
Abstract
PRKAG2 syndrome is a rare autosomal dominant inherited disorder that is characterized by cardiac hypertrophy, ventricular pre-excitation and conduction system abnormalities. There is little knowledge in cardiovascular magnetic resonance (CMR) characteristics of PRKAG2 cardiomyopathy. This study investigated the genetic defect in a three-generation Chinese family with cardiac hypertrophy and ventricular pre-excitation using whole-exome sequencing. A novel missense mutation, c.1006 G > T (p.V336L), was identified in PRKAG2. This mutation had not been identified in the ExAC database, and the prediction result of MutationTaster indicated a deleterious effect. Furthermore, it cosegregated with the disease in the present family and was absent in unrelated 300 healthy controls. cDNA analysis did not detect any splicing defects, although the variant occurred in the first base of exon 9. CMR evaluation in five affected members showed diffuse hypertrophy in a concentric pattern, with markedly increased left ventricular mass above age and gender limits (median 151.3 g/m2, range 108.4–233.4 g/m2). Two patients in progressive stage and one patient with sudden cardiac death exhibited extensive subendocardial late gadolinium enhancement. In conclusion, molecular screening for PRKAG2 mutations should be considered in patients who exhibit cardiac hypertrophy coexisting with ventricular pre-excitation. CMR offers promising advantages for evaluation of PRKAG2 cardiomyopathy.
Similar content being viewed by others

Introduction
PRKAG2 syndrome (PS) is a rare, early-onset autosomal dominant inherited disorder, and is characterized by cardiac hypertrophy, ventricular pre-excitation (VPE) and progressive conduction abnormalities1,2,3. Little is known about the prevalence of this disorder. Murphy RT et al.3 estimated it to be 1% in patients with both hypertrophy cardiomyopathy (HCM) and premature sinoatrial or atrioventricular conduction disease. To date, the reported patients with genetically confirmed PS are less than 2004.
Since the first reports of PRKAG2 mutations in 20011, 2, 5, PRKAG2 defects have been identified as the pathogenic molecular basis of PS. To date, 21 mutations associated with PS have been identified. The PRKAG2 gene codes for the γ2 regulatory subunit of AMP-activated protein kinase (AMPK)6. AMPK is known as a fuel gauge that is ubiquitously expressed in eukaryotic cells, and which modulates cellular energy homeostasis by switching on ATP-generating pathways and switching off anabolic pathways in response to cellular stress7. The AMPK-γ2 subunit is most abundantly expressed in heart and is responsible for regulating AMPK activity by competitively binding either ATP or AMP6, 8.
Identification of PRKAG2 defects provides a new insight into the molecular basis of unexplained left ventricular hypertrophy (LVH) beyond mutations in genes encoding the sarcomeric proteins. Distinct from sarcomeric HCM, the primary feature of myocardial histopathology in PRKAG2 cardiomyopathy is widespread intracellular vacuolation filled with glycogen, instead of myofiber disarray and interstitial fibrosis3, 9,10,11. Although cardiovascular magnetic resonance (CMR) has been widely used to evaluate myocardial morphology and function in sarcomeric HCM12, 13, and is increasingly used in the assessment of metabolic cardiomyopathies14, only a few cases with PS have been reported with CMR findings11, 15,16,17.
In this study, we investigated a three-generation Chinese family with cardiac hypertrophy and VPE. A novel PRKAG2 heterozygous mutation was detected by whole-exome sequencing, and the diagnosis of PS was established in affected family members. We also conducted a phenotype analysis using CMR findings in five patients in the present family, and performed a review of the reported PS cases with CMR findings, to add more knowledge to the CMR characteristics in PRKAG2 cardiomyopathy.
Results
Clinical features
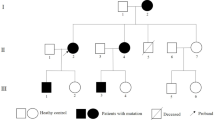
Figure 1 presents the pedigree of a three-generation Chinese family with cardiac hypertrophy and VPE, which was consistent with autosomal dominant inheritance. Totally, there are 7 clinically affected subjects in this family, including two deceased members (individuals I-1 and III-5). The proband (individual II-3), a 53-year-old female, was referred to our hospital for family screening because of her son’s unexplained cardiac hypertrophy and sudden cardiac death (SCD) a month earlier. Abnormal recordings on resting ECG had been observed since a physical examination at age 32. Two years ago, she presented with paroxysmal palpitation and type B WPW syndrome was suggested by her resting ECG (Fig. 2A). An electrophysiological study was performed and showed a right overt accessory pathway (7 o’clock on the tricuspid annulus) which was successfully ablated. Echocardiography showed high-normal thickness in the interventricular septum (IVS, 12 mm) and left ventricular posterior wall (LVPW, 11 mm). On admission, she presented with a short PR interval and widened ORS interval by ECG, suggestive of VPE (Fig. 2B). Echocardiography showed IVS and LVPW hypertrophy with a maximum thickness of 20 mm.

Pedigree of the family with PRKAG2 syndrome. Squares indicate males and circles indicate females. Filled symbols indicate the disease phenotype in affected individuals, i.e., cardiac hypertrophy (left half filled), ventricular pre-excitation (right-upper quadrant filled), or conduction system disease (right-lower quadrant filled). Open symbols represent unaffected individuals, symbols with dots represent mutation carriers without clinical manifestation, shading represents an uncertain clinical status and no sample was available. An arrow denotes the indexed subject and slants denote dead individuals. + indicates PRKAG2 p.V336L mutation positive, and − indicates PRKAG2 p.V336L mutation negative.

ECGs from the proband. (A) Resting ECG before ablation shows ventricular pre-excitation and delta waves (PR interval, 52 ms; QRS interval, 100 ms), with major QRS complex up waves of complexes in the V1 lead and major QRS complex down waves in V5–6 leads, indicative of a diagnosis of type B WPW syndrome. (B) Two years after ablation, short PR intervals (PR interval, 82 ms), enlarged ORS complexes (QRS interval, 190 ms), and deeply inverted T waves were detected in V5–6 leads. The arrow points to the delta wave.
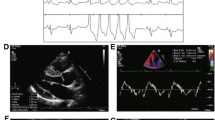
The proband’s son (individual III-5) died suddenly at age 24. He had a 7-year history of cardiac hypertrophy, with complaints of dizziness and amaurosis fugax after intense activity. Severe concentric LVH and mild right ventricular hypertrophy, with left ventricular outflow tract obstruction and impaired systolic function (left ventricular ejection fraction, LVEF, 46%), were demonstrated by CMR (Fig. 3). A 12-lead ECG performed two weeks before his death showed mild sinus bradycardia (heart rate, 56 bpm), high-voltage QRS complexes and abnormal ST-T interval changes. The proband’s father (individual I-1) died at 75 years old. He had a history of cardiac hypertrophy, atrial fibrillation and cerebral hemorrhage, and a permanent pacemaker was implanted at age 72.

Normal and contrast-enhanced CMR images in individual III-5 who suffered from SCD at age 24. Two chamber and four chamber cine images show remarked LVH and mild RVH (▲), with apical involvement (*) (A,B). Basal, mid, and apical LV short-axis images show hypertrophy in almost all segments, with a maximal LV wall thickness of 33 mm and the maximal RV wall thickness of 14 mm (C–E). Arrows indicate late gadolinium enhancement. LV hypertrophic regions exhibit patchy, subendocardial and midmyocardial late gadolinium enhancement (F–J) (white and black arrows). SCD, sudden cardiac death; LVH, left ventricular hypertrophy; RVH, right ventricular hypertrophy; LV, left ventricle.
Patient III-8, aged 24 years, developed hypertension eight years ago. Abnormalities in the kidneys, adrenal glands and renal arteries were excluded by ultrasound examinations. Echocardiography showed apical hypertrophy with a maximum thickness of 25 mm. The patient’s mother (individual II-7) had high-voltage QRS complexes in 12-lead ECG, but no cardiac hypertrophy or other abnormalities were detected by echocardiographic examination. Both patients II-5 and II-9, aged 49 and 45 years, respectively, had asymptomatic severe cardiac hypertrophy confirmed by CMR (see Supplementary Fig. S1). Patient III-6 had an 8-year history of asymptomatic cardiac hypertrophy, and had complained of fatigue after moderate activity over the past six months. The clinical details of the patients studied are shown in Table 1.
Identification of a novel PRKAG2 missense mutation
Whole-exome sequencing identified a heterozygous single-base alteration at position 1006 (c.1006 G > T) in PRKAG2 (Fig. 4A), which resulted in a substitution of the 336th evolutionally conserved valine for leucine (p.Val336Leu) (Fig. 4B). This mutation has not been identified in the ExAC database. And In silico prediction (MutationTaster) reported a deleterious effect. The mutation was confirmed in all five living patients. Individual II-7 was detected with the mutation as well. Because the disorder presented with autosomal dominant inheritance in the current family, and the daughter of individual II-7 was obviously affected, we inferred that she was an asymptomatic mutation carrier. Furthermore, this mutation was not found in other unaffected family members or 300 unrelated healthy controls. Collectively, these results suggested that the p.Val336Leu mutation in the PRKAG2 gene was potentially causative in the present family with cardiac hypertrophy and VPE. And the diagnosis of PS might be established in the affected members.

Genetic analysis identified a novel missense mutation V336L in a family with PRKAG2 syndrome. (A) DNA sequencing chromatogram shows a heterozygous G > T transition at nucleotide 1006 of PRKAG2. (B) Three-dimensional model and schematic of the AMPK-γ2 subunit and all identified PRKAG2 mutations (mutations with grey symbols represent possible disease-causing mutation, mutations with black symbols represent disease-causing mutation; data obtained from Human Gene Mutation Database, www.hgmd.org). Residue V336 is located adjacent to the CBS1 domain and is highly conserved across species. Orange triangle symbols point to AMP binding sites and brown ones point to ATP binding sites (data obtained from UniProtKB, www.uniprot.org). (C) PRKAG2 cDNA sequencing of the proband shows that the mutation does not cause exon skipping.
Notably, although the PRKAG2 gene was included in our targeted sequencing panel, a previous genetic analysis of the index case did not identify the variation. We reevaluated the target region capture sequencing data using IGV after the mutation screening by whole-exome sequencing, and found that the exon 9 sequence, in which the mutation was located, was not covered in the previous sequencing.
cDNA analysis
Because it is the first nucleotide of exon 9 in the PRKAG2 gene that is altered, we evaluated the effect of this mutation on mRNA splicing. The sequence analysis of PRKAG2 cDNA fragments from the proband containing a region between Exon 7–11 did not reveal any splicing defects (Fig. 5C).

Distribution of left ventricular hypertrophy in patients with PRKAG2 p.V336L mutations in the current family. A, II-3; B, II-5; C, II-9; D, III-5; and E, III-6. Upper and lower values indicate absolute (mm) and standardized (z-score) end-systolic maximal wall thickness in segments. Each segment is labeled with gray scaling according to z-score.
CMR characteristics
A CMR study was performed in the five affected family members (individuals II-3, II-5, II-9, III-5, and III-6). Detailed CMR characteristics are presented in Table 2. The proband was the only one who had preserved age, gender, and BSA standardized left ventricular end-diastolic volume (LVEDV), stoke volume (SV), and LVEF. The other four patients had much higher indexed LVEDV, and two patients (II-5 and III-6) had concurrently elevated indexed SV. Apparent impaired LVEF was observed in patient III-5, which decreased to 46% (z-score, −4.67). Notably, all studied patients had left ventricular (LV) mass above age and gender limits, and four patients (aged 49, 45, 24, and 28 years) had remarkably elevated LV mass (208.9 g/m2 [z-score, 15.87], 151.3 g/m2 [9.09], 233.4 g/m2 [18.75], 149.7 g/m2 [11.56]).
All five patients had conspicuous LV hypertrophy in an concentric pattern (Fig. 5), with a median maximal wall thickness of 31.6 mm (range 25–33.2 mm) and z-score of 12.5 (range 9.9–14.6) (Table 2). Patient II-3 had a characteristic spade-like configuration of the cavity (see Supplementary Fig. S1), with a maximal wall thickness of 25 mm located in the apical anterior segment (Fig. 5A). Patients II-5 and II-9 had high-normal LVED dimensions, and hypertrophy predominantly in the mid-anterior and mid-anteroseptal segments (Fig. 5B and C). Patient III-5, who had the most markedly increased LV mass, had hypertrophy predominantly in the mid-lateral segments with a maximal wall thickness of 33.2 mm (z-score, 13.47) (Fig. 5D). The right ventricle was also involved with a maximal wall thickness of 13.9 mm located in the mid-anterior segment. Prominent trabeculae were observed in the mid-distal part of the lateral wall of patient III-6 (see Supplementary Fig. S1), whose maximal wall thickness was located in the mid-inferolateral segment (Fig. 5E).
LGE was present in three of the five patients (60%) examined by CMR (Fig. 3 and Supplementary Fig. S1). Hyperenhancement primarily focused on the mid-distal hypertrophic regions, and all three LGE positive patients presented with a subendocardial pattern. Transmural hyperenhancement within the apical regions was also revealed in patients II-5 and II-9, while ill-defined LGE throughout the LV myocardium was observed in patient III-5 (Fig. 3F–J).
Discussion
In this study, we investigated the genetic defect and CMR characteristics in a three-generation Chinese family with cardiac hypertrophy and VPE. A novel missense mutation of the PRKAG2 gene (c.1006 G > T, p.V336L) was identified by whole-exome sequencing. And the diagnosis of PS was made in the affected members. Myocardial morphology and function were evaluated form CMR findings of five affected members.
AMPK is an important energy-sensing enzyme that is activated by increases in the AMP/ATP ratio, and is deeply involved in cellular ATP metabolism7. AMPK exists as a heterotrimeric complex composed of a catabolic α subunit and regulatory β and γ subunits with multiple isoforms for each subunit7. Two isoforms of the γ subunit (γ1and γ2) are expressed in cardiac tissue, with predominance of γ2 subunit8. The AMPK-γ2 subunit, which is encoded by PRKAG2, contains four tandem repeats of sequence motifs, known as CBS repeats (CBS 1–4), which assemble to generate four potential nucleotide-binding sites for adenosine-containing ligands, such as AMP, ADP and ATP18. Activation of AMPK is triggered by AMP binding to the γ subunit, which causes the conformational change of α subunit, promotes phosphorylation, and inhibits Thr172 dephosphorylation within the α subunit kinase domain19. Most of the reported disease-causing mutations occur in the highly conserved CBS domain region, disturbing the normal interaction between AMPK-γ2 subunit and adenosine-containing ligands. The mutation identified in the present family is located adjacent to the CBS1 domain, and replace the highly conserved valine, which potentially affect the tridimensional structure of AMPK and result in enzyme activity modification4. A recently reported causative mutation lies in the region between CBS1 and CBS2 as well, suggesting this domain interval has a critical function16. Moreover, a disease-causing mutation in the same position (V336) as that of the present family was recently identified in a three-generation pedigree20. All four family members carrying that mutation (p.V336A) showed severe left ventricular hypertrophy, obvious electrophysiological abnormalities, and a progressive course. The conspicuous phenotype seen with the substitution of this amino acid in two families suggests that this residue play an important role.
The molecular mechanisms underlying PRKAG2 phenotype are not clearly evident. Inconsistent results are obtained from previous studies regarding to the effect of the PRKAG2 mutation on the regulation of AMPK activity. In vitro, some studies showed impaired binding between AMP and AMPK-γ2 subunit but increased basal AMPK activity21, 22, while other studies showed reduction in both AMP binding and AMPK activity6, 23. In mouse models, variable AMPK activity is observed. AMPK is constitutively activated in the N488I hearts24, while AMPK activity in the R302Q hearts is low25. The R531G hearts have normal AMPK activity at 1 week, and decrease in the activity is observed after glycogen accumulation26. Increased glycogen content is usually observed in the cardiac histopathological biopsies from patients with PS and model mice. In the TGN488I mouse model, aberrantly activated AMPK may increase glucose uptake by accelerating translocation and expression of glucose transporters, and subsequently, persistent allosteric activation of glycogen synthase by abundant glucose-6-phosphate promotes glycogen storage18.
Electrocardiographic abnormalities are common in PS, particularly short PR interval, which is largely attributed to excessive glycogen accumulation in myocytes4, 27. During cardiogenesis, collagen-containing fibrous rings separate the atria and ventricle, and can be disrupted by glycogen-filled myocytes, resulting in VPE and reciprocating arrhythmias24, 28. Atrioventricular bypass tract, which was observed in the proband in the current PS family, is the most described accessory pathway in PS cases. Besides, nodoventricular and fasciculoventricular pathways are also observed in patients with PS, suggesting an important role for PRKAG2 in development of the cardiac conduction system29,30,31.
It is reported that more than 50% of PS patients present with LVH4, yet excessive glycogen deposition may not totally account for the cardiac hypertrophy. A recent study showed that the contribution of glycogen to increased heart mass was <5% in TGT400N mice which recapitulates symptoms of patients with PRKAG2 T400N mutation32. Proliferation and hypertrophy pathways are found to be involved in the development of PRKAG2 cardiomyopathy. In the TGT400N mouse model, Banerjee et al.32 demonstrated that early activation of the NF-κB and Akt signaling pathway, which are triggered by inappropriate AMPK activation, mediated cardiac hypertrophy. And recently, Kim and his colleagues suggested that the FoxO and mTOR signaling cascades were involved in activation of the proliferation and hypertrophy pathway related to AMPK-γ2 mutation33.
To date, 21 mutations have been reported to be associated with PS (Fig. 4B), and most of these are missense mutations, with the most frequently reported mutations being p.R302Q and p.N488I4. The clinical phenotypes are various among the PRKAG2 mutation families. Some affected families only present with VPE and conduction system disease with the absence of cardiac hypertrophy2, 34, while in other affected families, cardiac hypertrophy is the primary feature1, 3. Even in the same family, affected members sharing with the same mutation can have different manifestations. Cardiac hypertrophy is a prominent feature in the current family, which is found in all affected members. A patient presented with biventricular hypertrophy, while only the left ventricle is involved in other affected family members. In comparison with cardiac hypertrophy, VPE is found in 4 members, and only two affected members have conduction system dysfunction. Mutations causing PS have a high penetrance which is estimated to be 99%4. Individual II-7 with the p.V336L mutation in the present family fails to be found with cardiac hypertrophy, VPE and any conduction system abnormalities, and she is inferred as an asymptomatic mutation carrier.
The role of CMR in the evaluation of metabolic cardiomyopathies has been emerged these years. To our knowledge, only nine patients with PS have been reported with CMR findings11, 15,16,17 (Table 3), and most of them are case reports. In this study, we provide additional information in CMR characteristics of PRKAG2 cardiomyopathy from five affected family members. We found that LV hypertrophy is diffuse (≥10 segments [≥62% of LV]) with a concentric pattern in five patients, although marked hypertrophy was observed in certain segments. Most recently, Yogasundaram et al.17 reported a 31 year-old male PS patient with concentric LVH, also demonstrated by CMR. Concentric hypertrophy is the most common pattern in metabolic and infiltrative disorders35. However, a recent CMR study showed an eccentric pattern in six patients with PRKAG2 defects16. As two patients with remarkably increased LV mass presented with diffuse hypertrophy, hypertrophy in the interventricular septum was much more predominant. The other four patients exhibited a non-symmetric mid-infero-lateral pattern of hypertrophy. In two previous reports, asymmetric hypertrophy was confirmed by CMR in two PS patients11, 15. Existence of multiple morphological variants, as in the case of HCM36, is a possible explanation. In a cohort of 32 PRKAG2 mutation carriers with LVH, an echocardiography study revealed different hypertrophy patterns, including concentric (59%), asymmetric (38%), and distal (3%) hypertrophy3. Interestingly, four patients examined by CMR in the current family exhibited increased end-diastolic and end-systolic volumes, with concentric hypertrophy. This may imply a progressive disease course, as impairment of LVEF following SCD occurred in one patient, while enlargement of LV was observed in other two patients.
One of the most prominent CMR characteristics of PRKAG2 cardiomyopathy in the present study was global subendocardial hyperenhancement in mid-distal hypertrophic regions, with locally transmural hyperenhancement in the apical region. Global transmural or subendocardial LGE is the most frequent observation in cardiac amyloidosis37. Nevertheless, the basal subendocardium was not involved in our patients, while in other reports, PS patients with non-global hyerenhancement were shown11, 16, 17. Extensive LGE in HCM is suggested to be an independent prognostic marker for SCD or appropriate implantable cardioverter-defibrillator discharges38. In the current study, systolic function was impaired and a SCD event occurred in one patient with biventricular involvement of LGE. The other two patients with positive LGE developed enlarged LV. Additionally, three PS patients with mild to extensive LGE were reported to undergo pacemaker implantation16, 17. Myocardial infarction also occurred in a PS patient with localized gadolinium uptake in the interventricular septum11. Therefore, we propose that LGE may serve as a risk marker for SCD and other adverse outcomes. However, because of the small size of PS patients with CMR examinations, further studies are needed to determine the clinical relevance between presence and extent of LGE and adverse outcome in patients with PRKAG2 defects.
In summary, we report a novel PRKAG2 mutation that is associated with PS in a Chinese family. Molecular screening for PRKAG2 mutations should be considered in patients who exhibit cardiac hypertrophy coexisting with VPE. CMR offers advantages for better characterization of PRKAG2 cardiomyopathy in PS and is a valuable aid for diagnosis. PS cardiac hypertrophy may present with multiple morphologies, but with dominance of a concentric pattern. CMR may play a potential role in assessment of risk in patients with PS.
Methods
Ethics statements
This study was approved by the Ethics Committee of Fuwai Hospital. All participants provided written informed consent. The methods were performed in accordance with the approved guidelines.
Clinical evaluation
A Chinese family with a HCM phenotype was identified and investigated. Family members were evaluated by collecting a detailed medical history, physical examination, 12-lead electrocardiogram (ECG), 24-hour ambulatory ECG, and transthoracic echocardiography. When appropriate, CMR was conducted in patients with cardiac hypertrophy. Cardiac hypertrophy was defined by echocardiography or CMR as increased LV wall thickness ≥15 mm in one or more myocardial segments35. VPE was defined by a short PR interval (<120 ms)20. Presence of a short PR interval, widened ORS interval (>120 ms) and an abnormal QRS vector (delta-wave) was classified as Wolff-Parkinson-White (WPW) syndrome20.
CMR images were transferred to a workstation (Siemens Medical Systems) for analysis. In each patient, a 17 segment model was made from three short axis sections, according to American Heart Association criteria39. Left atrial and LV end-diastolic diameter, LV volumes, LVEF, LV mass, and maximal thickness of hypertrophied segments were measured. All parameters were standardized to age, gender, body surface area (BSA) and normal references (z-scores)40, 41. Late gadolinium enhancement (LGE) was qualitatively determined for each myocardial segment by reviewing all short and long axis contrast-enhanced images. LGE patterns were classified as subendocardial, subepicardial, mid-myocardial, or transmural (≥75% of any segmental wall thickness) by visual analysis42.
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Midi Kit (QIAGEN, Hilden, Germany), according to standard protocols. The proband had previously been screened using a targeted sequencing panel containing 64 candidate genes reported to be causative of inherited cardiomyopathy43. No pathogenic mutations were detected. Next, three affected individuals (II-9, III-6, and III-8) and one unaffected individual (II-2) were selected for whole-exome sequencing. The Agilent SureSelect Human All Exon V5 capture kit (Agilent, Santa Clara, CA, USA) was used for exome capture. Samples from the family quartette were multiplexed on a single lane and 101-bp, paired-end sequencing was performed using Illumina’s HiSeq4000 platform (Illumina, Inc, San Diego, CA, USA) to an average depth of 141×.
Sequence data were aligned to the human reference genome (GRCh37/hg19) with BWA followed by sorting and marking of duplicate reads using Picard (version 2.4, http://broadinstitute.github.io/picard/). Local realignment of insertions/deletions (indels) and base quality score recalibration were performed using GATK (version 3.6, https://www.broadinstitute.org/gatk/). GATK was also used to call and filter variants within a genome–wide region. The resultant variants were annotated with ANNOWAR44 and sequentially filtered using the following criteria: (1) variants with an East Asian minor allele frequency ≥0.01 in the databases for 1000 Genomes Project (http://browser.1000genomes.org) and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org) were removed; (2) variants in exons and splicing sites were retained; (3) certain types of variants were retained, including nonsynonymous, frameshift, nonframeshift insertion/deletion, stopgain and unknown; (4) variants in genes expressed in the heart according to the Human Protein Atlas (www.proteinatlas.org)45; and (5) variants that passed manual confirmation using the Integrative Genomics Viewer (IGV, http://software.broadinstitute.org/software/igv)46 were retained. Effects of variants on splicing signals were evaluated using the Human Splicing Finder (version 3.0, www.umd.be/HSF3/HSF.html)47.
Candidate variants were validated by Sanger sequencing. Three hundred unrelated healthy control samples were subjected to variant examination.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from peripheral blood leukocytes collected from individuals I-2 and II-3 using TRIzol LS reagent (Life Technologies, Grand Island, NY, USA). Reverse transcription reaction was performed using the GoScript Reverse Transcription System (Promega, Madison, WI, USA), according to the manufacturer’s protocol. cDNA sequence containing the mutation site was amplified by PCR using a forward primer located in exon 7 of the PRKAG2 gene (5′- TCGTTCCAACCAGTTCAAAGC-3′), and a reverse primer located in exon 11 (5′-GTTCTGCTTCATGAAGGCAGG-3′). The resulting PCR products were analyzed by Sanger sequencing.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Blair, E. et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10, 1215–1220, doi:10.1093/hmg/10.11.1215 (2001).
Gollob, M. H. et al. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 104, 3030–3033, doi:10.1161/hc5001.102111 (2001).
Murphy, R. T. et al. Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol. 45, 922–930, doi:10.1016/j.jacc.2004.11.053 (2005).
Porto, A. G. et al. Clinical Spectrum of PRKAG2 Syndrome. Circ Arrhythm Electrophysiol. 9, e003121, doi:10.1161/CIRCEP.115.003121 (2016).
Gollob, M. H. et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 344, 1823–1831, doi:10.1056/NEJM200106143442403 (2001).
Scott, J. W. et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 113, 274–284, doi:10.1172/JCI19874 (2004).
Hardie, D. G. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 33, 1–7, doi:10.1016/j.ceb.2014.09.004 (2015).
Cheung, P. C., Salt, I. P., Davies, S. P., Hardie, D. G. & Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 346(Pt 3), 659–669, doi:10.1042/bj3460659 (2000).
Liu, Y. et al. Identification of a novel de novo mutation associated with PRKAG2 cardiac syndrome and early onset of heart failure. PLoS One 8, e64603, doi:10.1371/journal.pone.0064603 (2013).
Arad, M. et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 109, 357–362, doi:10.1172/JCI14571 (2002).
Sternick, E. B., de Almeida Araujo, S., Rocha, C. & Gollob, M. Myocardial infarction in a teenager. Eur Heart J. 35, 1558–1558, doi:10.1093/eurheartj/ehu015 (2014).
Maron, M. S. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 14, 13, doi:10.1186/1532-429X-14-13 (2012).
Maron, B. J. & Maron, M. S. The Remarkable 50 Years of Imaging in HCM and How it Has Changed Diagnosis and Management: From M-Mode Echocardiography to CMR. JACC Cardiovasc Imaging. 9, 858–872, doi:10.1016/j.jcmg.2016.05.003 (2016).
Mavrogeni, S., Markousis-Mavrogenis, G., Markussis, V. & Kolovou, G. The Emerging Role of Cardiovascular Magnetic Resonance Imaging in the Evaluation of Metabolic Cardiomyopathies. Horm Metab Res. 47, 623–632, doi:10.1055/s-0035-1555913 (2015).
Fabris, E. et al. Cardiac hypertrophy, accessory pathway, and conduction system disease in an adolescent: the PRKAG2 cardiac syndrome. J Am Coll Cardiol. 62, e17, doi:10.1016/j.jacc.2013.02.099 (2013).
Poyhonen, P. et al. Cardiovascular magnetic resonance findings in patients with PRKAG2 gene mutations. J Cardiovasc Magn Reson. 17, 89, doi:10.1186/s12968-015-0192-3 (2015).
Yogasundaram, H., Paterson, I. D., Graham, M., Sergi, C. & Oudit, G. Y. Glycogen Storage Disease Because of a PRKAG2 Mutation Causing Severe Biventricular Hypertrophy and High-Grade Atrio-Ventricular Block. Circ Heart Fail. 9, doi:10.1161/CIRCHEARTFAILURE.116.003367 (2016).
Zaha, V. G. & Young, L. H. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 111, 800–814, doi:10.1161/CIRCRESAHA.111.255505 (2012).
Hardie, D. G., Schaffer, B. E. & Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 26, 190–201, doi:10.1016/j.tcb.2015.10.013 (2016).
Thevenon, J. et al. High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations. Europace, doi:10.1093/europace/euw067 (2016).
Burwinkel, B. et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet. 76, 1034–1049, doi:10.1086/430840 (2005).
Hawley, S. A. et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565, doi:10.1016/j.cmet.2010.04.001 (2010).
Akman, H. O. et al. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res. 62, 499–504, doi:10.1203/PDR.0b013e3181462b86 (2007).
Arad, M. et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation. 107, 2850–2856, doi:10.1161/01.CIR.0000075270.13497.2B (2003).
Sidhu, J. S. et al. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation. 111, 21–29, doi:10.1161/01.CIR.0000151291.32974.D5 (2005).
Davies, J. K. et al. Characterization of the role of gamma2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am J Physiol Heart Circ Physiol. 290, H1942–1951, doi:10.1152/ajpheart.01020.2005 (2006).
Gollob, M. H. Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem Soc Trans. 31, 228–231, doi:10.1042/doi:10.1042/ bst0310228 (2003).
Patel, V. V. et al. Electrophysiologic characterization and postnatal development of ventricular pre-excitation in a mouse model of cardiac hypertrophy and Wolff-Parkinson-White syndrome. J Am Coll Cardiol. 42, 942–951, doi:10.1016/S0735-1097(03)00850-7 (2003).
Govindan, M., Ward, D. & Behr, E. A rare connection: fasciculoventricular pathway in PRKAG2 disease. J Cardiovasc Electrophysiol. 21, 329–332, doi:10.1111/j.1540-8167.2009.01578.x (2010).
Sternick, E. B. et al. Clinical, electrocardiographic, and electrophysiologic characteristics of patients with a fasciculoventricular pathway: the role of PRKAG2 mutation. Heart Rhythm. 8, 58–64, doi:10.1016/j.hrthm.2010.09.081 (2011).
Tan, H. L. et al. Nodoventricular accessory pathways in PRKAG2-dependent familial preexcitation syndrome reveal a disorder in cardiac development. Circ Arrhythm Electrophysiol. 1, 276–281, doi:10.1161/CIRCEP.108.782862 (2008).
Banerjee, S. K., McGaffin, K. R., Huang, X. N. & Ahmad, F. Activation of cardiac hypertrophic signaling pathways in a transgenic mouse with the human PRKAG2 Thr400Asn mutation. Biochim Biophys Acta. 1802, 284–291, doi:10.1016/j.bbadis.2009.12.001 (2010).
Kim, M. et al. Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res. 114, 966–975, doi:10.1161/CIRCRESAHA.114.302364 (2014).
Charron, P., Genest, M., Richard, P., Komajda, M. & Pochmalicki, G. A familial form of conduction defect related to a mutation in the PRKAG2 gene. Europace. 9, 597–600, doi:10.1093/europace/eum071 (2007).
Authors/Task Force, M. et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 35, 2733–2779, doi:10.1093/eurheartj/ehu284 (2014).
Noureldin, R. A. et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 14, 17, doi:10.1186/1532-429X-14-17 (2012).
Syed, I. S. et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 3, 155–164, doi:10.1016/j.jcmg.2009.09.023 (2010).
Chan, R. H. et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 130, 484–495, doi:10.1161/CIRCULATIONAHA.113.007094 (2014).
Cerqueira, M. D. et al. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation. 105, 539–542, doi:10.1161/hc0402.102975 (2002).
Maceira, A. M., Prasad, S. K., Khan, M. & Pennell, D. J. Normalized left ventricular systolic and diastolic function by steady state free precession cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 8, 417–426, doi:10.1080/10976640600572889 (2006).
Kawel, N. et al. Normal left ventricular myocardial thickness for middle-aged and older subjects with steady-state free precession cardiac magnetic resonance: the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging. 5, 500–508, doi:10.1161/CIRCIMAGING.112.973560 (2012).
Wan, J. et al. Varied distributions of late gadolinium enhancement found among patients meeting cardiovascular magnetic resonance criteria for isolated left ventricular non-compaction. J Cardiovasc Magn Reson. 15, 20, doi:10.1186/1532-429X-15-20 (2013).
Wu, W. et al. Novel Phenotype-Genotype Correlations of Restrictive Cardiomyopathy With Myosin-Binding Protein C (MYBPC3) Gene Mutations Tested by Next-Generation Sequencing. J Am Heart Assoc. 4, doi:10.1161/JAHA.115.001879 (2015).
Yang, H. & Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 10, 1556–1566, doi:10.1038/nprot.2015.105 (2015).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science. 347, 1260419–1260419, doi:10.1126/science.1260419 (2015).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192, doi:10.1093/bib/bbs017 (2013).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67–e67, doi:10.1093/nar/gkp215 (2009).
Acknowledgements
The authors thank the family who participated in this study. This work was supported by CAMS Innovation Fund for Medical Sciences (2016-I2M-1-002), the National Key Research and Development Program of China (2016YFC1300100), Beijing Municipal Science and Technology Commission (Z151100003915078), National Natural Science Foundation of China (81600305), Beijing Outstanding Talent Training Project (2016000020124), PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (3332015108), and PUMC Graduate Innovation Fund.
Author information
Authors and Affiliations
Contributions
X.L.Z. and X.Z. designed and supervised the research. Y.Z., Y.K.Y., X.M., P.F., L.P.W., T.T. and Y.X.L. collected samples and clinical information. K.Q.Y. and Y.Z. performed the experiments. K.Q.Y., C.X.L., J.C.L. and T.L. performed data analysis. K.Q.Y. wrote the manuscript. C.X.L., X.Z. and X.L.Z. modified the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, KQ., Lu, CX., Zhang, Y. et al. A novel PRKAG2 mutation in a Chinese family with cardiac hypertrophy and ventricular pre-excitation. Sci Rep 7, 2407 (2017). https://doi.org/10.1038/s41598-017-02455-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02455-z
- Springer Nature Limited
This article is cited by
-
Phenotypic expression and clinical outcomes in a South Asian PRKAG2 cardiomyopathy cohort
Scientific Reports (2020)
-
Familial Atrial Enlargement, Conduction Disorder and Symmetric Cardiac Hypertrophy Are Early Signs of PRKAG2 R302Q
Current Medical Science (2020)
-
Cardiac MR manifestations in two cases of PRKAG2 mutations in a Chinese family
The International Journal of Cardiovascular Imaging (2020)