
Abstract
The planthopper Nilaparvata muiri is a sister species to N. lugens (Hemiptera: Delphacidae), a notorious insect pest in Asian rice fields. N. muiri and N. lugens have a different host preference despite the similarities in many biological features. To better understand the adaptive evolution of planthoppers, comprehensive genomic information on N. muiri and N. lugens are urgently needed. In this study, we used ultra-low input PacBio HiFi libraries and Hi-C sequencing technologies to assemble a reference genome of a single N. muiri at the chromosomal level. The genome size was determined to be 531.62 Mb with a contig N50 size of 2.47 Mb and scaffold N50 size of 38.37 Mb. Totally, 96.61% assembled sequences were anchored to the 15 pseudo-chromosomes. BUSCO analysis yielded an Insecta completeness score of 98.6%. A total of 22,057 protein-coding genes were annotated, and 168.16 Mb repetitive sequences occupying 31.63% of genome were pinpointed. The assembled genome is valuable for evolutionary and genetic studies of planthoppers, and may provide sights to pest control.
Similar content being viewed by others

Background & Summary
Rice (Oryza sativa L.) serves as the staple food for over half the world’s population and about 80% of the Asian population1. It faces significant production challenges due to an array of phytophagous pests, among which rice planthoppers (<5 mm in body length) have emerged as the dominant herbivores in rice ecosystems across most Asian countries, replacing larger herbivores like stem borers2. Planthoppers, including the Nilaparvata lugens, Sogatella furcifera, and Laodelphax striatellus, infest rice plants by feeding on the vascular tissue and transmit various virus diseases. Heavy planthopper infestation causes complete wilting and drying of rice plants, referred to as hopperburn, leading to serious yield losses3,4,5.
However, not all members of the planthopper family pose a threat to rice. Nilaparvata muiri is a common planthopper species with a small size (3–5 mm) widely distributed in the rice regions of southern China, Japan, Korea, and Vietnam6,7,8. N. muiri shares a morphological resemblance with its sister species N. lugens but exhibits different feeding behaviours (Fig. 1a). Both species undergo five nymphal stages, with newly hatched first-instar nymphs look like miniature adults except for the absence of wings and sexual immaturity, and grow gradually with increasing stage. However, in contrary to that N. lugens exclusively feeds on rice plants, N. muiri predominantly consumes Poaceae grasses including Leersia hexandra and Leersia sayanuka, which commonly reside besides rice paddy fields9. Of note, N. muiri can complete its lifecycle on rice plants, but lacks sustained reproductive capacity on this plant. This event indicates that N. muiri performs a specific host-plant preference that differs from its notorious relative N. lugens9. The differences between N. muiri and N. lugens extend to their reproductive organs, with distinguishable features in the genitalia of both sexes. Specifically, N. muiri has inner margin of female lateral lobe with a spatulated process at base compared with that in N. lugens, and males have parameres with slightly different shapes, suggesting divergent evolutionary paths (Fig. 1b).

Host specificity and phenotypic distinction between N. muiri and N. lugens. (a) Host selection and feeding patterns of N. muiri and N. lugens. Solid arrows indicate the primary host plants, with N. lugens feeding exclusively on rice O. sativa and N. muiri on the grass L. sayanuka. The dashed line suggests N. muiri can complete its lifecycle on rice plants, but lacks sustained reproductive capacity. Bars, 500 μm. (b) Morphological differentiation in the reproductive organs of N. lugens and N. muiri. The red arrows point to distinctive reproductive structures, N. muiri has inner margin of female lateral lobe with a spatulated process at base compared with that in N. lugens, and males have parameres with slightly different shapes. Bars, 200 μm.
In contrary to the limited knowledge of N. muiri, extensive studies have been conducted on N. lugens because of its economic importance. In the past half century, N. lugens’s outbreaks have re-occurred approximately every three years, with the annual outbreak area amounting to approximately 10–20 million hectares of rice, resulting in millions of tons of losses in Asia10,11. Features contributing to the success of N. lugens may include its mystical capacity to live on a sole host plant and to overcome host plant resistance, association with high fecundity, long-distance migration, and resistance to most pesticides12. To better understand the adaptive evolution of planthoppers, comprehensive genomic information on N. lugens and its relative species are urgently needed.
Previously we completed the genome assembling of N. lugens12. In the present study, our focus shifts to N. muiri. Initial analysis with pooled samples revealed a challenge: a high heterozygosity rate of 2.6% (Table 1), indicating a complication of genome sequencing and assembly of this species. To circumvent this challenge, the genome of N. muiri was de novo assembled based on low genomic DNA from a single individual, using an ultra-low input approach and assembled at the chromosome-level assisted by the Hi-C (High-throughput chromosome conformation capture) technique (Fig. 2). This novel genomic resource may enable us to uncover pivotal genes linked to host selection, dietary preferences, and adaptive strategies between N. muiri and N. lugens, which may ultimately be useful for the management of rice planthoppers.

The workflow overview of N. muiri chromosome-level genome assembly.
Methods
Insect
N. muiri was originally collected from rice paddies in Jinhua, Zhejiang Province, China, in 2022. The population subsequently reared in laboratory incubators, fed with the grass L. sayanuka. The walk-in incubators were maintained at a temperature of 26 °C, a photoperiod of 16:8 hours (light:dark), and a relative humidity of 65%.
Estimation of genome size by flow cytometry
Genome size of the N. muiri was estimated using flow cytometry13. Briefly, 20 heads of adult males were pooled into 500 µl of Galbraith buffer and then chopped using anatomical blades. Homogenized samples were filtered through a 6.25 μm nylon mesh to prepare a nuclear suspension, followed by centrifuge at 3,000 rpm for 5 min at 4 °C. The nuclei were resuspended in 400 μl of 1 × phosphate buffered saline (PBS) and chilled on ice for 5–10 min. Propidium iodide (Sigma-Aldrich, USA) and RNaseA (Thermo Fisher Scientific, USA) were added to the suspension, and samples were stained in darkness for 1–2 h at 4 °C. The fruit fly Drosophila melanogaster Canton-S strain with a standard genome size of 1 C = 176.4 Mb was used as the reference species. Flow cytometry revealed that the genome size of N. muiri males was approximately 505.145 Mb, 2.864 times larger than in D. melanogaster (Fig. 3a and Table 2).

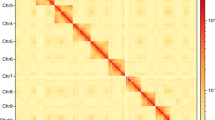
Characteristics of the N. muiri genome. (a) Flow cytometry results of N. muiri adult males. The ordinate is the number of nuclei, and the abscissa is the fluorescence of the nucleus. The fruit fly D. melanogaster was used as a reference. (b) N. muiri genome size estimation by k-mer distribution (k = 19). The x-axis represents the k-mer depth and the y-axis is the corresponding frequency. (c) Hi-C heatmap of N. muiri. The scale bar shows all interaction frequency of 15 chromosomes. (d) Circos plot of the N. muiri genome assembly.
Estimation of genome size by k-mer analysis
To conduct a genome survey using K-mer analysis, 40 fifth-instar male nymphs were collected for paired-end sequencing of libraries (350 bp insert size) using the Illumina Novaseq X Plus platform. To obtain clean reads, raw data was filtered by removing low-quality reads, short reads, adapter sequences, and polyG tails. Subsequently, we conducted a 19-mer frequency distribution analysis using Jellyfish v2.3.014. Genome size, heterozygosity, and duplication rate were estimated using GenomeScope v2.015 (Fig. 3b). The estimated genome size of N. muiri was 440.793 Mb, which is smaller than 505.145 Mb estimated using flow cytometry. This inconsistency was frequently observed in highly heterozygous insects16. Given the high heterozygosity of 2.6% (Table 1) derived from the k-mer analysis, the discrepancies between the genome assembly, k-mer analysis, and flow cytometry estimations are likely due to the high heterozygosity.
Ultra-low input DNA sequencing
A single adult male N. muiri was processed for HiFi library construction. The insect was initially washed twice with 75% ethanol, followed by washing twice with PBS, before dissection to remove the gut. Genomic DNA (gDNA) was extracted using FineOut animal tissue DNA kit (Genfine, China), and its integrity was assessed using the Femto pulse system. gDNA (20 ng) was fragmented using the Megaruptor 3 (Diagenode, USA), followed by purification with ProNex beads (Promega, USA). The fragmented DNA was then amplified using a two-step PCR process: the first round with 13 cycles of 98 °C denaturation, 62 °C annealing, and 72 °C extension; the second round with 13 cycles of 98 °C denaturation, 60 °C annealing, and 68 °C extension. The amplified DNA (>500 ng) was used for SMRTbell library construction with the Pacific Biosciences SMRTbell Express Template Prep Kit 2.0 (Pacific biosciences, USA). Size selection of the constructed library was conducted on the BluePippin system, targeting an insert size of 10 Kb. Primer annealing was followed by the binding of the SMRTbell templates to DNA polymerase. The final library sequencing was performed on the Annor Revio platform, and the output data was visualized using SMRTlink v12.0 (PacBio, USA). A total of 78.89 Gb HiFi reads were obtained and further used for de novo assembly. We assembled the genome using hifiasm v0.19.8-r60317. HaploMerger218 (https://github.com/mapleforest/HaploMerger2) was employed to remove heterozygosity from the first assembly. The NCBI non-redundant nucleotide database (NT) was used to identify and eliminate possible contaminating sequences. The genome size of the pseudo-haplotype assembly was 531.60 Mb with a contig N50 of 2.80 Mb.
Hi-C library construction and sequencing
A total of 30 fifth-instar male nymphs were pooled for the construction of Hi-C library to explore chromatin interactions across the genome. The samples were crosslinked with a 2% formaldehyde solution at room temperature for 10 min, and then added with glycine solution (2.5 M) priority to quality control19. Hi-C libraries were constructed and sequenced using Illumina Novaseq X plus platform. The polished HIFI reads were assembled into a chromosomal level based on Hi-C reads using Juicer v1.620 (https://github.com/aidenlab/juicer) and YahS21 (https://github.com/c-zhou/yahs). The visualization of contigs and scaffolds was achieved using Juicebox v1.11.0822. The final genome assembly of N. muiri was 531.62 Mb with a contig N50 of 2.47 Mb (Tables 3), and 96.61% contigs were anchored to 15 pseudo-chromosomes (Fig. 3c,d and Table 4).
Transcriptome sequencing
For assisting gene annotation, we prepared transcriptomes of N. muiri from eggs, nymphs (1st, 2nd, 3rd, 4th, and 5th), adult females and males. In addition, seven representative tissues including antennae, head, fat body, digestive gut, cuticle, ovary, and testis were collected for RNA-sequencing (RNA-seq). Total RNAs were extracted using RNAiso Plus (Takara, China) according to the manufacturer’s protocol. RNA-seq libraries were constructed using the VAHTS Universal V6 RNA-seq library prep kit for Illumina (Vazyme Biotech, China) following the manufacturer’s instructions. The libraries were then sequenced on the Illumina Novaseq X plus platform with paired-end reads of 150 bp, and the obtained reads (Table 5) were used for subsequent genome annotation.
Gene prediction and annotation
RepeatMasker v4.1.223 (http://repeatmasker.org/RepeatMasker) was applied to identify the repetitive sequences, encompassing interspersed repeats and transposable elements (TEs). Then RepeatModeler v2.0.324 (http://www.repeatmasker.org/RepeatModeler) and Repbase library25 (https://www.girinst.org/repbase) assisted to cluster the repeats by building a de novo repeat library. A total of 168,166,532 bp (31.63% of the genome) repetitive elements were identified, of which 27.09% was TEs, including long interspersed nuclear elements (LINEs) (2.78%), long terminal repeats (LTRs) (1.46%), short interspersed nuclear elements (SINEs) (0.49%), and DNA elements (DNAs) (3.08%) (Table 6).
Prediction of protein-coding genes was based on ab initio gene predictions, homology-based predictions, and transcriptome-based predictions. Initially, all the transcriptome data from various tissues and individuals at different developmental stages were assembled utilizing Trinity v2.15.126 (https://github.com/trinityrnaseq). Subsequently, the ab initio prediction was carried out with the assistance of Augustus v3.4.027 (https://github.com/Gaius-Augustus/Augustus). The HISAT2 v2.1.028 (https://github.com/DaehwanKimLab/hisat2) was employed to align the transcriptome data to the genome, followed by homology-based prediction using GeneWise v2.4.129 (https://www.ebi.ac.uk/Tools/psa/genewise). To generate a comprehensive protein-coding gene set, we used the GETA v2.6.1 pipeline (https://github.com/chenlianfu/geta) to integrate annotations from the above approaches. Overall, A total of 22,057 protein-coding genes were predicted (Table 3), with 97.7% of the complete BUSCO genes covered. The average length of gene, coding sequences (CDSs), and intron sequences was 10,677.91 bp, 1,565.91 bp, and 8342.15 bp, respectively.
For functional annotation, we quired our predicted protein-coding genes against the NCBI non-redundant databases (NR) (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), the NT database (https://www.ncbi.nlm.nih.gov/nucleotide/), and the eggNOG database30. The result showed that 95.69% of the protein-coding genes had significant hits in the functional annotation database (Table 7).
Synteny analysis
For synteny analysis, the genome data of the N. lugens was obtained from NCBI with accession number of GCA_014356525.1. To identify the synteny information, MCScanX v231 with default parameters was performed, and the result was visualized with TBtools32. This analysis identified 19,143 collinear genes out of a total of 46,958 genes, resulting in a collinearity rate of 40.77%. The collinear gene blocks showed that N. muiri had a remarkably high synteny with N. lugens (Fig. 4a).

Chromosomal synteny and phylogenetic analysis. (a) Chromosomal synteny of N. muiri and N. lugens. Numbers in the rectangles represent chromosomes of each genome. (b) A maximum likelihood tree based on N. muiri and 14 other Hemiptera species. The numbers indicate the divergence time. All nodes are supported by 1,000 bootstrap replicates. The scale at the bottom represents the divergence time.
Orthologue and phylogenetic analyses
The protein sequences of other 14 Hemiptera species, including N. lugens, S. furcifera, L. striatellus, Riptortus pedestris, Pyrrhocoris apterus, Oncopeltus fasciatus, Rhodnius prolixus, Cimex lectularius, Gerris buenoi, Myzus persica, Acyrthosiphon pisum, Aphis gossypii, Phenacoccus solenopsis, and Diaphorina citri, were downloaded from InsectBase 2.0 (http://v2.insect-genome.com/). Only the longest transcript of each gene was kept to represent the coding gene. OrthoFinder v2.5.533 with default parameters was used to determine the orthologs and paralogs, yielding 24,960 orthologous gene families from a total of 323,572 genes. All identified 223 single-copy orthologs were aligned using MAFFT v7.52034 and trimmed utilizing trimAl v1.4.135. The maximum likelihood tree was constructed using IQ-TREE v2.2.336 with 1,000 ultrafast bootstrap replicates, and D. melanogaster was used as an outgroup. The divergence time between different species was estimated using MCMCtree (PAML v4.10.7 package)37 based on the fossil records acquired from the TimeTree database38 (http://www.timetree.org/) using the approximate likelihood calculation method (A. gossypii vs. A. pisum 23.0–64.5 million years ago (MYA), D. citri vs. A. pisum 211.9–351.0 MYA, and R. prolixus vs. C. lectularius 108.0–212.9 MYA). The results of the phylogenetic analysis indicated that N. muiri and N. lugens clustered into a single clade within the sub-order Auchenorrhyncha, and diverged at approximately 11.49 MYA (Fig. 4b).
Data Records
The genome assembly utilized PacBio and Hi-C sequencing techniques, and the data have been stored in the NCBI Sequence Read Archive with the accession numbers SRR28434365 and SRR28434364 under accession number SRP49754239. Additionally, the transcriptome data used for genome annotation are available in the NCBI Sequence Read Archive with accession numbers ranging from SRR28434329 to SRR2843436339. The genome assembly has been deposited in the National Center for Biotechnology Information (NCBI), under the genome accession number of JBCEXI00000000040. The genome annotation files are available in Figshare under a DOI number of https://doi.org/10.6084/m9.figshare.2560846841.
Technical Validation
The final genome assembly of N. muiri was 531.62 Mb with a contig N50 of 2.47 Mb, and 96.61% contigs were anchored to 15 pseudo-chromosomes. The BUSCO results showed that 98.6% of the complete Insecta genes (single-copied gene: 97.1%, and duplicated gene: 1.5%) were captured by the genome. Additionally, the gene annotation result was assessed by BUSCO, which indicated that 97.7% genes (single-copied gene: 96.4%, and duplicated gene: 1.3%) were complete. Overall, these assessments reflect the high quality of the genomic assembly.
Code availability
Software for data analyses was mentioned in Methods. The core code and parameters are available at https://github.com/zhuozhuoliu/Nilaparvata_muiri.git.
References
FAO, F. Food and agriculture organization of the United Nations. Rome http://faostat.fao.org (2018).
Heong, K. L. & Hardy, B. (Eds.). Planthoppers: New Threats to The Sustainability of Intensive Rice Production Systems in Asia (International Rice Research Institute, 2009).
Backus, E. A., Serrano, M. S. & Ranger, C. M. Mechanisms of hopperburn: An overview of insect taxonomy, behaviour, and physiology. Annu. Rev. Entomol. 50, 125–151 (2005).
Cheng, J. Rice planthoppers in the past half century in China. In Rice planthoppers: Ecology, management, socio economics and policy (Zhejiang university press, 2015).
He, P. et al. Evolution and functional analysis of odorant‐binding proteins in three rice planthoppers: Nilaparvata lugens, Sogatella furcifera, and Laodelphax striatellus. Pest Manag. Sci. 75(6), 1606–1620 (2019).
Ding, J. A review of studies on classification and species of the genus Nilaparvata distant (Homoptera: delphacidae). J. Nanjing Agric. Univ. 78–85 (1981).
Luo, J. et al. Population dynamics of Nilaparvata lugens and its two sibling species under black light trap. Chin. J. Rice Sci. 24, 315–319 (2010).
Zheng, X. et al. Use of banker plant system for sustainable management of the most important insect pest in rice fields in China. Sci. Rep. 7(1), 45581 (2017).
Cui, Y. et al. Host plants of Nilaparvata muiri China and N. bakeri (Muir), two sibling species of N. lugens (Stål). Chin. J. Rice Sci. 27(1), 105 (2013).
Catindig, J. L. A. et al. Planthoppers: New threats to the sustainability of intensive rice production systems in Asia (eds KL Heong & B. Hardy) 191–220 (International Rice Research Institute, 2009).
Cheng, J. A. In Planthoppers: New Threats to The Sustainability of Intensive Rice Production Systems in Asia (eds KL Heong & B. Hardy) 157–178 (International Rice Research Institute, 2009).
Xue, J. et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 15, 521 (2014).
Zhao, X. et al. A chromosome‐level genome assembly of rice leaffolder, Cnaphalocrocis medinalis. Mol. Ecol. Resour. 21(2), 561–572 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Elliott, T. A. & Gregory, T. R. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos. Trans. R. Soc. B 370, 20140331 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Huang, S. et al. HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579.
Nagano, T. et al. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 16, 175 (2015).
Durand, N. C. et al. Juicer provides a One-Click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2022).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6, 256–258.e1 (2018).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2015).
Smit, A., & Hubley, R. RepeatModeler. http://www.repeatmasker.org/RepeatModeler/ (2008–2015).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 1–6 (2015).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439 (2006).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. methods 12, 357–360 (2015).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Huerta-Cepas, J. et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, 309–314 (2019).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 1–14 (2012).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202 (2020).
Emms, D. M., Kelly, S. & OrthoFinder Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Kumar, S., Stecher, G., Suleski, M. & Blair Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819 (2017).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRP497542 (2024).
NCBI GenBank https://identifiers.org/insdc.gca:GCA_041146465.1 (2024).
Figshare https://doi.org/10.6084/m9.figshare.25608468 (2024).
Ye, Y. et al. Chromosome‐level assembly of the brown planthopper genome with a characterized Y chromosome. Mol Ecol Resour. 21(4), 1287–1298 (2021).
Hu, Q. et al. Chromosome-level assembly, dosage compensation and sex-biased gene expression in the small brown planthopper, Laodelphax striatellus. Genome Biol Evol. 14(11), pevac160 (2022).
Ye, Y. et al. Chromosome-level Genome Assembly and Sex-specific Differential Transcriptome of the White-backed Planthopper, Sogatella furcifera. Curr Genomics. 23(6), 400 (2023).
Acknowledgements
We thank Mr. Run-Guo Shu (Zhejiang University) for assisting flow cytometry analysis. This research was supported by Key Research and Development Program of Zhejiang Province (2023SNJF034), and National Natural Science Foundation of China (32272519).
Author information
Authors and Affiliations
Contributions
H.J.X. convinced and designed the study. Z.Q.L. and P.Y.Z. reared the N. muiri, Z.Q.L. and N.T.X. collected the samples. Z.Q.L., J.L.Z., Y.W., X.Z., and Y.X.Y performed bioinformatics analysis. Z.Q.L. wrote the draft manuscript. H.X.X. and H.J.X. modified the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, ZQ., Zhu, PY., Xu, NT. et al. Chromosome-level genome assembly from a single planthopper Nilaparvata muiri (Hemiptera: Delphacidae). Sci Data 11, 937 (2024). https://doi.org/10.1038/s41597-024-03812-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03812-0
- Springer Nature Limited