
Abstract
Cancer-induced cachexia, characterized by muscle wasting, is a lethal metabolic syndrome with undefined etiology. Current consensus is that multiple factors contribute to cancer-induced muscle wasting, and therefore therapy requires combinational strategies. Here, we show that Toll-like receptor 4 (TLR4) mediates cancer-induced muscle wasting by directly activating muscle catabolism as well as stimulating an innate immune response in mice bearing Lewis lung carcinoma (LLC), and targeting TLR4 alone effectively abrogate muscle wasting. Utilizing specific siRNAs we observed that LLC cell-conditioned medium (LCM)-treated C2C12 myotubes underwent a rapid catabolic response in a TLR4-dependent manner, including activation of the p38 MAPK−C/EBPβ signaling pathway as well as the ubiquitin-proteasome and autophagy-lysosome pathways, resulting in myotube atrophy. Utilizing a reporter cell-line it was confirmed that LCM activated TLR4. These results suggest that LLC-released cachexins directly activate muscle catabolism via activating TLR4 on muscle cells independent of immune responses. Critically, LLC tumor-bearing TLR4−/− mice were spared from muscle wasting due to a blockade in muscle catabolic pathways. Further, tumor-induced elevation of circulating TNFα and interleukin-6 (IL-6) was abolished in TLR4−/− mice. These data suggest that TLR4 is a central mediator and therapeutic target of cancer-induced muscle wasting.
Similar content being viewed by others


Introduction
Cancer-induced cachexia, or cancer cachexia, is a highly complex metabolic syndrome characterized by progressive muscle wasting. Prominent clinical features of cachexia are weight loss, inflammation, insulin resistance, and increased muscle protein breakdown. About half of all cancer patients develop cachexia, and cachexia is directly responsible for about 30% of all cancer-related deaths1. The precise etiology of cancer cachexia is unknown. The current consensus is that multiple factors contribute to cancer cachexia, and therapy requires combinational strategies2, 3.
A key feature of cancer cachexia is systemic inflammation and anti-inflammation strategies are considered central to the therapy of cancer cachexia. Multiple inflammatory cytokines such as TNFα, interleukin-6 (IL-6) and interleukin-1 (IL-1) are elevated in cachectic cancer hosts and are known to promote muscle catabolism, thus are considered therapeutic targets4. Cancer-induced muscle catabolism involves activation of proteolysis mediated by the ubiquitin-proteasome pathway (UPP)5 in muscle cells through cytokine-activated signaling molecules including NF-κB6, 7, p38 MAPK8,9,10 or JAK-STAT3 pathways9, 11. In addition, another major cellular proteolytic system, the autophagy-lysosome pathway (ALP), may also contribute to muscle wasting in animal models of cancer12,13,14 and cancer patients15,16,17. However, the underlying signaling mechanism that mediates cancer-induced activation of ALP is unknown.
The involvement of multiple humoral factors and intracellular signaling pathways in cancer-induced muscle wasting makes it difficult to intervene effectively in cancer-induced muscle wasting. However, we reasoned that if we could identify the origin of systemic inflammation in cancer hosts, effective therapy might be achieved by targeting the potential “master mediator” of inflammation. Although certain types of cancer can release cytokines, the bulk of circulating cytokines in cancer hosts appears released by host tissues as a response to cancer18. Cancer can generate danger-associated molecular patterns (DAMPs) that activate TLR419, 20, a plasma membrane receptor that plays a central role in innate immunity21. Previously Cannon et al. observed that mice with nonfunctional TLR4 due to a TLR4 double mutation (TLR4d/d) were resistant to cachexia induced by squamous cell carcinoma SCCF VII cells, and suggested that TLR4 mediated muscle wasting by increasing circulating IL-1β22. However, the detailed mechanism through which TLR4 mediates cancer-induced muscle wasting remains unknown. Given that TLR4 is expressed by skeletal muscle cells23, 24, it is possible that cancer directly stimulates muscle catabolism by activating TLR4 on muscle cells. In addition, cancer-induced activation of TLR4 on immune cells would increase their synthesis and release of inflammatory cytokines that promote muscle catabolism. Thus, we tested the hypothesis that TLR4 is a key mediator of cancer-induced muscle wasting due to its integration of catabolic signaling through activating muscle protein degradation pathways directly and increasing cytokine release indirectly. Utilizing in vitro as well as in vivo approaches, we found evidence in the present study that supports our hypothesis. Our data suggest that TLR4 is a central mediator of cancer-induced muscle protein degradation through the UPP and the ALP, and thus may be a key therapeutic target of cancer cachexia.
Results
LLC cell-conditioned medium activates protein degradation pathways in C2C12 myotubes through TLR4
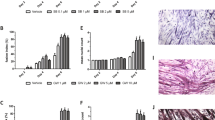
We previously showed that treating cultured myotubes with LLC cell-conditioned medium (LCM) recapitulates the muscle catabolism seen in LLC tumor-bearing mice through the activation of the p38 MAPK-C/EBPβ signaling pathway which upregulates E3 ubiquitin ligases atrogin18 and UBR225. In addition, LCM activates autophagy in cultured myotubes13. These data indicate a direct activation of muscle catabolism by cancer-released cachexins independent of host response. However, the membrane receptor(s) through which LCM activates UPP- and ALP-mediated muscle catabolism was unknown. To test the hypothesis that the catabolic effect of LCM on muscle cells is specifically mediated by TLR4, we knocked down TLR4 or TLR2 in C2C12 myotubes using specific siRNAs. LCM treatment of myotubes transfected with scrambled siRNA (control) activated downstream effectors of TLR4, p38 MAPK (phosphorylation of T181/Y182) and NF-κB (phosphorylation of p65), as expected. In addition, we observed activation of transcription factor C/EBPβ (phosphorylation of T188) in 1 h (Fig. 1A), upregulation of ubiquitin ligases UBR2 and atrogin1, and activation of autophagy (indicated by increased LC3-II) in 8 h (Fig. 1B). In addition, by treating myotubes with lysosome inhibitor chloroquine we observed a further increase in LC3-II, indicating that LCM increased autophagy flux (Fig. 1C). Activation/upregulation of these catabolic mediators resulted in myofibrillar protein myosin heavy chain (MHC) loss (Fig. 1D) as well as myotube atrophy (Fig. 1E) in 72 h. TLR4 knockdown in myotubes abolished all the catabolic response to LCM and preserved myotube mass. In contrast, TLR2 knockdown did not alter the catabolic effect of LCM (Fig. 1). To verify whether LCM actually activated TLR4, a TLR4 reporter cell-line, HEK293 cells that overexpress TLR4 (HEK-Blue hTLR4, InvivoGen), was treated with LCM and resulted in activation of TLR4-controlled reporter gene, secreted embryonic alkaline phosphatase (SEAP, Fig. 2). Thus, LCM induces muscle catabolism through direct activation of TLR4 on muscle cells.

LLC cell-conditioned medium activates myotube catabolism through TLR4. C2C12 myoblasts were transfected with indicated siRNAs. After incubation in differentiation medium for 96 h, myotubes were treated with Lewis lung carcinoma cell-conditioned medium (LCM) or control medium. Activation of NF-κB (p65), p38 MAPK and C/EBPβ was evaluated by Western blot analysis of cell lysate in 1 h (A). Levels of UBR2, atrogin1 and LC3-II were evaluated by Western blot analysis of cell lysate in 8 h (B). Autophagy flux increase was demonstrated by measuring LC3-II levels in myotubes pre-treated with 20 μM of lysosome inhibitor chloroquine (CQ, C). Levels of myosin heave chain (MHC) were evaluated by Western blot analysis of cell lysate in 72 h (D). TLR2 and TLR4 knockdown was monitored at each of the time points. Myotubes treated for 72 h were immunofluorescence-stained for MHC and their diameter was measured (E). Data was analyzed by ANOVA. *Denotes a difference (P < 0.05).

LLC cell-conditioned medium activates TLR4 in reporter cells. TLR4 reporter cell line HEK-Blue hTLR4 (InvivoGen) was treated with LCM or control medium for 24 h and TLR4 activation was measured as the enzymatic activity of secreted embryonic alkaline phosphatase (SEAP) in arbitrary unit (a.u.). Data was analyzed by Student t test. *Denotes a difference (P < 0.05).
TLR4 is critical to LLC tumor-induced muscle catabolism in mice
To determine whether LLC tumor induces muscle wasting in vivo through activation of TLR4-mediated protein degradation, we utilized the existing TLR4−/− mice26 that were in the same C57BL/6 background as LLC to examine the role of TLR4 in cancer cachexia. In 21 days of LLC cell inoculation tumor-bearing wild-type mice developed muscle wasting as measured by changes in body and muscle mass, muscle strength (grip strength) and muscle proteolysis (tyrosine release from EDL muscle). However, TLR4−/− mice were spared from muscle wasting, without altered tumor growth (Fig. 3A). Consequently, muscle fibers in tumor-bearing TLR4−/− mice maintained normal histology and mass as measured by cross sectional area in contrast to tumor-bearing wild-type mice in which muscle fibers shrank and interstitial space increased (Fig. 3B). Further analysis of the activity of the catabolic pathways revealed that TLR4 deficiency prevented tumor-induced activation of p38 MAPK and NF-κB (p65), upregulation of ubiquitin ligases UBR2 and atrogin1, activation of autophagy, and loss of MHC (Fig. 4A). These results were similar to the response of TLR4-deficient myotubes to LCM described above. To verify whether autophagosome formation was increased in cachectic muscle of LLC tumor-bearing mice in a TLR4-dependent manner, we overexpressed GFP-LC3 in mouse TA and observed increased GFP-LC3-labeled autophagosomes in LLC tumor-bearing WT but not TLR4−/− mice (Fig. 4B). These data suggest that TLR4 is critical to cancer-induced muscle wasting due to its mediation of muscle protein degradation through the UPP and ALP.

TLR4−/− mice are resistant to LLC tumor-induced muscle wasting. Wild-type C57BL/6 and TLR4−/− mice (7-week old male) were inoculated with LLC cells. Muscle wasting was evaluated in 21 days by measuring tumor mass, body weight change (excluding tumor weight), grip strength, tyrosine release from EDL, muscle weight (TA and EDL) (A) as well as muscle fiber cross-sectional area (B). Bar = 100 μm. Data was analyzed by ANOVA (A) or Chi-square analysis (B). *Denotes a difference (P < 0.05).

LLC tumor-induced muscle catabolism is dependent on TLR4. (A) TLR4 is required for the activation of catabolic pathways in muscle of LLC-bearing mice. TA muscle collected from mice described in Fig. 3 was analyzed by Western blotting for markers of TLR4-mediated signaling and muscle catabolism. Data was analyzed by ANOVA. *Denotes a difference (P < 0.05). (B) Autophagosome formation is activated in cachectic muscle of LLC-bearing mice in a TLR4-dependent manner. The TA of wild type C57BL/6 or TLR4−/− mice that had been implanted with LLC cells (controlled by PBS injection) were transfected with GFP-LC3 or GFP-expressing plasmid on day 14. In 7 days, autophagosome formation was evaluated by fluorescence microscopy examination of frozen sections of TA. Bars represent 50 μM.
TLR4 mediates LLC tumor-induced increase in circulating cytokines
Systemic activation of TLR4 increases cytokine synthesis and release from various host cells as an innate immune response21. We postulated that TLR4 knockout prevents the increase in circulating cytokines that could stimulate muscle catabolism such as TNFα, IL-6 and IL-1β, and determined the effect of TLR4 deficiency on circulating cytokines in LLC tumor-bearing mice. We found that serum TNFα and IL-6 levels were elevated in wild-type LLC tumor-bearing mice that developed cachexia, while IL-1β level remained unchanged. However, TNFα and IL-6 levels did not increase in TLR4−/− tumor-bearing mice (Fig. 5). These data suggest that TLR4 plays a central role in mediating tumor-induced muscle wasting by activating muscle catabolism directly as well as by increasing the release of inflammatory cytokines to enhance muscle catabolism indirectly. Thus, perturbing TLR4 signaling systemically may prevent muscle wasting by abrogating both its direct and indirect effects.

TLR4 is required for LLC tumor-induced elevation of circulating catabolic cytokines in mice. Sera collected from mice described in Fig. 3 on day 21 of LLC tumor implant were analyzed for catabolic cytokines TNFα, IL-6 and IL-1β by multiplex immunoassay (Bio-Rad). Data was analyzed by ANOVA. *Denotes a difference (P < 0.05).
Discussion
The current study reveals that TLR4 is a central mediator of muscle catabolism in a mouse lung cancer model in vitro and in vivo due to its coordinate activation of the UPP and the ALP. Importantly, our data demonstrate that LLC directly activates muscle catabolism through the activation of TLR4 on muscle cells independent of host input. In addition, elevation of circulating TNFα and IL-6 in LLC tumor-bearing mice is dependent on TLR4. These data suggest that tumor-induced activation of TLR4 is responsible for both the systemic inflammation and muscle catabolism. These observations explain why previous clinical trials using anti-cytokine strategies had only limited success on intervening muscle wasting in cancer patients27,28,29 and suggest anti-TLR4 strategies could achieve much better results. Our findings open the door to a new therapeutic strategy that targets only a single plasma membrane receptor to alleviate systemic inflammation and abrogate muscle wasting in cancer hosts. Thus, cancer cachexia may become manageable by using a single reagent that perturbs TLR4 signaling.
Observed dependence of LCM-induced myotube catabolism on TLR4 and the rapid activation of TLR4 downstream effector p38 MAPK within 1 h indicate that LLC directly activates muscle catabolism without the need for new protein synthesis/release by host tissues. Thus, cancer-released cachexins instead of host responses to cancer are likely the primary cause of cancer-induced muscle wasting. However, the identity of LLC-released cachexin(s) that activates TLR4 is currently unknown. Identification of the cachexin(s) responsible for TLR4 activation is therefore warranted.
Our data suggest that TLR4 is a specific mediator of cancer-induced muscle catabolism. Despite the fact that LLC activates TLR2 via releasing versican18 and muscle cells express TLR223, 24, our data indicates that TLR2 is not responsible for the catabolic effect of LLC on muscle. The difference between the intracellular signaling mechanism of TLR2 and TLR4 is that TLR2 signaling is dependent on MyD88 while TLR4 signaling involves TRIF in addition to MyD8821. Thus, the catabolic effect of LLC is likely being mediated by TRIF, which is consistent with the previous observations that MyD88 does not mediate LLC or LPS-induced muscle wasting30. On the other hand, TLR7 that also relies on MyD88 for signaling mediates muscle cell death in cancer cachexia31.
Recent clinical studies of cancer cachexia patients showed diverse patterns of increase in circulating cytokines. TNFα was most consistently increased in these patient populations, followed by IL-6, with IL-1β being less consistently increased32, 33. Our finding on the increase of serum TNFα and IL-6, but not IL-1β, in LLC tumor-bearing mice is similar to these patient data. On the other hand, whether cytokine increase in cancer patients is dependent on TLR4 is unknown.
The observed in vivo effects of TLR4 deficiency on cancer cachexia may involve other organs in addition to muscle and immune system. TLR4 is expressed and mediates inflammatory response in many types of cells. In addition, TLR4-mediated elevation of inflammatory cytokines exerts detrimental effects on various organs. For example, they can stimulate lipolysis in adipocytes and cause loss of fat tissue, which frequently takes place in cancer cachexia34. The product of excessive lipolysis, free fatty acids, could in turn activate TLR435 to enhance muscle wasting. Further, TLR4 mediates insulin resistance36, 37, which is another feature of cancer cachexia. Therefore, TLR4 activation is likely to exert extensive pathological impact on various organs and thus a key contributor to the etiology of cancer cachexia.
In addition to cancer cachexia, TLR4 may mediate cachexia associated with other diseases. We have previously demonstrated that TLR4 mediates lipopolysaccharide (LPS)-induced muscle wasting38. In addition, TLR4 plays important roles in the etiology of diabetes and nephropathy39, thus, may contribute to cachexia associated with these diseases. Taken together, we showed in the present study a central role of TLR4 in mediating cancer-induced muscle wasting by activating muscle protein degradation pathways, which opens the door for combating cachexia by targeting a single plasma membrane receptor.
Methods
Cell cultures
Murine C2C12 myoblasts (American Type Culture Collection) were cultured in growth medium (DMEM supplemented with 10% fetal bovine serum) at 37 °C with 5% CO2. At 85–90% confluence, myoblast differentiation was induced by incubation for 96 h in differentiation medium (DMEM supplemented with 4% heat-inactivated horse serum) to form myotubes. Preconditioned medium from cultures of Lewis lung carcinoma cells (obtained from National Institute of Cancer, Frederick, MD) or non-tumorigenic human lung epithelial cell line NL20 (obtained from ATCC) for control were collected from 48 h cultures and centrifuged, the supernatant was used to treat C2C12 myotubes (25% final volume in fresh medium) when indicated. The conditioned medium was replaced with fresh one every 24 h. All cell culture experiments were independently replicated 3 times as indicated (N = 3).
Animal use
Experimental protocols were approved in advance by the institutional Animal Welfare Committee at the University of Texas Health Science Center at Houston. All methods were performed in accordance with the relevant guidelines and regulations. For LLC-induced cancer cachexia model, 100 μl LLC cells (5 × 101), or an equal volume of PBS (control) was injected subcutaneously into the right flanks of 7-week-old male C57BL/6 (Jackson Laboratories, Bar Harbor, ME) or TLR4−/− mice in the C57BL/6 background (a gift from Dr. S. Akira of Osaka University, Osaka, Japan)26. Development of cachexia was monitored by tumor size, body weight and grip strength. Mice were euthanized on day 21 of LLC implant. Tibialis anterior (TA) and extensor digitorum longus (EDL) muscles were then collected immediately for analyses. Each experimental group contained 6 mice (N = 6). To monitor autophagosome formation the TA of wild type C57BL/6 or TLR4−/− mice that had been implanted with LLC cells (or injected with PBS as control) were transfected with GFP-LC3 or GFP-expressing plasmid40 (100 μg in 25 μl PBS) on day 14 as previously described41. In 7 days, when cachexia was developed frozen cross sections (10 μm) of TA were prepared from euthanized mice with anti-fade mounting solution containing DAPI.
Transfection of siRNA
Predesigned siRNAs specific for TLR2 and TLR4 were purchased form Sigma-Aldrich (IDS: SASI_Mm01_00135214 and SASI_Mm01_00139037, respectively). Control siRNA was purchased from ThermoFisher Scientific. The siRNAs were introduced into C2C12 myoblasts using the jetPRIME reagent (Polyplus-transfection Inc., Illkirch, France) according to the manufacturer’s protocol. In 24 h, myoblasts were differentiated and experiments were started in another 96 h when myotubes were formed.
Western blot analysis
Western blot analysis was carried out as described previously10. Antibodies to total and/or phosphorylated p38MAPK (T181/Y182) as well as p-C/EBPβ (Thr-188) were from Cell Signaling Technology (Beverly, MA). Antibody to total C/EBPβ, TLR2, TLR4, phosphorylated and total p65 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to atrogin1/MAFbx was from ECM Biosciences (Versailles, KY). Antibodies to UBR2 and LC3-II were obtained from Novus Biologicals (Littleton, CO). Anti-MHC antibody (MF-20) was from R&D Systems (Minneapolis, MN). Data was normalized to α-Tubulin (antibody was from Development Studies Hybridoma Bank at the University of Iowa, Iowa City, IA) or GAPDH (antibody was from Millipore, Billerica, MA).
Fluorescence microscopy and histology study
C2C12 myotubes were stained with anti-MHC antibody (MF-20) and FITC-conjugated secondary antibody, and examined using a Zeiss Axioskop 40 microscope and a Zeiss Axiocam MRM camera system controlled by Axiovision Release 4.6 imaging software. Acquired images were analyzed for myotube diameter using the method of Menconi et al.42 with modifications. Briefly, myotube diameters were measured in a total of 100 myotubes from ≥10 random fields using computerized image analysis (Scion Image, Frederick, MD, USA) at 3 points along their length. The average of diameters at the 3 points was used for each myotube. The measurements were conducted in a blind fashion. Cross-sectional area of H&E stained muscle sections was quantified by using the ImageJ software (National Institute of Health). Frozen sections of mouse TA that expressed GFP or LC3-GFP were examined with fluorescence microscopy as described above for C2C12 myotubes.
Tyrosine release assay
Tyrosine release was measured as described previously8.
In vitro TLR4 activation assay
HEK-Blue™-hTLR4 cells (InvivoGen, San Diego, CA) are HEK293 cells co-transfected with the human TLR4, MD-2 and CD14 co-receptor genes, and an inducible SEAP (secreted embryonic alkaline phosphatase) reporter gene that is under the control of NF-κB and AP-1. HEK-Blue-hTLR4 cells and wild type HEK293 cells (as control) were treated with LCM for 24 h. SEAP activity in culture media was measured by using QUANTI-Blue as chromogenic substrate at 630 nm in Synergy 2 Multi-Mode Microplate Reader (Biotek Instruments) following manufacturer’s protocol.
Bio-Plex® Multiplex Immunoassay
Sera from LLC tumor-bearing mice were analyzed for specific cytokines utilizing Bio-Plex Pro™ Mouse Cytokine Th17 Panel A 6-Plex Group l (Bio-Rad Laboratories, Hercules, CA) according to manufacturer’s protocol. Cytokine concentrations were determined using the Bio-Plex 200 reader (software version 6.0, Bio-Rad Laboratories).
Statistical analysis
Data were presented as the mean ± S.D., and analyzed by the SigmaStat software (Systat Software, San Jose, CA) with Student t test (data with two groups) or one-way ANOVA (data with more than two groups) followed by a multiple comparison test chosen by the software. Chi-square analysis was carried out by using R to compare the distributions of muscle fiber cross-sectional area among various groups. A P value < 0.05 was considered to be statistically significant.
References
Fearon, K. C., Glass, D. J. & Guttridge, D. C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell metabolism 16, 153–166, doi:10.1016/j.cmet.2012.06.011 (2012).
Fearon, K. Cachexia: Treat wasting illness on multiple fronts. Nature 529, 156, doi:10.1038/529156b (2016).
Dingemans, A. M., de Vos-Geelen, J., Langen, R. & Schols, A. M. Phase II drugs that are currently in development for the treatment of cachexia. Expert Opin Investig Drugs 23, 1655–1669, doi:10.1517/13543784.2014.942729 (2014).
Argiles, J. M., Lopez-Soriano, F. J. & Busquets, S. Counteracting inflammation: a promising therapy in cachexia. Crit Rev Oncog 17, 253–262 (2012).
Baracos, V. E., DeVivo, C., Hoyle, D. H. & Goldberg, A. L. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol 268, E996–1006 (1995).
Cai, D. et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119, 285–298, doi:10.1016/j.cell.2004.09.027 (2004).
Li, Y. P. et al. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J 17, 1048–1057, doi:10.1096/fj.02-0759com (2003).
Zhang, G., Jin, B. & Li, Y. P. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. The EMBO journal 30, 4323–4335, doi:10.1038/emboj.2011.292 (2011).
Puppa, M. J., Gao, S., Narsale, A. A. & Carson, J. A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 28, 998–1009, doi:10.1096/fj.13-240580 (2014).
Li, Y. P. et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19, 362–370, doi:10.1096/fj.04-2364com (2005).
Bonetto, A. et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 303, E410–421, doi:10.1152/ajpendo.00039.2012 (2012).
Penna, F. et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 182, 1367–1378, doi:10.1016/j.ajpath.2012.12.023 (2013).
Bohnert, K. R. et al. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J 30, 3053–3068, doi:10.1096/fj.201600250RR (2016).
Talbert, E. E., Metzger, G. A., He, W. A. & Guttridge, D. C. Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J Cachexia Sarcopenia Muscle 5, 321–328, doi:10.1007/s13539-014-0141-2 (2014).
Op den Kamp, C. M. et al. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 98, 738–748, doi:10.3945/ajcn.113.058388 (2013).
Stephens, N. A. et al. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 6, 53–61, doi:10.1002/jcsm.12005 (2015).
Aversa, Z. et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 6, 30340, doi:10.1038/srep30340 (2016).
Kim, S. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106, doi:10.1038/nature07623 (2009).
Hernandez, C., Huebener, P. & Schwabe, R. F. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene, doi:10.1038/onc.2016.104 (2016).
Wang, W., Zhang, Y., Yang, L. & Li, H. The innate immune signaling in cancer and cardiometabolic diseases: Friends or foes? Cancer Lett, doi:10.1016/j.canlet.2016.06.004 (2016).
Takeda, K. & Akira, S. Toll-like receptors in innate immunity. Int Immunol 17, 1–14, doi:10.1093/intimm/dxh186 (2005).
Cannon, T. Y. et al. The effect of altered Toll-like receptor 4 signaling on cancer cachexia. Arch Otolaryngol Head Neck Surg 133, 1263–1269, doi:10.1001/archotol.133.12.1263 (2007).
Lang, C. H., Silvis, C., Deshpande, N., Nystrom, G. & Frost, R. A. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock 19, 538–546, doi:10.1097/01.shk.0000055237.25446.80 (2003).
Boyd, J. H. et al. Toll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-kappaB and calcineurin. Infect Immun 74, 6829–6838, doi:10.1128/IAI.00286-06 (2006).
Zhang, G., Lin, R. K., Kwon, Y. T. & Li, Y. P. Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 27, 2893–2901, doi:10.1096/fj.12-222711 (2013).
Hoshino, K. et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162, 3749–3752 (1999).
Wiedenmann, B. et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol 6, 18–25 (2008).
Goldberg, R. M. et al. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 13, 2856–2859 (1995).
Gordon, J. N. et al. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut 54, 540–545, doi:10.1136/gut.2004.047563 (2005).
Braun, T. P. et al. Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 27, 3572–3582, doi:10.1096/fj.13-230375 (2013).
He, W. A. et al. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci USA 111, 4525–4529, doi:10.1073/pnas.1402714111 (2014).
Fujiwara, Y. et al. Metabolomics evaluation of serum markers for cachexia and their intra-day variation in patients with advanced pancreatic cancer. PLoS One 9, e113259, doi:10.1371/journal.pone.0113259 (2014).
Bilir, C., Engin, H., Can, M., Temi, Y. B. & Demirtas, D. The prognostic role of inflammation and hormones in patients with metastatic cancer with cachexia. Med Oncol 32, 56, doi:10.1007/s12032-015-0497-y (2015).
Tsoli, M., Swarbrick, M. M. & Robertson, G. R. Lipolytic and thermogenic depletion of adipose tissue in cancer cachexia. Semin Cell Dev Biol 54, 68–81, doi:10.1016/j.semcdb.2015.10.039 (2016).
Huang, S. et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53, 2002–2013, doi:10.1194/jlr.D029546 (2012).
Shi, H. et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116, 3015–3025, doi:10.1172/JCI28898 (2006).
Song, M. J., Kim, K. H., Yoon, J. M. & Kim, J. B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun 346, 739–745, doi:10.1016/j.bbrc.2006.05.170 (2006).
Doyle, A., Zhang, G., Abdel Fattah, E. A., Eissa, N. T. & Li, Y. P. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J 25, 99–110, doi:fj.10-164152 (2011).
Wada, J. & Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol 12, 13–26, doi:10.1038/nrneph.2015.175 (2016).
Xu, Y. et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144, doi:10.1016/j.immuni.2007.05.022 (2007).
Zhang, G. & Li, Y. P. p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet Muscle 2, 20, doi:10.1186/2044-5040-2-20 (2012).
Menconi, M., Gonnella, P., Petkova, V., Lecker, S. & Hasselgren, P. O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem 105, 353–364, doi:10.1002/jcb.21833 (2008).
Acknowledgements
This work is supported by an R01 grant from National Institute of Arthritis and Musculoskeletal and Skin Diseases to Y-PL (AR063786). JMG is supported by Department of Veterans Affairs MERIT grants BX000507 and CX000174, and National Institute of Aging grant AG040583. We thank S. Akira (Osaka University, Osaka, Japan) for sharing the TLR4−/− mice, Nicholas Justice (UTHealth) for helping with multiplex immunoassays and D. Lewis (UTHealth) for critiquing the manuscript.
Author information
Authors and Affiliations
Contributions
Y.-P.L. and G.Z. designed research and wrote the manuscript. G.Z., Z.L., H.D. conducted experiments, analyzed data and generated figures. J.M.G. conducted experiments. H.M. analyzed data.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Liu, Z., Ding, H. et al. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci Rep 7, 2273 (2017). https://doi.org/10.1038/s41598-017-02347-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02347-2
- Springer Nature Limited
This article is cited by
-
The myokine Fibcd1 is an endogenous determinant of myofiber size and mitigates cancer-induced myofiber atrophy
Nature Communications (2022)
-
Enrichment of Newly Synthesized Proteins following treatment of C2C12 Myotubes with Endotoxin and Interferon-γ
Inflammation (2022)
-
Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia
Oncogenesis (2021)
-
IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models
Nature Communications (2020)
-
T. gondii infection induces IL-1R dependent chronic cachexia and perivascular fibrosis in the liver and skeletal muscle
Scientific Reports (2020)