illumina paired-end sequenced DNA reads. This analysis was conducted before genome assembly to estimate the genome size and the level of heterozygosity. Briefly, quality-filtered reads were subjected to a 21-mer frequency distribution analysis employing Jellyfish v2.2.108. For a read of length L, the number of k-mer produced is (L - 21 + 1). Therefore, the genome size (G) is estimated by the formula: G = Knumber / Kdepth, where Knumber represents the total number of k-mer produced and Kdepth represents the peak value of k-mer depth. Furthermore, the overall genomic properties were inferred by GenomeScope v1.09. The preliminary genome survey of D. aiolomorphi revealed a low level of heterozygosity level (0.19%) within a substantial genome, 988,63 Mb. This estimated genome size was used to evaluate the integrity of the subsequent assembly (Fig. 1, Supplementary Table S1).

The K-mer distribution of the D. aiolomorphi genome. len, genome haploid length; uniq, genome unique length; het, heterozygosity; kcov, genome coverage; err, read error rate; dup, duplicated sequence.
The primary assembly of the clean reads obtained from the Nanopore platform was conducted using nextDenovo v2.5.010, and subsequently corrected using Canu v2.1.111. Illumina paired-end sequenced DNA reads were then utilized to polish and enhance the genome assembly using nextPolish v1.4.012. To eliminate haplotigs and contig overlaps in the de novo assembly, purge_dups v1.2.5 (https://github.com/dfguan/purge_dups) was employed. Finally, the primary assembly yielded 147 scaffolds with 1,084.58 Mb in genome size, 18.13 Mb in contig N50 and 224.87 Mb in scaffold N50.
Chromosome Hi-C assembly
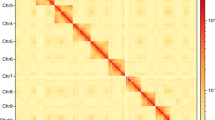
The High-through Chromosome Conformation Capture (Hi-C) method13 was utilized to anchor accurately position hybrid scaffolds onto chromosomes. Genomic DNA was extracted from the thorax of an individual D. aiolomorphi for the Hi-C library. This library, along with the sequencing data was processed via the Illumina Novaseq/MGI-2000 platform. The procedure yielded high-quality clean reads of 110.44 Gb of raw data (Table 1). All subsequent analyses were then applied to these clean reads. The clean Hi-C paired-end reads were initially mapped to the primary assembly using Bowtie2 v2.3.214. Then, HiC-Pro v2.8.115 was utilized to identify valid alignments, simultaneously filtering out multiple hits and singletons alignments. Finally, Lachesis16 was employed to cluster, order and orient the scaffolds. Following Lachesis analysis, 1,083.41 Mb of reads were allocated to five pseudochromosomes, amounting to 99.89% of the final assembly (Fig. 2, Table 2).

Overview of the genomic features of the D. aiolomorphi genome. (a) Genome-wide all-by-all Hi-C interaction identified five pseudochromosome link groups of the Diomorus aiolomorphi genome; (b) Genomic features of the D. aiolomorphi genome. Tracks from outside to inside (a-e) are as follows: pseudochromosomes, GC contents, repeat density, gene density and collinearity between the pseudochromosomes.
Assessment of the genome assembly
To assess the completeness and accuracy of the final assembly of D. aiolomorphi genome, Benchmarking Universal Single Copy Orthologs (BUSCO) v5.2.217 with the insect_obd10 database and hymenoptera_obd10 database were utilized. The assessments yielded high BUSCO scores of 97.3% and 91.1%, respectively (Fig. 3, Supplementary Table S2-3). Additionally, to ascertain the integrity of the genome assembly, the five pseudochromosomes from the final assembly were aligned to the Nt library to evaluate the genome assembly using BLAST v2.5.018. Among the 5 chromosomes, 60% (3 pseudochromosomes) showed similarity to Nasonia vitripennis, 20% (1 pseudochromosomes) to Eretmocerus sp. and 20% (1 pseudochromosomes) to Torymus sp. These results suggest the pseudochromosomes sequences are free from sequences of non-target organisms, contaminants, or symbionts presented in the DNA library (Supplementary Table S4).

The BUSCO summary of the D. aiolomorphi genome. The x axis represents the percentage of BUSCOs and the y axis represents BUSCO datasets.
Repetitive element annotation
In the D. aiolomorphi genome, transposon element (TE) were identified using the Extensive de novo TE Annotator (EDTA) v1.9.619. Tandem Repeats Finder (TRF) v4.0920 facilitated the identification of tandem repeats. Based on these findings, a de novo repeat database was consequently generated using RepeatModeler v2.0.221. The known repeats in Dfam database22 were combined with the results of TE detection and the de novo repeat database, creating a reference library that was clustered using Cd-hit v4.8.123 to eliminate redundant sequences. After combining and clustering, comprehensive repeat and TE detection was conducted using RepeatMasker v4.1.2 (https://www.repeatmasker.org/). The genome was found to have a total of 762.12 Mb repetitive sequences, accounting for 70.27% of the genome. Long Terminal Repeat (LTR) elements and DNA transposons emerged as the most predominant types of repeats, representing 24.40% and 22.60% of the genome, respectively (Table 3).
Protein-coding genes annotation
Transcriptome sequencing, homologous gene search and de novo prediction were employed to infer the protein-coding genes (PCGs) in the D. aiolomorphi genome, which were then integrated to generate a final gene set. Initially, transcriptome reads were aligned using Hisat2 v2.2.124 and assembled with StringTie v2.1.725. Meanwhile, Trinity v2.8.526 was utilized for de novo assembly of transcriptome reads. Subsequent mapping of the transcriptome assembly to the genome for gene structural prediction by PASA v2.3.327. For the identification of homologous gene sets, sequences from various insects, manually annotated in the Universal Protein Resource database (UniProt, https://www.uniprot.org/) and National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/), were aligned to the D. aiolomorphi genome using Exonerate v2.4.028 and Gemoma v1.7.129. The process of de novo gene prediction involved three separate programs, Augustus v3.3.330, SNAP v2.54.331 and GeneMark-ETP v4.6532. A non-redundant consensus of gene structures was then generated by combining all results using EVidenceModeler v1.1.133. To annotate gene functions, the identified PCGs were aligned to various databases, including Nt, Nr, Swiss-Prot and TrEMBL, employing Diamond v2.0.534 with an e-value threshold of 1e-5. Protein classification and domain search were performed using eggNOG-mapper v2.1.435 and InterProScan v5.8.036. Finally, a total of 18,011 protein-coding genes were predicted, with 17,829 genes (98.99%) functionally annotated (Table 4).
Non-coding RNA annotation
To identify noncoding RNA, BAsic Rapid Ribosomal RNA Predictor (BARRNAP) v0.9 and tRNAScan-SE v2.0.537 were executed for predicting rRNA and tRNA, respectively. Infernal v1.1.238 was used to identify the remaining noncoding RNA based on the alignment with the Rfam library39. Finally, 539 noncoding RNAs (ncRNAs) were predicted, including 57 micro-RNAs (miRNAs), 104 ribosomal RNAs (rRNAs), 21 small nuclear RNAs (snRNAs), 15 small nucleolar RNAs (snoRNAs), and 344 transfer RNAs (tRNAs) (Supplementary Table S5).
Data Records
The MGI, ONT, RNA-seq and Hi-C sequencing data used for the genome assembly were deposited in the NCBI Sequence Read Archive (SRA) database with accession numbers SRR2688253040, SRR2688252941, SRR2688253142 and SRR2688252843, respectively, under the BioProject accession number PRJNA1036143. The chromosome assembly was deposited at GenBank with accession number JAXKQO00000000044. Genome annotation information was deposited in the Figshare database45.
Technical Validation
To ensure the reliability and integrity of the genomic data, we implemented rigorous preprocessing protocols on various datasets (Illumina sequencing system protocol: https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/novaseq/1000000019358_17_novaseq-6000-system-guide.pdf; Nanopore sequencing system protocol: https://a.storyblok.com/f/196663/x/a2ee9a9945/j2586-promethion-24-combined-qsg_170x250mm_rev2-final.pdf), including Illumina paired-end sequenced DNA raw short-reads, Nanopore sequenced DNA raw long-reads, Illumina paired-end sequenced RNA raw reads and Illumina paired-end Hi-C sequences. This preprocessing was carried out using fastp v.0.21.646, a widely recognized tool in genomic studies. The primary objective of this preprocessing step was to filter out low-quality sequences (Quality scores < 20), adapter sequences, reads containing Poly-N and sequences shorter than 30 bp. Following these stringent filtering criteria, we successfully obtained clean reads, which were subsequently stored in the fastq/fasta format.
Code availability
If no detailed parameters were mentioned, all software and tools in this study were performed according to those manuals and protocols of the applied bioinformatics software. No specific code or script was used in the study.
References
Shibata, E. Spatial density-independent parasitism of the inquiline, Diomorus aiolomorphi (Hymenoptera: Torymidae), on the bamboo gall maker, Aiolomorphus rhopaloides (Hymenoptera: Eurytomidae). Appl. Entomol. Zool. 41, 493–498 (2006).
Shibata, E. Sampling procedure for density estimation of bamboo galls induced by Aiolomorphus rhopaloides (Hymenoptera: Eurytomidae) in a bamboo stand. J. For. Res. 8, 0123–0126 (2003).
Shibata, E. Oviposition site preference of the bamboo gall maker, Aiolomorphus rhopaloides (Hymenoptera: Eurytomidae), on bamboo in terms of plant-vigor hypothesis. Appl. Entomol. Zool. 40, 631–636 (2005).
Takahashi, F. & Mizuta, K. Life cycles of a eurytomid wasp, Aiolomorphous rhopaloides, and three species of wasps parasitic on it. Japanese J. Appl. Entomol. Zool. 15, 36–43 (1971).
Askew, R. R. On the biology of the inhabitants of oak galls of Cynipidae (Hymenoptera) in Britain. Trans. Soc. Br. Entomol. 14, 237–268 (1961).
Sanver, D. & Hawkins, B. A. Galls as habitats: the inquiline communities of insect galls. Basic Appl. Ecol. 1, 3–11 (2000).
Kamijio, K. A new species of the genus Diomorus Walker (Hymenoptera: Torymidae). Insecta Matsumurana. 27, 16-17 (1964).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Hu, J., Fan, J. P., Sun, Z. Y. & Liu, S. L. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289–293 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Ou, S. J. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021).
Li, W. Z. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 22, 1658–1659 (2006).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6, 31 (2005).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, 9 (2016).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 19, II215–II225 (2003).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP: automatic gene finding in eukaryotic genomes in consistency with extrinsic data. bioRxiv. https://doi.org/10.1101/2023.01.13.524024 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Zdobnov, E. M. & Apweiler, R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 9 (2001).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 16 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137 (2015).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR26882530 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR26882529 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR26882531 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR26882528 (2023).
NCBI GenBank. https://identifiers.org/nucleotide:JAXKQO000000000 (2023).
Ruizhong, Y. et al. Protein-coding genes annotation of the Diomorus aiolomorphi genome. figshare https://doi.org/10.6084/m9.figshare.24785745 (2023).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, 884–890 (2018).
Acknowledgements
This study was supported by the Key International Joint Research Program of National Natural Science Foundation of China (31920103005), the General Program of National Natural Science Foundation of China (32070467), the National Key Research and Development Program of China (2023YFD1400600), the Provincial Key Research and Development Plan of Zhejiang (2021C02045) and the Key Project of Laboratory of Lingnan Modern Agriculture (NT2021003).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision: Pu Tang and Xuexin Chen; Software: Ruizhong Yuan, Qiuyu Qu, Shiji Tian, Yu Jin, Jiabao Gong & Xiqian Ye; Investigation: Ruizhong Yuan, Qiuyu Qu, Zhaohe Lu & Xiansheng Geng; Writing – Original Draft Preparation: Ruizhong Yuan & Qiuyu Qu; Writing – Review & Editing: Ruizhong Yuan, Qiuyu Qu, Pu Tang & Xuexin Chen; Visualization: Ruizhong Yuan & Qiuyu Qu; Funding Acquisition: Pu Tang & Xuexin Chen.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuan, R., Qu, Q., Lu, Z. et al. A chromosome-level genome assembly of the gall maker pest inquiline, Diomorus aiolomorphi Kamijo (Hymenoptera: Torymidae). Sci Data 11, 944 (2024). https://doi.org/10.1038/s41597-024-03779-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03779-y
- Springer Nature Limited