
Abstract
Auxin response factors (ARFs) are the transcription factors that regulate auxin responses in various aspects of plant growth and development. Although genome-wide analysis of ARF gene family has been done in some species, no information is available regarding ARF genes in chickpea. In this study, we identified 28 ARF genes (CaARF) in the chickpea genome. Phylogenetic analysis revealed that CaARFs can be divided into four different groups. Duplication analysis revealed that 50% of CaARF genes arose from duplication events. We analyzed expression pattern of CaARFs in various developmental stages. CaARF16.3, CaARF17.1 and CaARF17.2 showed highest expression at initial stages of flower bud development, while CaARF6.2 had higher expression at later stages of flower development. Further, CaARF4.2, CaARF9.2, CaARF16.2 and CaARF7.1 exhibited differential expression under different abiotic stress conditions, suggesting their role in abiotic stress responses. Co-expression network analysis among CaARF, CaIAA and CaGH3 genes enabled us to recognize components involved in the regulatory network associated with CaARFs. Further, we identified microRNAs that target CaARFs and TAS3 locus that trigger production of trans-acting siRNAs targeting CaARFs. The analyses presented here provide comprehensive information on ARF family members and will help in elucidating their exact function in chickpea.
Similar content being viewed by others

Introduction
Auxin or indole-3-acetic acid (IAA), a crucial phytohormone, plays a vital function in regulation of numerous aspects of growth and development in plants. Many studies have shown role of auxin in regulation of several biological processes, such as apical dominance, embryo patterning, formation of lateral root, shoot elongation, tropic responses and vascular differentiation1,2,3,4. Auxin response factors (ARFs) are important transcription factors that can activate or repress the expression of early/primary auxin response genes [Auxin/Indole-3-acetic acid (Aux/IAA), Small Auxin Up RNA (SAUR) and Gretchen Hagen 3 (GH3)] via binding with auxin response elements (AuxREs, TGTCTC) or some variation of these elements (TGTCCC or TGTCAC) in their promoters5,6,7. Recently, microarray experiments indicated that AtARF1 and AtARF5 prefer to bind TGTCGG elements as compared to AuxRE TGTCTC8. TGTCGG appeared to be the preferred DNA binding motif of ARF2 and ARF5 in a “cistrome” analysis as well9.
ARF proteins contain three domains, i.e. a N-terminal B3-like DNA binding domain (DBD), C-terminal PB1 (Phox and Bem1) domain contained within a region that was previously called motif III/IV, and a middle region (MR), which is responsible for gene activation/repression8, 10,11,12,13,14,15.
ARF gene family has been analyzed in many plants, such as Arabidopsis, rice, Medicago and tomato2, 10, 16,17,18. Function of many ARF proteins have also been studied in plants. For instance, arf1 and arf2 loss-of-function mutants in Arabidopsis exhibited abnormal abscission of floral organs and senescence in leaf tissues19, while arf3/ett illustrated abnormal floral meristem patterning and gynoecium development20. In addition, arf5 mutant revealed abnormality in formation of embryo axis and vascular strands21. Furthermore, response of hypocotyl to blue light and auxin stimulus was found to be affected by mutation in AtARF7 22. However, double mutants of arf6/arf8 were found to have infertile closed buds with short petals and stamen filaments besides bearing undehisced anthers23. Auxin-dependent lateral root development was found to be hampered in double mutants of arf7/arf1924. The antisense OsARF1 rice transgenic lines exhibited altered organ size, curled leaves, poor vigor and reduced growth, indicating its role in development of somatic and reproductive tissues25. In addition, OsARF16 was found to be involved in iron and phosphate starvation responses in rice26, 27.
Chickpea (Cicer arietinum L.) is a model legume crop rich in dietary proteins and fibers for humans and animals. Since ARFs orchestrate various developmental processes in plants and their genetic manipulation holds potential for generating better yielding crops17, 28,29,30, it is important to study this gene family in chickpea. In this study, we identified members of ARF gene family in the chickpea genome. Duplication analysis revealed expansion of ARF gene family in chickpea via segmental duplication. Comprehensive gene expression profiling revealed significant differential expression of several ARF genes across diverse tissue types, indicating functional divergence of this gene family. A co-expression network of chickpea ARF, IAA and GH3 genes was generated and candidate genes involved in developmental processes were identified. These analyses along with prediction of ARFs as targets of miRNA and tasiRNA will help in understanding auxin response in chickpea.
Results and Discussion
Discovery of ARF gene family in chickpea
To define members of ARF gene family in chickpea, protein sequences of 23 known ARFs in Arabidopsis (AtARFs) were used to identify their homologs in chickpea genome using BLAST searches. In addition, chickpea proteome was used to perform HMM (Hidden Markov Model) profile searches. The list of putative members of ARF gene family obtained from the above two approaches were merged to generate a unique gene list. To confirm the presence of ARF domain, protein sequences of putative ARF gene family members of chickpea were analyzed in Pfam and SMART databases. In total, 28 protein sequences were confirmed as ARFs in chickpea (CaARFs). Naming of chickpea ARF genes was done in accordance to their evolutionary relationship with Arabidopsis ARFs (Table S1).
The size of CaARF proteins ranged from 279 (CaARF5.2) to 1089 (CaARF19) amino acids. Their isoelectric points varied from 5.55 (CaARF5.1) to 8.56 (CaARF10.2), indicating that different CaARF proteins may function in different microenvironments.
Determination of gene structure and evolutionary relationship
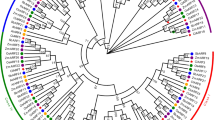
An unrooted phylogenetic tree was constructed using protein sequence alignment of ARF gene family members from Arabidopsis (23) and chickpea (28) for examining evolutionary relationship among them (Fig. 1). Phylogenetic analysis grouped ARF proteins into four major classes, I (A and B), II, III and IV (Fig. 1). Group IA consisted of five CaARF proteins and five AtARFs. Notably, group IB lacked any CaARF protein and was comprised of only eight AtARFs (Fig. 1). Six ARFs were clustered in group II (four from chickpea and two from Arabidopsis). Group III harbored 17 members (12 CaARFs and five AtARFs), whereas clustering of 10 members (seven CaARFs and three AtARFs) was observed in group IV. Most of CaARF paralog pairs identified from phylogenetic tree analysis were present on duplicated chromosomal blocks (Fig. 1). Similarity of sequences between paralog pairs was quite high, indicating that these genes might be involved in governing similar functions. Furthermore, we identified at least 14 conserved motifs in the CaARF protein sequences using MEME (Fig. 2, Table S2a,b). Among these, four motifs (1, 2, 9 and 11) were found to be associated with N-terminal B3-domain, six motifs (4, 6, 7, 12, 13 and 14) associated with middle ARF domain, while motifs 8 and 10 were associated with C-terminal PB1 domain. In addition, we identified two novel motifs (motifs 3 and 5) located at the N-terminus of most of CaARF protein sequences (Fig. 2). Their function needs to be recognized. Corroborating our predictions, members grouped together in the phylogenetic tree revealed similar motif organization with few exceptions (for example, motifs 3 and 5 were absent in CaARF5.2, 5.3, 5.4 and 5.5 as compared to CaARF5.1), signifying their functional coherence (Fig. 2). Further, we analyzed exon-intron organization of all CaARFs genes. Although the number of exons varied from 2 to 17 (Fig. 2), members of the same group represented similar exon-intron structure.

Phylogenetic relationship among ARF proteins from chickpea and Arabidopsis. The deduced full-length amino acid sequences of chickpea (CaARF) and Arabidopsis (AtARF) genes were aligned by MUSCLE and the phylogenetic tree was constructed by MEGA (v7.0) using the neighbour joining (NJ) method. The numbers on the nodes represent bootstrap values from 100 replicates. Grouping of ARFs has been indicated in different colors.

Gene structure and motif organization of ARF genes in chickpea. Left panel illustrates the exon–intron organization of ARF genes. Exons and introns are represented by boxes (blue) and lines (black), respectively. Right panel shows motif organization in ARF proteins. Motifs 1, 2, 9 and 11 represent B3-domain; Motifs 4, 6, 7, 12, 13 and 14 represent ARF-domain; Motifs 8 and 10 represent PB1-domain; Motifs 3 and 5 are unknown novel motifs.
Genomic location and gene duplication events in CaARF gene family
Physical position of 27 CaARF genes was found on seven chromosomes of chickpea (except chromosome 8), while one CaARF gene (CaARF17.1) was present on a scaffold (Fig. 3). The number of ARF genes on different chromosomes ranged from one to seven (Fig. 3). One ARF gene was located on chromosome 3 (CaARF9.2), two genes on chromosome 2 (CaARF8.1 and 17.2) and chromosome 5 (CaARF5.4 and 8.2) each, three genes on chromosome 7 (CaARF7.1, 9.3 and 16.3), six genes on chromosome 1 (CaARF1, 3.2, 6.1, 10.2, 4.2 and 7.2) and chromosome 4 (CaARF16.2, 16.1, 5.3, 5.2, 5.5 and 5.1) each, and seven ARF genes were located on chromosome 6 (CaARF2, 3.1, 4.1, 6.2, 9.1, 10.1 and 19) (Fig. 3).

Mapping of duplicated CaARF genes on chickpea chromosomes. Grey ribbons indicate collinear relationship among the blocks in whole genome and blue ribbons show CaARF paralogs. The chickpea chromosomes are arranged in a colored arc and the size of each arc corresponds to the size of respective chromosome (Mb). Genes with black asterisk represent segmentally duplicated genes and red asterisks highlight the tandemly duplicated genes.
Duplication analysis revealed expansion of ARF gene family in chickpea. Among the 28 ARF genes, 14 (50%) arose from duplication events, including twelve genes (CaARF3.1/3.2, CaARF10.1/10.2/16.1/16.2, CaARF4.1/4.2, CaARF7.1/7.2 and CaARF8.1/8.2) representing segmental duplications (42.8%) and two genes (CaARF5.2/5.3) representing tandem duplications (7.1%) (Fig. 3). In Arabidopsis, 14 (60.8%) out of 23 ARFs arose from duplication events, representing seven segmentally (30.4%) and seven tandemly (30.4%) duplicated genes31. All seven tandemly duplicated ARF genes (30.4%) of Arabidopsis belonged to group IB31, which is absent in chickpea (this study) and other plants2, 17, 18, 28,29,30. In phylogenetic tree, all the duplicated CaARF gene pairs were found to be clustered together (Fig. 3). Furthermore, higher sequence similarity between duplicated gene pairs suggested that they are likely to participate in regulation of similar biological processes. For example, CaARF5.2 and CaARF5.3 representing tandem duplication had 48.5% similarity, while CaARF8.1 and CaARF8.2 representing segmental duplication showed 84.2% similarity at the protein sequence level. In group III, one pair of tandemly duplicated genes (CaARF5.2/5.3) and one pair of segmentally duplicated genes (CaARF8.1/8.2) were present. The members grouped within subfamilies II and IV, represented segmental duplications (Figs 1 and 3). Thus, the expansion of chickpea ARF gene family may be attributed mainly due to segmental duplications, which is consistent with results found in other plants18, 30, 32.
Further, the approximate date of CaARF gene duplication events was estimated via determining Ks and Ka values along with Ka/Ks ratio (Table S3). Ka/Ks ratio of <1 suggests purifying selection, while Ka/Ks ratio >1 indicates possibility of positive selection33, 34. Further, Ka/Ks ratio gives insight into the selection pressure on amino acid substitutions. Usually, selection pressure suggests selective advantage for the altered amino acid residues in a protein and is essential for understanding functional residues and shift in protein function35. The tentative date for segmental gene duplications ranged from 46.7 Mya (Ks = 0.56) for the paralogous pair CaARF8.1/8.2 to 117.7 Mya (Ks = 1.42) for CaARF10.2/16.1. Ka/Ks ratio for all the CaARF paralogous pairs was less than 1, suggesting that they are under strong purifying selection. Among these, Ka/Ks ratio of three paralogous pairs (CaARF10.2/16.2, CaARF3.1/3.2 and CaARF4.1/4.2) was found to be more than 0.3, suggesting possibility of significant functional divergence after duplication. Five paralogous pairs (CaARF10.2/16.1, CaARF10.1/10.2, CaARF7.1/7.2, CaARF8.1/8.2 and CaARF10.1/16.1) showed Ka/Ks ratio less than 0.3, suggesting their functional conservation.
Differential expression and promoter analysis of CaARF genes
To investigate putative roles of CaARF genes, expression pattern of all CaARFs was studied in diverse organs/tissues representing vegetative and reproductive stages using RNA-seq data36, 37. Many of CaARFs exhibited a distinct tissue-specific expression pattern (Fig. 4). For example, CaARF1, 2, 9.1 and 9.2, belonging to group IA, exhibited higher transcript accumulation in root as compared to any other tissue, indicating that they may function in root development (Fig. 4a, Table S4). Interestingly, CaARF1 and 2 are putative orthologs of Arabidopsis, AtARF1 and 2 (group IA), respectively, that were also found to have higher expression in primary root tips38. CaARF16.3, 17.1 and 17.2, which belong to group IV showed higher expression specifically in FB1 stage of flower development (Fig. 4a, Table S4). AtARF8 is known to regulate flower maturation and fertilization in Arabidopsis23, 39. Interestingly, its putative orthologs in chickpea, CaARF8.1 and CaARF8.2, showed higher expression at later stages of flower development. (Fig. 4a, Table S4). CaARF8.1 and CaARF8.2 were found to be segmentally duplicated, suggesting that their subfunctionalization after duplication might have resulted into partitioning of their function in different aspects of reproductive development in chickpea. AtARF6 have been reported to be involved in flower maturation along with AtARF8 23, 39. Its ortholog, CaARF6.2, also exhibited higher expression at later stages of flower development (Fig. 4a, Table S4). AtARF3/ETTIN is known to integrate function of AGAMOUS (AG) and APETALA2 (AP2) in floral meristem determinacy40. Interestingly, its ortholog CaARF3.2 exhibited greater transcript accumulation in shoot apical meristem (SAM, Fig. 4). In addition, AtARF3/ETTIN interacts with KANADI proteins to form a functional complex required for leaf polarity41. Furthermore, AtARF5/MONOPTEROS has been reported to regulate flower formation21. Its putative orthologs in chickpea, CaARF5.1, 5.3, 5.4 and 5.5, showed enhanced expression at flower bud stages (Fig. 4a, Table S4). CaARF5.3 and CaARF5.5 were found to be expressed specifically but at very low levels at FB1 (FPKM = 0.02) and FB3 (FPKM = 1.62) stages, respectively. Likewise, CaARF6.2, 7.1, 8.2, 10.1, 16.1, 16.2, and 19, which clustered together (Fig. 4a, Table S4), showed increased transcript accumulation at different stages of flower development, suggesting their possible role during flower development. The expression profile of at least five CaARF genes (CaARF5.3, 5.5, 9.2, 10.1 and 16.3) was analyzed through RT-qPCR to validate the RNA-seq results. The expression patterns obtained via RT-qPCR were well correlated with that of RNA-seq (Fig. 4a,b).

Expression profiles of CaARF genes in different tissue/organs. (a) Heatmap showing the normalized RNA-seq expression (FPKM) transformed into Z-score. Hierarchical clustering was conducted in R using the pheatmap package with dissimilarity metrics based on Euclidean distance with complete linkage rule. Color key at the bottom represents row wise Z-score. (b) RT–qPCR analysis of CaARF genes in various tissue/stages of development. Expression of germinating seedling (GS) was taken as a reference to determine relative mRNA level in all other tissues for each gene. Error bars indicate standard error (SE) of mean. GS, germinating seedling; ML, mature leaf; YL, young leaf; SAM, shoot apical meristem; FB1–4, stages of flower bud; FL1–5, stages of flower; YP, young pod.
The temporal and spatial gene expression is influenced by the presence of different cis-regulatory elements in the promoters, where transcription factors can bind. We analyzed cis-regulatory elements in the promoter sequences of CaARF genes using PlantPAN 2.0 database42. Most of CaARF promoter sequences revealed the presence of canonical TATABOX 1 and CAATBOX 1 (Table S5). In addition, a variety of light signalling related elements, such as GBOXLERBCS, GT1GMSCAM4 and BOXIIPCCHS were found to be present in CaARF promoters (Table S5) indicating their possible role in crosstalk between auxin and light signalling pathway43. Further, many hormone-responsive elements, namely, ABREATRD22, TCA1MOTIF, T/GBOXATPIN2, GAREAT, WBOXATNPR1, GCCCORE, C2GMAUX28, D1GMAUX28 and D3GMAUX28 were also detected in their promoters (Table S5), suggesting that CaARFs are regulated by auxin and other hormones.
Differential expression of ARF genes under abiotic stress
Promoter sequences of CaARFs exhibited the presence of several abiotic stress response related elements, including dehydration-responsive element (DRECRTCOREAT), low temperature-responsive element (LTRE1HVBLT49), ABA-responsive element (ABREATCONSENSUS, ABREATRD22, ABRELATERD1) and C-repeat binding factor (CBFHV) (Table S5), indicating their role in abiotic stress responses as well. Therefore, to understand the role of CaARFs in abiotic stress responses, we analyzed the RNA-seq data for abiotic stress-treated chickpea tissues44. We detected four genes, including CaARF4.2, CaARF7.1, CaARF9.2 and CaARF16.2, which exhibited significant differential expression (fold change ≥ 2 and p-value ≤ 0.05) under different abiotic stresses (Fig. 5a, Table S6). For instance, CaARF4.2 was significantly up-regulated under salt and cold stresses in root (Fig. 5a, Table S6), suggesting its involvement in salinity and cold stress responses. CaARF9.2 and CaARF16.2 were significantly up-regulated during desiccation and cold stresses in root samples (Fig. 5a, Table S6). In shoot, CaARF7.1 was significantly up-regulated under desiccation (Fig. 5), suggesting its role in desiccation stress response. The differential expression of these CaARF genes under abiotic stress was analyzed via RT-qPCR as well (Fig. 5b). Although the expression profile of the CaARF genes in most of the samples could be validated via RT-qPCR, some differences between RNA-seq and RT-qPCR results were also observed (Fig. 5a,b). For instance, the differential expression of CaARF4.2, CaARF9.2 and CaARF16.2 in roots under cold stress revealed by RNA-seq, was not observed via RT-qPCR. Likewise, higher expression of CaARF9.2 was observed in roots under salinity stress via RT-qPCR, which was not obvious in the RNA-seq results. It would be interesting to validate the mechanistic roles of these CaARFs in abiotic stress responses in chickpea.

Expression profiles of CaARF genes under abiotic stress conditions. (a) Heatmap showing differential expression of CaARFs based on RNA-seq data. Color key at the bottom represents log2 fold change. (b) RT–qPCR analysis of CaARF genes under various stress treatments. Root and shoot control were taken as a reference to determine relative mRNA level under stress conditions. Error bars indicate standard error (SE) of mean. DS, desiccation; SS, salinity; CS, cold stress, CT, control.
Co-expression network of CaARF, CaIAA and CaGH3 genes
ARFs have been demonstrated to play a crucial role in plant development by interacting with other proteins especially with Aux/IAAs13, 14. ARF proteins activate or repress expression of auxin-responsive genes, which indicate the existence of a complex regulatory network between ARFs and other auxin-responsive genes. The middle region of ARF proteins is responsible for activation or repression depending on composition of amino acids45. The middle domain of repressor ARFs is enriched in proline, serine and threonine, whereas activators are enriched in glutamine45. Based on transactivation assay or middle domain prediction, five ARFs have been identified as activators (ARF5–ARF8 and ARF19) and rest as repressors except four truncated proteins (ARF3, ARF13, ARF17 and ARF23) in Arabidopsis10. Co-expression analysis is an important approach to explore potential function of genes. To understand the possible regulatory relationship of CaARFs with CaIAAs and CaGH3s, we performed co-expression network analysis using RNA-seq data36, 37 via Weighted Gene Co-expression Network Analysis (WGCNA)46 and detected highly interconnected modules of co-expressed genes (Fig. 6a,b). Normalized FPKM (fragments per kilobase of the transcript per million mapped reads) expression values in different tissues/organs36, 37 for CaARF, CaIAA and CaGH3 genes were used as input for construction of co-expression network. Pearson correlation coefficient (PCC) with β = 8 (soft threshold power) was used to calculate an adjacency matrix. We detected five modules containing different sets of CaARF, CaIAA and CaGH3 genes based on their coexpression (Fig. 6a,b). Among all, turquoise module was the largest containing 15 genes, followed by blue (10 genes), brown (9 genes), yellow (6 genes) and green (5 genes) modules (Fig. 6b). Since all the modules have different set of genes and expression patterns, it is understandable that different cellular/developmental events might be the result of specific auxin response arising from differential transcriptional activity of ARF, Aux/IAA and GH3 family proteins.

Co-expression network analysis of CaARF, CaIAA and CaGH3 genes. (a) Cluster dendrogram showing correlation (0 to 1) between the modules. A high degree of correlation between modules is indicated by dark red color. (b) Module tissue correlation. Each row corresponds to a module. The number of genes in each module is indicated on the left. Each column corresponds to specific tissue labeled below. The color of each cell at row-column intersection indicates the correlation coefficient from −1 to 1. A high degree of correlation between a specific module and the tissue-type is indicated by dark red.
It has also been shown that under high auxin concentration, ARFs form homodimers and activate auxin-responsive gene expression45. Several activator ARFs have been reported to interact with many Aux/IAA proteins in Arabidopsis and rice45, 47, 48. Co-expression analysis showed positive and negative correlation among the expression profiles of many of the CaARF, CaIAA and CaGH3 members within modules. For instance, in green module, expression of CaARF19 was found to be positively correlated with CaIAA7, CaIAA13, CaGH3-3 and CaGH3-4 (Fig. 7). Interaction between AtARF19 and several Aux/IAA proteins have been reported in Arabidopsis47. AtARF8 was found to negatively regulate free IAA level by controlling expression of GH3 genes in Arabidopsis49. The expression of AtGH3-5, DFL1/AtGH3-6 and AtGH3-17 was found to be decreased in arf8 loss-of-function mutants and increased in AtARF8 over expressing plants49, indicating that AtARF8 can activate GH3 gene expression. We hypothesize that CaARF19 can bind to the AuxRE elements residing in the promoter regions of CaIAA7, CaIAA13, CaGH3-3 and CaGH3-4 genes and induce their expression.

Co-expression modules and network of CaARF, CaIAA and CaGH3 genes. Left panel shows heatmap depicting the expression of all genes included in each module. Color range from blue (lowest expression) to yellow (highest expression) represent expression level. Right panel shows co-expression network of genes in each module. Network is visualized in ViSANT. Spheres (nodes) represent genes, and green and red color lines (edges) represent positive and negative correlation, respectively.
In brown module, CaARF7.1 and CaARF7.2 were positively correlated with CaIAAs and negatively correlated with other genes (CaGH3-6, 11, 12 and CaARF5.5), indicating that CaIAAs may form heterodimer with CaARF7.1 or CaARF7.2 for repressing the expression of downstream genes. In Arabidopsis also, several Aux/IAA proteins have been found interacting with AtARF7 too47. Similar correlations between CaIAAs, CaARFs and CaGH3s were observed for yellow and turquoise modules too (Fig. 7). Furthermore, CaARF1, 2, 9.2, 9.3 and 16.1, were negatively correlated with CaIAAs (CaIAA6 and 21) and CaGH3s (CaGH3-1, 2 and 8) in blue module, implying that higher expression of repressor CaARFs can inactivate downstream genes. It has been hypothesized that repressor ARFs can inhibit the activator ARF(s) via sequestering them by heterodimerization, with/without the binding of Aux/IAA protein(s) to the activator ARFs, or repressor ARFs might compete with activator ARFs for AuxRE binding sites, individually or as heterodimers, with or without the recruitment of other proteins such as TOPLESS50. A few CaARF proteins did not harbor one or the other of the three well-characterized functional domains (B3, ARF and CTD/PB1) as shown in Fig. 2. It will be interesting to elucidate the regulatory network of these CaARFs with different domain composition.
All the CaARF genes are yet to be functionally characterized. Via studying the expression patterns of CaARFs in diverse tissue types and construction of co-expression networks of CaARFs, we have created a framework for understanding the function of these uncharacterized genes in chickpea for future studies.
Prediction of putative target CaARF genes of miRNAs and tasiRNA
MicroRNAs (miRNAs) are 21-nucleotide long RNAs that play an important role in regulation of gene expression in plants and animals51,52,53. In Arabidopsis, expression levels of AtARF6 and AtARF8 were found to be regulated by miR167 54, whereas AtARF10, AtARF16 and AtARF17 were targeted by miR160 55. To study potential regulation of CaARF genes by miRNAs, we predicted target CaARF genes of the known miRNAs in chickpea56, 57. We found that CaARF6.1 and CaARF6.2, and CaARF8.1 were targeted by miR167 in chickpea also. In addition, CaARF10.1 and CaARF10.2, CaARF16.1, CaARF16.2 and CaARF16.3, and CaARF17.1 and CaARF17.2 were detected as targets of miR160. These results suggest that regulation of ARFs seems to be conserved in Arabidopsis and chickpea. Earlier reports also demonstrated conservation of miR160 and miR167–target interactions throughout plant evolution58,59,60,61. In addition, we found that CaARF3.2 was targeted by Cat-miR395h, whereas CaARF5.2, CaARF5.3 and CaARF5.5 were targeted by a novel miRNA, Cat-NovmiR4, identified in our previous study56. In Arabidopsis, 3′ cleavage products of AtARF10, AtARF16 and AtARF17 were detected in many tissues, indicating that miRNA160 regulates these ARFs post-transcriptionally62. Further, Mallory et al.62 documented that miR160 directed post-transcriptional regulation of AtARF17 is necessary for proper expression of certain GH3-like early auxin-responsive genes. The plants expressing a miRNA-resistant version of AtARF17 showed enhanced AtARF17 mRNA level and altered accumulation of auxin-responsive GH3 mRNAs encoding for auxin-conjugating proteins. These expression changes were found to be correlated with many developmental defects, like defects in embryo, leaf symmetry and leaf shape, premature inflorescence development, reduction in petal size, abnormal stamen, sterility, and root growth defects62.
Further, we investigated miR390-TAS3-ARF pathway in chickpea. In plants, miR390 slices TRANS-ACTING SIRNA 3 (TAS3) transcripts to produce tasiRNAs that regulate ARF genes. In Arabidopsis, TAS3-derived tasiARF targets are, AtARF2, AtARF3 and AtARF4 63. This miR390-TAS3-ARF pathway performs crucial function in regulation of plant growth and development, including lateral root growth, leaf morphogenesis, developmental timing and patterning64,65,66,67,68. For the identification of TAS3 locus in chickpea, we annotated genomic loci harbouring miR390 target sites and tasiRNAs that target ARF gene(s). We identified one genomic locus (TCONS_00009070), with two target sites of miR390 and two tasiRNAs in phased manner targeting CaARFs (CaARF3.1, 3.2, 4.1 and 4.2) (Fig. 8). Interestingly, TAS3-derived tasiRNA targets in chickpea, CaARF3.1, 3.2, 4.1 and 4.2, belonged to same phylogenetic clade as reported for Arabidopsis (AtARF3 and 4) too61. Taken together, these analyses suggested a role of small RNA pathways in regulation of CaARFs and biogenesis of tasiRNAs, known for regulation of their ARF targets.

Target sites of tasiRNA in CaARF genes. Diagram showing TAS3 locus with miRNA390 target sites (blue and purple boxes) and tasiARF biogenesis site (red box). tasiARF targets on CaARFs are shown by red lines.
In conclusion, we identified 28 chickpea ARF genes and established the classification and evolutionary relationship among these genes via phylogenetic tree, gene structure and conserved protein motif analyses. Expression analyses highlighted the involvement of CaARF genes in flower development and response to abiotic stress. Furthermore, co-expression network analysis indicated ARF-mediated regulation of Aux/IAA and GH3 genes and suggested their role in developmental processes. We revealed regulation of CaARF genes via identifying targets of miRNAs and tasiRNAs. The data presented here will provide solid foundation for future studies on the functional characterization of ARF genes and ARF-mediated signal transduction pathways in chickpea.
Methods
Identification of putative ARF genes in chickpea genome
Kabuli chickpea (Cicer arietinum L.) genome annotation was downloaded from Legume Information system (LIS; http://cicar.comparative-legumes.org/)69. For identification of ARF gene family members, BLASTP and HMM profile searches were employed. For BLASTP, 23 Arabidopsis ARF protein sequences were taken as query and searched in the chickpea proteome with e-value cut-off ≤1e−5. For HMM search, HMM profile of ARF domain (PF00025) was downloaded from Pfam database (http://pfam.sanger.ac.uk/search). The gene ids obtained from these two searches were combined to make a non-redundant list and their protein sequences were analyzed in Pfam and SMART databases for the presence of ARF domain. Properties of chickpea ARF proteins were analyzed using ExPASy server (http://web.expasy.org/compute_pi/).
Prediction of gene structure and motifs, and phylogenetic analysis
Exon/intron organization of the ARF genes was determined using Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/) using GFF file. The motifs of chickpea ARF protein sequences were analyzed via MEME programme70 (http://meme-suite.org/tools/meme) with motif length 6–100 and number of sites 2–120 with maximum number of motifs set to 15. For phylogenetic analysis, protein sequence of ARFs, from chickpea and Arabidopsis were aligned using MUSCLE multiple sequence alignment tool with default settings. Unrooted phylogenetic tree construction was done by Neighbour-Joining (NJ) method using MEGA (v7) software with following parameters: JTT model, pair-wise gap deletion and 100 bootstrap.
Localization of genes on chromosomes and gene duplication analysis
Information about location of chromosomes was obtained from the genome annotation GFF file. Tandem and segmental duplication of chickpea ARF genes were analyzed using MCScanX software. The Ka (nonsynonymous substitution rate) and Ks (synonymous substitution rate) were calculated using perl script add_ka_and_ks_to_synteny.pl from MCSanX. The Ks value was used to calculate the tentative date of duplication event (T = Ks/2λ) assuming clock-like substitution of 6.05 × 10−9 substitutions/synonymous site/year for chickpea. Mapping of genes and segmentally duplicated regions on the chickpea chromosomes was done using Circos tool71.
RNA-seq data for gene expression analysis of chickpea ARFs and RT-qPCR
To study gene expression profiles of chickpea ARFs, we used RNA-seq data from previous studies36, 37. RNA-seq data from 17 tissues/organs, including germinating seedling (GS), root (R), shoot (S), stem (ST), mature leaf (ML), young leaf (YL), shoot apical meristem (SAM), stages of flower bud (FB1-4; where FB1, FB2, FB3 and FB4 represent 4 mm, 6 mm, 8 mm and 8–10 mm size flower buds, respectively), stages of flower (FL1-5; where FL1 = young flower with closed petals, FL2 = flower with partially opened petals, FL3 = mature flower, FL4 = mature flower with opened and faded petal and FL5 = drooped flower with senescing petals) and young pod (YP), were analyzed as described earlier72. Expression analysis under abiotic stress was performed as described72, where root and shoot tissues were analyzed under control and stress (desiccation, salt and cold) conditions, using RNA-seq data as described44.
Various tissue samples used in RNA-seq analysis were collected as described36, 37. Total RNA was extracted from tissues using Tri Reagent (Sigma-Aldrich) followed by synthesis of cDNA using 3 µg of total RNA as described72. Primer pairs used in qPCR were designed using Primer Express software according to manufacturer’s guidelines (Applied Biosystem, USA) and are listed in Table S7. qPCR reactions for each tissue sample were performed in at least two biological replicates and three technical replicates for each biological replicate employing ABI 7500 system (Applied Biosystems) as described earlier73. Elongation factor-1 alpha (EF-1α) was used as endogenous control for normalization of transcript levels across the tissue samples.
Co-expression network construction
Weighted gene correlation network analysis (WGCNA)46 method was employed for constructing a co-expression network among CaARFs, CaIAAs 72 and CaGH3s. GH3 genes in the kabuli chickpea genome (CaGH3s) were identified as described previously74 (Table S8). WGCNA presents a systematic method for examining possible related genes acting in a common pathway, using gene expression data46. Firstly, adjacency matrix between CaARF, CaIAA and CaGH3 genes was calculated using expression data on the basis of Pearson correlation coefficient46. The following formula depicts how adjacent values between the two genes can be expressed46: aij = |cor(x i , x j ))|β, where a ij represents the adjacency value between gene i and gene j; cor(x i , x j ) is the Pearson correlation coefficient between gene i and j; β represents the weight value. TOM similarity algorithm was used to convert the adjacency matrix to a topological overlap (TO) matrix, which signifies gene correlation in a network. Hierarchical clustering was performed using dissimilarity matrix, the inverse matrix of TO value (1–TO), for representing genetic link network. The dynamic tree-cut algorithm was used for cutting the hierarchical clustered tree into branches corresponding to diverse modules46. Graphical representation of the network was done using VisANT75.
Promoter sequence analysis
Promoter sequences (1000 bp upstream from the start codon) of all ARF genes from chickpea were subjected to search in PlantPAN 2.0 database42 (http://plantpan2.itps.ncku.edu.tw/) for identification of cis-regulatory elements.
References
Woodward, A. W. & Bartel, B. Auxin, regulation, action, and interaction. Ann. Bot. 95, 707–735 (2005).
Kumar., R., Tyagi, A. K. & Sharma, A. K. Genome-wide analysis of auxin response factor (ARF) gene family from tomato and analysis of their role in flower and fruit development. Mol. Gen. Genet. 285, 245–260 (2011).
Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 61, 49–64 (2010).
Ljung, K. Auxin metabolism and homeostasis during plant development. Development. 140, 943–950 (2013).
Liu, Z. B., Ulmasov, T., Shi, X., Hagen, G. & Guilfoyle, T. J. Soybean GH3 promoter contains multiple auxin-inducible elements. Plant Cell 6, 645–657 (1994).
Ulmasov, T., Hagen, G. & Guilfoyle, T. J. ARF1, a transcription factor that binds to auxin response elements. Science 276, 1865–1868 (1997).
Hagen, G. & Guilfoyle, T. Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol. Biol. 49, 373–385 (2002).
Boer, D. R. et al. Structural basis for DNA binding specificity by the Auxin-dependent ARF transcription factors. Cell 156, 577–589 (2014).
O’Malley, R. C. et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 166, 1598 (2016).
Guilfoyle, T. J. & Hagen, G. Auxin response factors. Curr. Opin. Plant Biol. 10, 453–460 (2007).
Guilfoyle, T. J. & Hagen, G. Getting a grasp on domain III/IV responsible for auxin response factor–IAA protein interactions. Plant Sci. 190, 82–88 (2012).
Guilfoyle, T. J. The PB1 domain in auxin response factor and Aux/IAA proteins: a versatile protein interaction module in the auxin response. Plant Cell 27, 33–43 (2015).
Nanao, M. H. et al. Structural basis for oligomerization of auxin transcriptional regulators. Nat. Commun. 5, 3617 (2014).
Korasick, D. A. et al. Molecular basis for auxin response factor protein interaction and the control of auxin response repression. Proc. Natl. Acad. Sci. USA 111, 5427–5432 (2014).
Korasick, D. A. et al. Defining a two-pronged structural model for PB1 (Phox/Bem1p) domain interaction in plant auxin responses. J. Biol. Chem. 290, 12868–12878 (2015).
Liscum, E. & Reed, J. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 49, 387–400 (2002).
Wang, D. et al. Genome-wide analysis of the auxin response factor (ARF) gene family in rice (Oryza sativa). Gene 394, 13–24 (2007).
Shen, C. et al. Genome-wide identification and expression analysis of auxin response factor gene family in Medicago truncatula. Front. Plant. Sci. 6, 73 (2015).
Ellis, C. M. et al. AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development 132, 4563–4574 (2005).
Sessions, A. et al. ETTIN patterns the Arabidopsis floral meristem and reproductive organs. Development 124, 4481–4491 (1997).
Hardtke, C. S. & Berleth, T. The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J. 17, 1405–1411 (1998).
Harper, R. M. et al. The NPH4 locus encodes the auxin response factor ARF7, a conditional regulator of differential growth in aerial Arabidopsis tissue. Plant Cell 12, 757–770 (2000).
Nagpal, P. et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 132, 4107–4118 (2005).
Wilmoth, J. C. et al. NPH4/ARF7 and ARF19 promote leaf expansion and auxin-induced lateral root formation. Plant J. 43, 118–130 (2005).
Attia, K. A. et al. Antisense phenotypes reveal a functional expression of OsARF1, an auxin response factor, in transgenic rice. Curr. Issues Mol. Biol. 11, 29–34 (2009).
Shen, C. et al. OsARF16 is involved in cytokinin-mediated inhibition of phosphate transport and phosphate signaling in rice (Oryza sativa L.). PLoS One 9, e112906 (2014).
Shen, C. et al. OsARF16, a transcription factor, is required for auxin and phosphate starvation response in rice (Oryza sativa L.). Plant Cell Environ. 36, 607–620 (2013).
Haiyang, J., Wei, W., Yan, L., SuWen, Z. & BeiJiu, C. Genome-wide analysis and evolution of the auxin response factor (ARF) gene family in Sorghum bicolor. J. Anhui. AgricUniv. 37, 395–400 (2010).
Xing, H. et al. Genome-wide identification and expression profiling of auxin response factor (ARF) gene family in maize. BMC Genomics 12, 178 (2011).
Kalluri, U. C., Difazio, S. P., Brunner, A. M. & Tuskan, G. A. Genome-wide analysis of Aux/IAA and ARF gene families in Populus trichocarpa. BMC Plant Biol. 7, 59 (2007).
Remington, D. L., Vision, T. J., Guilfoyle, T. J. & Reed, J. W. Contrasting modes of diversification in the Aux/IAA and ARF gene families. Plant Physiol. 135, 1738–1752 (2004).
Le, B. et al. Genome-wide characterization and expression pattern of auxin response factor (ARF) gene family in soybean and common bean. Genes Genomics 38, 1165–1178.
Yang, Z. & Bielawski, J. P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503 (2000).
Hurst, L. D. The Ka/Ks ratio, diagnosing the form of sequence evolution. Trends Genet. 18, 486–487 (2002).
Morgan, C. C., Loughran, N. B., Walsh, T. A., Harrison, A. J. & O’Connell, M. J. Positive selection neighboring functionally essential sites and disease-implicated regions of mammalian reproductive proteins. BMC Evol. Biol. 10, 39 (2010).
Singh, V. K., Garg, R. & Jain, M. A global view of transcriptome dynamics during flower development in chickpea by deep sequencing. Plant Biotechnol. J. 11, 691–701 (2013).
Jain, M. et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 74, 715–729 (2013).
Rademacher, E. H. et al. A cellular expression map of the Arabidopsis AUXIN RESPONSE FACTOR gene family. Plant J. 68, 597–606 (2011).
Goetz, M., Vivian-Smith, A., Johnson, S. D. & Koltunow, A. M. AUXIN RESPONSE FACTOR8 is a negative regulator of fruit initiation in Arabidopsis. Plant Cell 18, 1873–1886 (2006).
Liu, X. et al. AUXIN RESPONSE FACTOR 3 integrates the functions of AGAMOUS and APETALA2 in floral meristem determinacy. Plant J. 80, 629–641 (2014).
Kelley, D. R., Arreola, A., Gallagher, T. L. & Gasser, C. S. ETTIN (ARF3) physically interacts with KANADI proteins to form a functional complex essential for integument development and polarity determination in Arabidopsis. Development 139, 1105–1109 (2012).
Chow, C. N. et al. PlantPAN 2.0: an update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants. Nucleic Acids Res. (D1) 44, D1154–D1160 (2016).
Jaillais, Y. & Chory, J. Unraveling the paradoxes of plant hormone signaling integration. Nat. Struct. Mol. Biol. 17, 642–645 (2010).
Garg, R., Bhattacharjee, A. & Jain, M. Genome-scale transcriptomic insights into molecular aspects of abiotic stress responses in chickpea. Plant Mol. Biol. Rep. 33, 388–400 (2015).
Tiwari, S. B., Hagen, G. & Guilfoyle, T. J. The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell 15, 533–543 (2003).
Langfelder, P. & Horvath, S. WGCNA, an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Vernoux, T. et al. The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol. Syst. Biol. 7, 508 (2011).
Piya, S., Shrestha, S. K., Binder, B., Stewart, C. N. Jr. & Hewezi, T. Protein–protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant Sci. 5, 744 (2014).
Tian, C. E. et al. Disruption and overexpression of auxin response factor 8 gene of Arabidopsis affect hypocotyl elongation and root growth habit, indicating its possible involvement in auxin homeostasis in light condition. Plant J. 40, 333–343 (2004).
Chandler, J. W. Auxin response factors. Plant Cell Environ. 39, 1014–1028 (2016).
Borges, F. & Martienssen, R. A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 16, 727–741 (2015).
Cao, D. et al. Regulations on growth and development in tomato cotyledon, flower and fruit via destruction of miR396 with short tandem target mimic. Plant Sci. 247, 1–12 (2016).
Li, C. & Zhang, B. H. MicroRNAs in control of plant development. J. Cell Physiol. 231, 303–313 (2016).
Wu, M.-F., Tian, Q. & Reed, J. W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133, 4211–4218 (2006).
Liu, P. P. et al. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 52, 133–146 (2007).
Jain, M., Chevala, V. V. & Garg, R. Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J. Exp. Bot. 65, 5945–5958 (2014).
Srivastava, S. et al. High throughput sequencing of small RNA component of leaves and inflorescence revealed conserved and novel miRNAs as well as phasiRNA loci in chickpea. Plant Sci. 235, 46–57 (2015).
Bartel, B. & Bartel, D. P. MicroRNAs, At the root of plant development? Plant Physiol. 132, 709–717 (2003).
Rhoades, M. et al. Prediction of plant microRNA targets. Cell 110, 513–520 (2002).
Axtell, M. J. & Bartel, D. P. Antiquity of microRNAs and their targets in land plants. Plant Cell 17, 1658–1673 (2005).
Finet, C., Berne-Dedieu, A., Scutt, C. P. & Marletaz, F. Evolution of the ARF gene family in land plants: old domains, new tricks. Mol. Biol. Evol. 30, 45–56 (2013).
Mallory, A. C., Bartel, D. P. & Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response Genes. Plant Cell 17, 1360–1375 (2005).
Allen, E., Xie, Z., Gustafson, A. M. & Carrington, J. C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221 (2005).
Adenot, X. et al. DRB4- dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr. Biol. 16, 927–932 (2006).
Garcia, D., Collier, S. A., Byrne, M. E. & Martienssen, R. A. Specification of leaf polarity in Arabidopsis via the trans-acting siRNA pathway. Curr. Biol. 16, 933–938 (2006).
Fahlgren, N. et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22, 1074–89 (2010).
Hunter, C. et al. Trans-acting siRNA-mediated repression of ETTIN and ARF4 regulates heteroblasty in Arabidopsis. Development 133, 2973–2981 (2006).
Marin, E. et al. miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 22, 1104–1117 (2010).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucl. Acids Res. 37, 202–208 (2009).
Krzywinski, M. et al. Circos, An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Singh, V. K. & Jain, M. Genome-wide survey and comprehensive expression profiling of Aux/IAA gene family in chickpea and soybean. Front. Plant Sci. 6, 918 (2015).
Garg, R., Sahoo, A., Tyagi, A. K. & Jain, M. Validation of internal control genes for quantitative gene expression studies in chickpea (Cicer arietinum L.). Biochem. Biophys. Res. Commun. 396, 283–288 (2010).
Singh, V. K., Jain, M. & Garg, R. Genome-wide analysis and expression profiling reveals diverse roles of GH3 gene family during development and abiotic stress responses in legumes. Front. Plant Sci. 5, 789 (2015).
Hu, Z., Snitkin, E. S. & DeLisi, C. VisANT: an integrative framework for networks in systems biology. Brief. Bioinform. 9, 317–325 (2008).
Acknowledgements
This work is financially supported by the Department of Biotechnology (DBT) Government of India under the Challenge Programme on Chickpea Functional Genomics (BT/AGR/CG-PhaseII/01/2014), core grant from the NIPGR and infrastructural facility by the Jawaharlal Nehru University, New Delhi. VKS acknowledges the receipt of research fellowship from the DBT.
Author information
Authors and Affiliations
Contributions
M.J. conceived and supervised the study. V.K.S. performed most of the analysis and drafted the MS. M.S.R. performed real-time PCR experiments. R.G. and M.J. participated in data interpretation and finalizing the M.S.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, V.K., Rajkumar, M.S., Garg, R. et al. Genome-wide identification and co-expression network analysis provide insights into the roles of auxin response factor gene family in chickpea. Sci Rep 7, 10895 (2017). https://doi.org/10.1038/s41598-017-11327-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11327-5
- Springer Nature Limited
This article is cited by
-
Elucidating the role of exogenous melatonin in mitigating alkaline stress in soybeans across different growth stages: a transcriptomic and metabolomic approach
BMC Plant Biology (2024)
-
MdARF8: An Auxin Response Factor Involved in Jasmonate Signaling Pathway in Malus domestica
Journal of Plant Growth Regulation (2023)
-
Genome-wide identification, in silico characterization and expression analysis of the RNA helicase gene family in chickpea (C. arietinum L.)
Scientific Reports (2022)
-
A comprehensive review of global alignment of multiple biological networks: background, applications and open issues
Network Modeling Analysis in Health Informatics and Bioinformatics (2022)
-
Dynamic changes of transcriptome of fifth-instar spodoptera litura larvae in response to insecticide
3 Biotech (2021)