
Abstract
Chronic kidney disease (CKD) is a global health problem with a genetic component. Genome-wide association studies have identified variants associated with specific CKD etiologies, but their genetic overlap has not been well studied. This study examined SNP associations across different CKD etiologies and CKD stages using data from 5,034 CKD patients of the German Chronic Kidney Disease study. In addition to confirming known associations, a systemic lupus erythematosus-associated risk variant at TNXB was also associated with CKD attributed to type 1 diabetes (p = 2.5 × 10− 7), a membranous nephropathy-associated variant at HLA-DQA1 was also associated with CKD attributed to systemic lupus erythematosus (p = 5.9 × 10− 6), and an IgA risk variant at HLA-DRB1 was associated with both CKD attributed to granulomatosis with polyangiitis (p = 2.0 × 10−4) and to type 1 diabetes (p = 4.6 × 10−11). Associations were independent of additional risk variants in the respective genetic regions. Evaluation of CKD stage showed a significant association of the UMOD risk variant, previously identified in population-based studies for association with kidney function, for advanced (stage ≥G3b) compared to early-stage CKD (≤stage G2). Shared genetic associations across CKD etiologies and stages highlight the role of the immune response in CKD. Association studies with detailed information on CKD etiology can reveal shared genetic risk variants.
Similar content being viewed by others


Introduction
The prevalence of chronic kidney disease (CKD) is high with >10% of the adult population affected in many countries1. Its genetic architecture is complex and incompletely understood. Genome-wide association studies (GWAS) have helped to gain insight into complex disease genetics2,3 by identifying single-nucleotide polymorphisms (SNPs) in >70 independent risk loci associated with the estimated glomerular filtration rate (eGFR), CKD disease risk and microalbuminuria (MA)4,5,6 as well as specific kidney diseases such as IgA7,8 or membranous nephropathy9 in case control studies. Because many of these specific kidney diseases are individually rare, only very few studies have collected sufficient numbers of patients with CKD attributed to various of these specific etiologies using one study design and protocol. Consequently, genetic risk variants identified in association with a specific etiology of CKD have so far not been examined for their association with CKD attributed to other etiologies. Capitalizing on data from the large German Chronic Kidney Disease (GCKD) study, we therefore aimed to systematically examine whether risk loci discovered for specific etiologies of CKD, especially for autoimmune conditions10, are associated with other CKD etiologies as well. Additionally, we aimed to examine whether risk loci discovered in the general population are also associated with advanced stages of CKD and with CKD in patients for whom the leading cause of disease was hypertension or diabetes, the most common causes of CKD.
Subjects and Methods
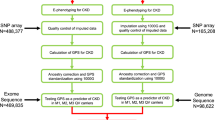
The GCKD study11,12 is an ongoing prospective observational study of 5,217 patients under nephrological care, followed for up to 10 years. At enrolment, all patients had CKD defined as an estimated glomerular filtration rate (eGFR) of 30–60 mL/min/1.73 m2 or either a urinary albumin-to-creatinine ratio (UACR) >300 mg/g or a protein-to-creatinine ratio >500 mg/g when eGFR was >60 mL/min/1.73 m2. The GCKD study was approved by local ethics committees and registered in the national registry of clinical studies (DRKS 0003971). All methods were carried out in accordance with relevant guidelines and regulations. Written informed consent was obtained from all subjects. Case groups for all analyses of specific CKD etiologies were derived based on the leading cause of CKD, which was determined by using standardized case report forms by the treating nephrologist. Serum creatinine was measured using an IDMS traceable gold-standard method. eGFR was calculated using the CKDepi equation13. Regardless of CKD etiology, two case groups of “advanced CKD” status were defined and included all patients with eGFR <45 ml/min/1.73 m² (stage G3b+ , n = 2245) or UACR ≥ 300 mg/g (stage A3, n = 1385), respectively. As controls, 1,655 GCKD patients for whom CKD etiology was assigned to a cause that could reasonably be assumed to differ from the case groups (nephrosclerosis, infections, tumor nephrectomies, interstitial nephritis and vascular diseases) were used as the control group for the etiology-specific analyses. For the examination of stage G3b+ and A3 CKD, GCKD patients with eGFR ≥ 45 ml/min/1.73 m² (n = 1006) and UACR < 30 mg/g (n = 2117) were selected as control groups, respectively.
In the GCKD study, 5,123 participants were genotyped for 2,612,357 markers at the Helmholtz Center Munich using the Illumina Infinium Omni 2.5 Exome-8 microarray (Illumina, GenomeStudio, Genotyping Module Version 1.9.4). Data cleaning was performed according to standard protocols14,15. Plink v1.90, R programming language and custom shell scripts were used during cleaning. Individual-level data were filtered for missingness (<3%) and mean heterozygosity (>2 SD). Sex checks were performed. These checks resulted in 57 individuals being filtered. Identity-by-descent (IBD) allele sharing measure calculations were used to check for unrecognized and cryptic relatedness. Across 1.28 × 107 evaluated pairs, we detected 11 with the proportion of alleles shared IBD of >0.1875 (between second and third degree relatives). For these pairs, we removed one individual prior to data analysis. Principal component analyses (PCA) using the software Eigenstrat SmartPCA16 were conducted to examine and account for population stratification. Outliers were removed in terms of genetic ancestry (automatic outlier detection, taking into account 10 PCs, deviation >8 SD). SNPs were filtered for callrate (>96%), minor allele frequency (>1%) and for deviation from the Hardy-Weinberg equilibrium (p > 1 × 10−5). Genotype imputation using the 1000 Genomes Phase 3 ALL reference panel17 was conducted according to standard protocols14,15. Imputation quality was assessed using the Impute2 “info” measure and was >0.8 for all SNPs. The final dataset after QC contained 5,034 individuals with genotypes for 2,337,794 SNPs.
A literature search was performed to assemble a table of all previously reported SNPs that were associated with either the kidney function measures eGFR and UACR, or with general or etiology-specific CKD risk in populations of European ancestry at genome-wide significance (p < 5 × 10−8) and with evidence for replication3.
Descriptive statistics were derived as percentages and frequency distributions for all categorical variables and mean and standard deviation or median and interquartile ranges for continuous variables depending on their distribution.
Thresholds of statistical significance were defined for each analysis using a Bonferroni correction to account for multiple testing and set to p < 0.05/n for two-sided hypothesis testing, with n as the number of tested SNPs multiplied by the number of phenotypes. For the analyses comparing etiologies, we used α = 0.05/(38*5) = 2.6 × 10−4, for the analyses comparing hypertensive and diabetic nephropathy, we used α = 0.05/(55*2) = 4.5 × 10−4. Power calculations have been performed using the software package Quanto18.
Multivariable adjusted logistic regression analyses was used to evaluate the association between CKD etiology and genotype dosage assuming an additive genetic model, and accounted for sex and age as covariates. Conditional analyses accounted for further SNPs in addition. We did not adjust the logistic models for principal components as the genetic ancestry of the study population was very homogenous because of the study design and data cleaning. Sensitivity analyses adjusting for prinicipal components were carried out to verify that results were robust. STATA 13.0 (StataCorp., College Station, TX) was used to perform all analyses.
Results
Association studies were performed using different case definitions as the outcome and genotype data from 38 + 55 = 93 selected SNPs3 as the exposure (Subjects and Methods). All GCKD patients were of European ancestry, 60% of them were male. Mean age at study entrance was 60 ± 12 years, and mean eGFR was 49.5 ± 18.1 ml/min/1.73 m² (Table 1).
We tested 38 known etiology-specific risk loci for association with additional CKD etiologies across the broad spectrum of CKD etiologies available in the GCKD study: IgA nephropathy (n = 366), membranous nephropathy (MN, n = 147), systemic lupus erythematosus (SLE, n = 128), granulomatosis with polyangiitis (GPA, n = 116) and type 1 diabetes mellitus (T1DM, n = 91; Supplementary Table 1). The proportion of patients with a renal biopsy supporting the diagnosis was 85% (641/757) for these case groups except for T1DM (Table 1). The respective case groups were compared to a control group, which consisted of n = 1655 patients for whom CKD etiology was assigned to a cause which could reasonably be assumed to differ from the case groups (see Subject and Methods). Several known associations7,9,10,19,20,21 were replicated: both HLA-DQA1 and PLA2R1 were strongly associated with MN (p = 2.4 × 10−22 and p = 6.7 × 10−8, respectively), STAT4 was associated with SLE (p = 9.7 × 10−5) and HLA-DPB1 with GPA (p = 1.7 × 10−11, Table 2, Supplementary Table 2), supporting an appropriate selection of the control group. These associations had consistent directions with previously reported associations. Only 4 of 38 (11%) previously reported etiology-specific risk loci showed significant associations after applying the Bonferroni threshold, but many of them had been found and reported from meta-analyses assembling a much larger number of cases. For 21 variants previously reported as associated with IgA nephropathy, 15 displayed associations in the same direction (i.e. the same risk allele) in our data, which is significantly more than expected by chance (p-binomial for observing 15 or more direction-consistent associations = 0.021). Of the 21 variants, 6 were nominally significant (p < 0.05). The effect sizes were similar on average (median effect size difference 0%, inter-quartile range −9% to 6%).
Interestingly, several SNPs previously reported as associated with one specific CKD-etiology were associated with one or more additional etiologies of CKD. While PLA2R1 and STAT4 were exclusively associated with the previously reported entities MN and SLE, respectively, TNXB and several genes in the HLA region were shared across several CKD etiologies (Table 2, Supplementary Table 3). For instance, the known SLE risk variant rs1150754 at TNXB was associated not only with CKD attributed to T1DM (OR = 2.53, p = 2.5 × 10−7), but also with MN (OR = 2.77, p = 1.9 × 10−11).
In order to investigate whether these newly emerging associations represented new findings or emerged because of co-occurrence with previously reported variants for that CKD etiology on shared haplotypes in the HLA region, conditional analyses were performed and linkage disequilibrium calculations were carried out (Table 3, Supplementary Tables 4 and 5). For MN, there were no new independently associated SNPs in the region beyond the original MN risk SNP rs2187668 at HLA-DQA1, suggesting that new associations between HLA risk variants for other CKD etiologies and MN were observed because of linkage disequilibrium with a known MN risk variant. Conversely, the association between CKD attributed to T1DM and rs1150754 was independent of previously reported risk variants for other CKD etiologies in the HLA-region (OR = 2.62, p-conditional = 1.0 × 10−6). In addition, the IgA risk locus at HLA-DRB1 was associated independently with GPA conditioned on the known GPA risk variant in HLA-DPB1 (p = 2.0 × 10−4, Table 3). Furthermore, TNXB (previously reported for SLE, p = 2.5 × 10−7 in GCKD) and HLA-DRB1 (IgA risk locus, p = 4.6 × 10−11 in GCKD) showed independent associations with CKD attributed to T1DM. These associations remained robust in effect size and direction when adjusting for additional previously reported risk variants for T1DM22,23,24,25,26,27 in the HLA region (Supplementary Table 6). LD calculations were consistent with the conditional analyses, with low LD observed between variants for which the effect was not attenuated in conditional analyses (Supplementary Table 4).
Next, two different categories of advanced CKD were examined for 55 SNPs: the first was defined as eGFR < 45 ml/min/1.73 m² (n = 2,245, CKD stage G3b) and the second as UACR ≥ 300 mg/g (n = 1,385, CKD stage A3). These cases were compared to a GCKD control group of 2,245 patients with eGFR > 60 ml/min/1.73 m² and 2,117 patients with UACR < 30 mg/g, respectively. Of the risk loci discovered in the general population, UMOD showed significant association (OR = 0.76 per T allele, p < 4.2 × 10−4) with CKD stage 3b (Supplementary Table 7) compared to the GCKD control group, after correction for multiple testing. The effect direction was consistent with observations from population-based studies, with the minor T allele associated with better eGFR and lower CKD risk.
In a second set of analyses, the association between the 55 population-based risk loci and CKD for which the treating nephrologists had determined diabetes (n = 653) or hypertension (n = 1086) as the leading cause were examined because they represent the majority of CKD cases in population-based studies. No significant association was detected for both hypertensive nephropathy and diabetic kidney disease (Supplementary Table 8).
Discussion
This study found genetic associations shared across specific etiologies of CKD. Several known CKD etiology-specific risk loci were replicated in the GCKD study, and risk variants at TNXB, HLA-DQA1, -DQB1 and -DRB1 were independently associated with additional CKD etiologies beyond the ones initially reported.
Based on post-hoc power calculations, our study had excellent power (>99%) to detect some true-positive associations such as the ones between MN nephropathy and the validated risk alleles at HLA-DQA1 and PLA2R1, as well as for some of the new and independent associations with additional disease entities such as the IgA variant rs660895 and T1DM nephropathy (99%) or the MN risk variant rs2187668 and an association with SLE nephropathy (90%). Nevertheless, power was moderate for other combinations, such as the association between the IgA risk variant rs660895 and GPA (50%). A priori power calculations were complicated by the fact that it is unclear whether the effect size of a genetic risk variant is the same or similar for different CKD etiologies. Power calculations were therefore provided across a range of case numbers, allele frequencies and effect sizes (Supplementary Table 9) to assess minimum detectable effect sizes. For example, for a case group of 100 patients, there was >80% power to detect an association signal for a SNP with a frequency of 30% and an OR of 2.0. We therefore cannot exclude that there are additional shared genetic risk variants that will only become apparent in future, larger studies that have assembled cases with CKD from different etiologies.
The HLA region is known for containing various risk loci for CKD of different etiologies. Several of them could be examined across CKD etiologies available in the GCKD study, and showed evidence of novel, additional associations that had not been identified at genome-wide significance in genome-wide association studies of these additional etiologies. The associations across CKD etiologies highlight the shared role of the adaptive immune response, and suggest some overlap between etiologies. For example, known risk variants for MN were independently associated with CKD attributed to SLE, which is interesting in light of the histopathological appearance of membranous nephropathy in lupus nephritis class V.
The shared genetic risk variant for CKD attributed to SLE and to T1DM is supported by a report that examined the co-existence of auto-immune diseases28. In this report, SLE and T1DM co-exist more often than expected based on the prevalence of both diseases. Several case reports describe different overlapping auto-immune diseases that affect the kidney, such as MN and IgA nephropathy29 or MN and further extra-renal autoimmune diseases such as colitis ulcerosa)30. More detailed and better powered studies focusing on SNP associations across sub-types of different autoimmune diseases are required to address these important questions in more detail. Furthermore, studies with larger case groups of the respective disease could examine co-incidences of two diseases with overlapping genetic risk factors such as MN/SLE and T1DM/GPA.
The T1DM risk loci found in our study can be interpreted in two ways: either as risk loci for T1DM itself, or as risk loci for diabetic nephropathy resulting from T1DM, or a combination of both. To resolve this question, a control group of T1DM patients without CKD would be required.
We additionally found that the UMOD locus known to be associated with kidney function in the general population was also associated with advanced CKD (stage G3b). Experimental evidence links genotype at the UMOD risk variant identified in GWAS to altered gene expression and salt-sensitive hypertensive CKD31. In support, the risk allele at the UMOD variant was significantly associated with stage G3b + CKD in our study, and the association with CKD attributed to hypertension was direction-consistent to the UMOD variant reported in previous GWAS of hypertension32. This finding illustrates associations across a spectrum of renal function from small changes in eGFR in population-based studies over advanced CKD in the GCKD study to severe renal phenotypes in monogenic diseases caused by loss of function mutations in UMOD 33.
Strengths of our study include the availability of CKD of different stages and from different etiologies in one study using the same standardized recruitment procedures, and the availability of genome-wide genotype data that allows for carrying out conditional analyses. Other CKD cohorts do not have access to comparable numbers of carefully phenotyped subgroups of patients with as many specific CKD etiologies. Nevertheless, because of limited sample size within subgroups, analyses in our study were restricted to the examination of a predefined number of candidate SNPs and the study of large and moderate genetic effects, rigorously accounting for multiple comparisons. Limitations include the absence of an internal control group of patients without CKD who suffer from the specific diseases that can give risk to CKD, such as T1DM patients without nephropathy.
In conclusion, genetic risk variants for specific etiologies of CKD were shared across some etiologies, suggesting a common mechanism by which the adaptive immune system may contribute to the shared etiologies. In addition, we found the known risk variant at the UMOD locus that is associated with CKD in the general population to also associate with advanced stage CKD (G3b+) in the GCKD study, supporting the presence of genetic risk across the spectrum of kidney function.
References
Levey, A. S. & Coresh, J. Chronic kidney disease. Lancet 379, 165–80 (2012).
Visscher, P. M., Brown, M. A., McCarthy, M. I. & Yang, J. Five Years of GWAS Discovery. The American Journal of Human Genetics 90, 7–24 (2012).
Wuttke, M. & Kottgen, A. Insights into kidney diseases from genome-wide association studies. Nat Rev Nephrol 12, 549–62 (2016).
Böger, C. A. CUBN is a gene locus for albuminuria. J Am Soc Nephrol 22, 555–570 (2011).
Köttgen, A. et al. New loci associated with kidney function and chronic kidney disease. Nature Genetics 42, 376–384 (2010).
Pattaro, C. Genetic Associations at 53 Loci Highlight Cell Types and Biologic Pathways for Kidney Function. Nat Commun 7, 10023 (2015).
Kiryluk, K. et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nature Genetics 46, 1187–1196 (2014).
Li, M. et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nature Communications 6, 7270 (2015).
Stanescu. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364, 616–26 (2011).
Sekula. Genetic risk variants for membranous nephropathy: extension and association with other chronic kidney disease aetiologies. Nephrology Dialysis Transplantation, in press (2016).
Eckardt, K. U. & Barthlein, B. et al. The German Chronic Kidney Disease (GCKD) study: design and methods. Nephrol Dial Transplant 27(4), 1454–1460 (2012).
Titze, S. et al. Disease burden and risk profile in referred patients with moderate chronic kidney disease: composition of the German Chronic Kidney Disease (GCKD) cohort. Nephrol Dial Transplant 30, 441–51 (2015).
KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease.
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat Protoc 5, 1564–73 (2010).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat Rev Genet 11, 499–511 (2010).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–9 (2006).
The 1000 Genomes Project Consortium. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Morrison, J. G. W. Quantov1.2. 4; http://biostats.usc.edu/Quanto.html (2009).
de Bakker, P. I. W. et al. Differential Genetic Associations for Systemic Lupus Erythematosus Based on Anti–dsDNA Autoantibody Production. PLoS Genetics 7, e1001323 (2011).
Gharavi, A. G. et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nature Genetics 43, 321–327 (2011).
Xie, G. et al. Association of Granulomatosis With Polyangiitis (Wegener’s) WithHLA-DPB1*04 and SEMA6A Gene Variants: Evidence From Genome-Wide Analysis. Arthritis & Rheumatism 65, 2457–2468 (2013).
Barrett, J. C. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41, 703–7 (2009).
Bradfield, J. P. et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet 7, e1002293 (2011).
Cooper, J. D. et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet 40, 1399–401 (2008).
Grant, S. F. et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes 58, 290–5 (2009).
Hakonarson, H. et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448, 591–4 (2007).
Tomer, Y. et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. J Autoimmun 60, 32–9 (2015).
Kota, S. K., Meher, L. K., Jammula, S. & Modi, K. D. Clinical profile of coexisting conditions in type 1 diabetes mellitus patients. Diabetes Metab Syndr 6, 70–6 (2012).
Nishida, M., Kato, R. & Hamaoka, K. Coexisting Membranous Nephropathy and IgA Nephropathy. Fetal Pediatr Pathol 34, 351–4 (2015).
Warling, O. et al. Overlap syndrome consisting of PSC-AIH with concomitant presence of a membranous glomerulonephritis and ulcerative colitis. World J Gastroenterol 20, 4811–6 (2014).
Trudu, M. et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19, 1655–60 (2013).
Padmanabhan, S. et al. Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet 6, e1001177 (2010).
Hart, T. C. et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 39, 882–92 (2002).
Acknowledgements
The GCKD study is funded by grants from the German Ministry of Education and Research (BMBF, grant number 01ER0804) and the KfH Foundation for Preventive Medicine. We are grateful for the willingness of the patients to participate in the GCKD study. The enormous effort of the study personnel of the various regional centres is highly appreciated. We thank the large number of nephrologists who provide routine care for the patients and collaborate with the GCKD study. A list of nephrologists currently collaborating with the GCKD study is available at http://www.gckd.org. The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding programme Open Access Publishing. GCKD Investigators are as follows.University of Erlangen-Nürnberg, Germany: Kai-Uwe Eckardt, Stephanie Titze, Hans-Ulrich Prokosch, Barbara Bärthlein, André Reis, Arif B. Ekici, Olaf Gefeller, Karl F. Hilgers, Silvia Hübner, Susanne Avendaño, Dinah Becker-Grosspitsch, Nina Hauck, Susanne A. Seuchter, Birgit Hausknecht, Marion Rittmeier, Anke Weigel, Andreas Beck, Thomas Ganslandt, Sabine Knispel, Thomas Dressel and Martina Malzer. Technical University of Aachen, Germany: Jürgen Floege, Frank Eitner, Georg Schlieper, Katharina Findeisen, Elfriede Arweiler, Sabine Ernst, Mario Unger and Stefan Lipski. Charité, Humboldt-University of Berlin, Germany: Elke Schaeffner, Seema Baid-Agrawal, Kerstin Petzold and Ralf Schindler. University of Freiburg, Germany: Anna Köttgen, Ulla T. Schultheiss, Simone Meder, Erna Mitsch, Ursula Reinhard and Gerd Walz. Hannover Medical School, Germany: Hermann Haller, Johan Lorenzen, Jan T. Kielstein and Petra Otto. University of Heidelberg, Germany: Claudia Sommerer, Claudia Föllinger and Martin Zeier. University of Jena, Germany: Gunter Wolf, Martin Busch, Katharina Paul and Lisett Dittrich. Ludwig-Maximilians University of München, Germany: Thomas Sitter, Robert Hilge and Claudia Blank. University of Würzburg, Germany: Christoph Wanner, Vera Krane, Daniel Schmiedeke, Sebastian Toncar, Daniela Cavitt, Karina Schönowsky and Antje Börner-Klein. Medical University of Innsbruck, Austria: Florian Kronenberg, Julia Raschenberger, Barbara Kollerits, Lukas Forer, Sebastian Schönherr and Hansi Weißensteiner. University of Regensburg, Germany: Peter Oefner, Wolfram Gronwald and Helena Zacharias. Department of Medical Biometry, Informatics and Epidemiology (IMBIE), University of Bonn: Matthias Schmid. The work of MW and AK was funded by the CRC 1140 Initiative and by KO 3598/3-1 (AK) of the German Research Foundation. UTS and MW were supported by the Else Kröner-Fresenius-Stiftung (2013_Kolleg.03), Bad Homburg, Germany. Genotyping was funded by Bayer Pharma AG. The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding programme Open Access Publishing.
Author information
Authors and Affiliations
Contributions
S.W., A.K. and M.W. designed this study. U.S., F.K., K.U.E. and A.K. were involved in the management of the GCKD study. B.H. and A.B.E. were involved with biobanking and/or genotyping. S.W. and M.W. developed statistical methods and performed the analyses. S.W., M.W. and A.K. interpreted the results. S.W., A.K. and M.W. drafted the manuscript. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wunnenburger, S., Schultheiss, U.T., Walz, G. et al. Associations between genetic risk variants for kidney diseases and kidney disease etiology. Sci Rep 7, 13944 (2017). https://doi.org/10.1038/s41598-017-13356-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13356-6
- Springer Nature Limited
This article is cited by
-
Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy
Nature Genetics (2023)
-
Membranous nephropathy: new pathogenic mechanisms and their clinical implications
Nature Reviews Nephrology (2022)
-
Impact of early-life diet on long-term renal health
Molecular and Cellular Pediatrics (2020)
-
The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis
Nature Communications (2020)
-
Gene-diet interactions associated with complex trait variation in an advanced intercross outbred mouse line
Nature Communications (2019)