
Abstract
The gastrointestinal (GI) microbiota acts a natural barrier to the proliferation of opportunistic pathogens. Candida glabrata is an opportunistic yeast pathogen that has adapted to colonize all segments of the human GI tract. We observed an increase in Escherichia coli, Enterococcus faecalis, and Bacteroides vulgatus populations, and a decrease in Lactobacillus johnsonii, Bacteroides thetaiotaomicron, and Bifidobacterium animalis in mice with DSS-induced colitis. This reduction was more pronounced for L. johnsonii during C. glabrata overgrowth. In addition, C. glabrata overgrowth increased mouse mortality and inflammatory parameters, and modulated the expression of intestinal receptors and signaling pathways. The C. glabrata cell wall underwent various changes during the course of C. glabrata colonization, and showed a significant increase in chitin. C. glabrata deficient in chitin synthase-3 induced fewer inflammatory parameters than the parental strain during intestinal inflammation. Oral administration of chitin attenuated the impact of colitis, and reduced the number of aerobic bacteria and C. glabrata overgrowth, while chitinase-3-like protein-1 increased. This study provides evidence that inflammation of the gut alters the microbial balance and leads to C. glabrata cell wall remodeling through an increase in chitin, which is involved in promoting persistence of C. glabrata in the gut.
Similar content being viewed by others


Introduction
The gastrointestinal (GI) microbiota acts a natural barrier to colonization and proliferation of opportunistic pathogens, decreasing the risk of intestinal infection and disease. Deregulation of the dynamic crosstalk between the microbiota, intestinal epithelial cells and immune cells is critically involved in the development of inflammatory bowel disease (IBD). IBD is a chronic inflammatory disease of the GI tract, which includes Crohn’s disease (CD) and ulcerative colitis (UC)1. CD and UC are distinguishable by the location of the inflammation and by the pattern of histologic alterations in the GI tract.
A recent study has revealed that CD patients have significantly higher quantities of fungal species than healthy subjects, and that this is positively correlated with high levels of anti-Saccharomyces cerevisiae antibodies (ASCA)2. Animal models have played a significant role in increasing our understanding of IBD pathogenesis, especially models of murine colitis3. Experimental studies have shown that either Candida albicans or C. glabrata aggravate intestinal inflammation induced by dextran sulfate sodium (DSS) in mice, and, conversely, that DSS colitis promotes fungal colonization4,5.
Like C. albicans, C. glabrata is an opportunistic fungal pathogen commonly found in the human GI tract. C. glabrata is a particular problem in immunocompromized patients where it can disseminate from the GI tract to cause invasive candidiasis (IC)6,7, which is associated with high rates of morbidity and mortality8,9.
The fungal cell wall is the predominant site of interaction between the fungus and its host. This cell wall consists of a complex structure of polysaccharides, proteins, and lipids7, but its composition is dynamic, responding to changes in the local environment7,10. Expansion of the fungal wall during growth involves permanent remodeling of the cell wall polysaccharide network, which is comprised of three major types of polysaccharide: mannans, β-glucans, and chitin. Chitin is a homopolymer of β1,4-N-acetylglucosamine (GlcNAc) and is essential for biological functions in fungi, including cell division11, forming the primary septum of all septa, hyphal growth12, and virulence13,14. Chitin synthesis in C. glabrata is carried out by chitin synthases15. Deregulation of chitin biosynthesis is a potential mechanism of virulence and resistance to antifungal treatments.
Chitin has been reported to have anti-ulcer16, anti-tumor17, and anti-inflammatory18 properties. Chitin is recognized by different receptors, triggering an immune response. Previous investigations have shown that NOD-2 and TLR-9 recognize chitin and act together to mediate an anti-inflammatory response via secretion of the cytokine, interleukin (IL)-1019.
In the present study, we investigated the impact of C. glabrata colonization on the gut microbiota diversity in a DSS-induced colitis model, and assessed how the C. glabrata cell wall is remodeled in order to persist in the gut environment. We also analyzed the effect of chitin deficiency on C. glabrata-host interactions in the DSS mouse model.
Results
Measurement of inflammatory parameters
Mice were administered an oral dose of C. glabrata WT and were exposed to DSS treatment for 2 weeks in order to induce acute colitis. Mice given C. glabrata WT only showed no signs of inflammation or mortality (Supplementary data Fig. 1 and Table 1). In contrast, mice given DSS or C. glabrata WT-DSS showed, from day 6, a decrease in body weight and mortality rates of 10% and 20%, respectively. The clinical score for inflammation was significantly higher in C. glabrata WT-DSS mice than in the DSS group, starting gradually from day 6, in which the first clinical symptoms of inflammation appeared, including diarrhea and bloody stools (Supplementary data Fig. 1B). The histologic score, which was based on the degree of inflammatory cell infiltration and the amount of tissue damage, was significantly higher in C. glabrata WT-DSS mice than in DSS-treated mice (Supplementary data Fig. 1C). Epithelial damage was observed along all parts of the colon mucosa, and leukocyte infiltrates, crypt abscesses and mucosal edema were more common in the colons from C. glabrata WT-DSS mice than in those from mice treated with DSS only (Supplementary data Fig. 1D).
Impact of C. glabrata colonization on the gut microbiota
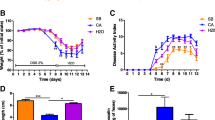
The number of C. glabrata CFUs and the changes in microbiota diversity were determined in freshly collected stool samples from tagged mice, using traditional culture methods based on agar plates (Fig. 1). A high number of C. glabrata CFUs was observed in both C. glabrata WT and C. glabrata WT-DSS groups on day 1 (Fig. 1A). In the absence of DSS, C. glabrata was eliminated from the mice from day 2. In contrast, the number of C. glabrata CFUs increased from day 2 in the C. glabrata WT-DSS group, and remained significantly higher than that in the C. glabrata WT group up to day 14 (P < 0.05).

C. glabrata colonization in mouse DSS-induced colitis. (A) Number of C. glabrata colony forming units (CFU) recovered from stools. Data are the mean ± SD of 16 mice per group. (B) Number of C. glabrata CFU recovered from the stomach, cecum, and colon. Data are the mean ± SD of 20 mice per group (P < 0.001). (C) Number of C. glabrata CFU recovered from the liver and kidneys. Data are the mean ± SD of 16 mice per group (P < 0.001).
To assess the extent of C. glabrata colonization in the GI tract, we assessed the number of yeasts adhering to the stomach, caecum, and colon (Fig. 1B). Significantly higher numbers of viable C. glabrata were detected in the stomach, caecum, and colon of C. glabrata WT-DSS mice than in mice colonized with C. glabrata WT (P < 0.001).
DSS-induced colitis promoted C. glabrata dissemination to the liver and kidneys (Fig. 1C). Regarding the microbiota diversity, cultures confirmed that the number of E. coli and E. faecalis colonies increased significantly from day 6 to day 14 in both the DSS and C. glabrata WT-DSS groups when compared to the C. glabrata WT or control groups, suggesting that irrespective of C. glabrata colonization, DSS-induced colitis promotes an increase in E. coli and E. faecalis populations in mice (Fig. 2A,C). In contrast to Bacteroides vulgatus populations, the number of B. thetaiotaomicron and Bacteroides sp. TP5 decreased significantly in both DSS and C. glabrata WT-DSS mice (Fig. 2B,D and E). In terms of anaerobic bacteria, the number of Bifidobacterium spp., in particular Bifidobacterium animalis, and L. johnsonii colonies was significantly reduced in both DSS and C. glabrata WT-DSS mice, but the reduction in the L. johnsonii population was significantly more pronounced in the C. glabrata WT-DSS group, suggesting that overgrowth of C. glabrata impacts on the L. johnsonii population during the development of colitis (Fig. 2F and G). In contrast, the Lactobacillus reuteri population revealed unpredictable changes and significant fluctuations over the course of DSS-induced colitis suggesting no association between C. glabrata-induced intestinal inflammation and L. reuteri levels (Fig. 2H).

Measurement of viable fecal microorganisms in DSS-induced colitis. The four groups consisted of controls (water), C. glabrata alone (Cg), DSS alone (D), and C. glabrata + DSS (CgD). Data are the mean ± SD of 16 mice per group. For all experiments, stool bacteria were isolated from mice on day 0 before C. glabrata challenge and DSS treatment. (A–H) Enumeration of E. coli, B. vulgatus, E. faecalis, Bacteroides spp. TP-5, B. thetaiotaomicron, L. johnsonii, Bifidobacterium spp., and L. reuteri CFUs in stool samples. Data are the mean ± SD of 16 mice per group (**P < 0.001, *P < 0.05).
Analysis of cytokines, signaling pathways, and receptor expression
To understand the mechanism by which changes in the microbiota modulate the inflammatory mediator responses during colitis and colonization with C. glabrata, pro-inflammatory cytokine expression (including IL-1β, and IL-6) was measured in the mice colons. Expression of IL-1β and IL-6 was significantly higher in the colons of C. glabrata WT-DSS mice than in DSS mice (Fig. 3A–D). Conversely, expression of these cytokines was significantly lower in the colons of C. glabrata WT mice and control groups. In terms of PPARγ and Myd88 expression, there were no significant differences between DSS and C. glabrata WT-DSS groups (Fig. 3E,G). To determine the activation/expression of receptors in response to C. glabrata sensing and colitis, the expression levels of TLR-4, TLR-9, and MBL-C were examined. DSS-induced colitis significantly increased TLR-4, TLR-9, and MBL-C expression in colon tissues. TLR-9 and MBL-C expression increased significantly in response to both C. glabrata and colitis, when compared to mice treated with DSS only (Fig. 3H–J), although TLR-4 expression decreased.

Cytokine and receptor expression in C. glabrata wild-type and DSS-induced colitis. (A and B) Relative expression levels of IL-1β, and IL-6 mRNA in mouse colons. (C and D) Protein levels of IL-1β and IL-6 in mouse colons. (F–J) Relative expression levels of Myd88, NF-κB, PPARγ, MBL-C, TLR-4, and TLR-9 mRNA in mouse colons. Data are the mean ± SD of 16 mice per group (*P < 0.05).
Remodeling of the C. glabrata cell wall after passage through the GI tract
To assess whether C. glabrata undergoes cell wall remodeling during fungal colonization in the DSS-induced colitis model, we analyzed the cell wall fitness in terms of α-mannans (α-mans), β-mans, and chitin in the stools of tagged mice over a 2 week period. This analysis was carried out using flow cytometry by measuring the median fluorescence intensity (MFI) (Fig. 4A,B and E).

Flow cytometry analysis of the expression of C. glabrata cell wall surface glycans after passage through the digestive tract. The expression of cell wall surface glycans was determined in C. glabrata using mAbs 5B2 and WGA, and concanavalin A immunofluorescent staining. (A,B and E) C. glabrata cell wall surface glycan expression analyzed in mice receiving only C. glabrata. (C,D,F) C. glabrata cell wall surface glycan expression analyzed in mice receiving C. glabrata and DSS. Black and orange peaks (controls) are either fluorescent staining or isotype mAb background fluorescence for each specific mAb or immunofluorescent stain investigated.
In the absence of DSS, C. glabrata was rapidly eliminated from the mouse gut; the fitness of the fungal cell wall was therefore only analyzed over 2 days. A significant increase in α-mans was observed, while the chitin level did not change significantly between day 0 and day 1 (Fig. 4).
In DSS-induced colitis, a significant increase in chitin and β-man levels was observed, while α-mans decreased significantly (Fig. 4C,D,F). Remodeling of the fungal cell wall was also assessed by confocal microscopy and was consistent with the flow cytometry analysis, indicating that both β-mans and chitin are involved in promoting persistence of C. glabrata in the gut.
Impact of chitin deficiency on C. glabrata virulence in the mouse colitis model
To assess the impact of chitin deficiency on persistence of C. glabrata in the gut, C. glabrata strains ΔChs1 (Cg ΔChs1), and Δchs3 (Cg ΔChs3) were used in the DSS-induced colitis model and were compared with their parental strain, HTL (Fig. 5). Phenotypically, no growth defect was found in any strain when grown on YPD medium (data not shown). Flow cytometry revealed a significant decrease in chitin levels in C. glabrata ΔChs3 (80–90%), while chitin levels were increased in C. glabrata ΔChs1 when compared to C. glabrata HTL (Supplementary data Fig. 2). Additionally, β-man levels were increased in C. glabrata ΔChs1 when compared to C. glabrata ΔChs3 or C. glabrata HTL (Supplementary data Fig. 2). These data were consistent with confocal microscopy, which revealed that C. glabrata ΔChs3 stained weakly with WGA, while C. glabrata ΔChs1 and C. glabrata HTL stained strongly with WGA, and this staining was particularly strong in C. glabrata ΔChs1.

Effect of chitin-deficient C. glabrata on inflammatory parameters. (A) Mouse survival. Results are expressed as percent survival from the time of C. glabrata HTL, C. glabrata ΔChs1, or C. glabrata ΔChs3 challenge and DSS treatment. The survival data were significantly different by the log-rank test (P < 0.05). (B) Mouse body weight. The data shown are the mean ± SD from two independent experiments. (C) Clinical analysis of DSS-induced colitis in mice. ***P < 0.001 for C. glabrata ΔChs1 + D vs. C. glabrata ΔChs3 + D and C. glabrata HTL + D. (D) Histologic scores. Data are the mean ± SD of 16 mice per group (*P < 0.001).
Mice were administered a unique inoculum of these C. glabrata strains orally. In the absence of DSS, neither mouse mortality, nor clinical inflammation was observed in groups receiving C. glabrata (Fig. 5).
In mice with DSS-induced colitis, mortality rates were recorded as 20% in DSS mice, as 20% in C. glabrata HTL-DSS, and as 62.5% in C. glabrata ΔChs1 mice. This suggests that C. glabrata ΔChs1 is more virulent than the other strains (Fig. 5A).
Significantly higher clinical and histologic scores for inflammation were observed in C. glabrata ΔChs1-DSS mice than in the C. glabrata ΔChs3-DSS and C. glabrata HTL-DSS groups (Fig. 5C,D). In addition, histologic sections of colons from C. glabrata ΔChs1-DSS mice revealed high levels of leukocyte infiltration, epithelial damage, and edema when compared to colons from C. glabrata ΔChs3-DSS or C. glabrata HTL-DSS mice. In terms of fungal colonization, all C. glabrata strains were eliminated within 2 days after challenge from mice without colitis. In mice with DSS-induced colitis, there was a significant increase in number of C. glabrata colonies detected in stools when the first clinical signs of inflammation appeared. Although C. glabrata ΔChs1 is highly virulent, the number of CFUs of this strain was lower than that of the C. glabrata HTL parental strain (Supplementary data Fig. 3).
To assess the impact of chitin deficiency on C. glabrata colonization of the gut, we measured the number of C. glabrata CFUs adhering to the stomach, caecum, and colons in the different groups of mice (Supplementary data Fig. 3B).
The number of C. glabrata ΔChs1 colonies was significantly higher in the stomachs of mice treated with DSS than in C. glabrata ΔChs3 and C. glabrata HTL-DSS treated mice. In terms of fungal dissemination from the gut to the organs, higher numbers of C. glabrata ΔChs1 colonies were observed while the C. glabrata ΔChs3 strain was unable to disseminate. A trend towards a large number of C. glabrata ΔChs1 colonies was observed in the kidneys of DSS-treated mice (Supplementary data Fig. 3C).
The impact of C. glabrata colonization on the gut microbiota was also investigated. The change in E. coli numbers was determined in freshly collected stool samples (Supplementary data Fig. 4). In contrast to mice not treated with DSS, where the population of E. coli remained relatively stable, the E. coli population increased significantly in the DSS, C. glabrata ΔChs1, and C. glabrata HTL groups, as inflammation progressed, when compared to C. glabrata ΔChs3 mice.
Similarly, E. faecalis CFUs were significantly lower in C. glabrata ΔChs3-DSS mice than in DSS, C. glabrata ΔChs1-DSS, or C. glabrata HTL-DSS mice (data not shown). In terms of the activation/expression of receptors and signaling pathways in response to C. glabrata sensing and colitis, MBL-C and Myd88 expression increased significantly, while the expression of TLR-4 and PPARγ decreased in the colons in response to C. glabrata ΔChs3 (Supplementary data Fig. 5).
Effect of oral administration of chitin on intestinal inflammation, C. glabrata overgrowth and the gut microbiota
Before conducting our experiments in mice, we determined the effect of chitin on transepithelial electrical resistance and permeability of Caco-2 cells and whether chitin can reduce the adherence of C. glabrata to Caco-2 cells. We used different concentrations of chitin (1, 3, 5, and 10 mg). We found that chitin concentrations greater than 1 mg reduced C. glabrata adherence to Caco-2 cells (Supplementary data Fig. 6). Furthermore, the addition of chitin to Caco-2 cells before adding C. glabrata promoted intestinal barrier function as measured by a significant increase in transepithelial electrical resistance (data not shown). To determine the effect of chitin on modulation of intestinal inflammation and microbiota diversity including C. glabrata, chitin purified from C. glabrata was administered orally (3 mg/dose) to mice for 5 days after C. glabrata challenge. No mortality was observed in mice that received chitin during DSS-induced inflammation and colonization with C. glabrata. Administration of chitin also decreased the clinical and histologic scores when compared to untreated mice (Supplementary data Fig. 7). Mice treated with chitin showed decreased levels of C. glabrata in the stools as well as in the stomach and colon (Supplementary data Fig. 8). In terms of bacterial biodiversity, chitin treatment re-established the anaerobic bacteria including L. reuteri, L. johnsonii, Bifidobacterium and Bacteroides spp. and reduced aerobic bacteria such as E. coli and E. faecalis (Fig. 6 and Supplementary data Fig. 9). In addition, TLR-8, dectin-1, NOD-2, and PPARg receptors were significantly activated as a result of chitin administration while the expression of TLR-4, TLR-5, TLR-7, and TLR-9 was decreased (Fig. 7).

Cultivable bacterial diversity after chitin treatment of mice with DSS-induced colitis. Data are the mean ± SD of 20 mice per group. For all experiments, stool bacteria were isolated from mice on day 0 before C. glabrata challenge and DSS treatment. (A–H) E. coli, E. faecalis, B. thetaiotaomicron, B. vulgatus, Bacteroides spp. TP-5, L. johnsonii, Bifidobacterium spp., and L. reuteri CFUs recovered from stools. Data are the mean ± SD of 16 mice per group (*P < 0.05).

Cytokine and receptor expression after chitin treatment of mice with DSS-induced colitis. (A–F). Relative expression levels of TLR-8, TLR-9, PPAR-γ, Dectin-1, NOD-2, and MBL-C mRNA in mouse colons. (G and H) Protein levels of IL-10 and chitinase-3-like-1 protein in mouse colons. (I) Relative expression levels of chitinase-3-like-1 protein mRNA in mouse colons. Data are the mean ± SD of 16 mice per group (*P < 0.05).
When mRNA and protein expression of chitinase-3-like protein-1 was assessed after chitin treatment, a significant increase in chitinase-3-like protein-1 was observed, correlated with a decrease in C. glabrata CFUs in the gut indicating that both mRNA and protein expression of chitinase-3 increased after chitin treatment (Fig. 7).
Discussion
Dysbiosis is a change in the normal gut microbiome with a breakdown of host- microbial crosstalk20. This change in the gut microbiome promotes the overgrowth of opportunistic pathogens that contribute to intestinal mucosal inflammation21,22. Patients with CD are more frequently colonized with Candida species than healthy subjects23. Experimental studies show that colonization with Candida species exacerbates intestinal inflammation in the DSS-induced colitis model3,4. These clinical and experimental observations reveal the major role of opportunistic yeasts in modulation of the host immune-inflammatory response. In the present study, we assessed how the opportunistic yeast C. glabrata changes its cell wall composition in order to persist in the gut, and how overgrowth of this fungus together with intestinal inflammation affects the gut microbiome through modulation of both cytokine production and pathogen-recognition receptor (PRR) stimulation. The study also explored the effect of the fungal cell wall component chitin on modulation of the gut microbiome and its biological activities that confer a benefit to the host in terms of reducing inflammation. We also investigated the changes in C. glabrata virulence related to chitin gene deletions in the DSS-induced colitis model. In this DSS model, which is used to experimentally mimic IBD, mice received a single inoculum of C. glabrata by oral gavage and C. glabrata colonization increased gradually when DSS-induced intestinal inflammation was maintained in the mice. Erosions and ulceration of the mucosal surface of the gut promoted overgrowth of C. glabrata, which in turn exacerbated inflammation. In contrast, C. glabrata was rapidly eliminated from the gut in the absence of DSS-induced colitis, around 2 days after fungal challenge.
In the present study, we observed that regardless of C. glabrata colonization, DSS-induced colitis triggered changes in the gut microbiome. We focused in particular on cultivable bacteria belonging to the phyla Firmicutes, Bacteriodetes, Proteabacteria, or Actinobacteria by using selective bacterial media since these bacteria are known to be involved in CD (i.e. E. coli and E. faecalis)24. We observed a decrease in Firmicutes and Bacteroidetes while Proteobacteria and Enterobacteria increased in mice that developed colitis. These data are consistent with the clinical study, which showed a decrease in Firmicutes and Bacteroidetes, and an increase in Proteobacteria and fungal load, in particular C. glabrata, in CD patients25. Kim et al. showed that the dual association of E. coli and E. faecalis, both commensal organisms, rapidly induced severe pancolitis with dysplasia26. In addition, E. coli numbers increased during colitis and had a high pro-inflammatory potential to trigger inflammation via TLR-427,28. E. faecalis is a facultative anaerobic bacterium that likely benefits from potentially increased oxygen availability in the inflamed intestine, in a manner similar to E. coli, which is strictly aerobic. Furthermore, E. coli can even exploit the environment in an inflamed gut to obtain a growth advantage when compared to anaerobic bacteria (Lactobacillus or Bifidobacterium). In response to epithelial cell death and tissue damage during intestinal inflammation, the dead cells could provide extra nutrients, such as ethanolamine, to support E. coli overgrowth29,30.
Aerobic culture of stomach and colon samples showed an increase in E. coli and E. faecalis populations in DSS-treated mice when compared to control mice. These data are consistent with clinical studies, which showed that aerobic cultures of biopsies obtained by colonoscopy from control colons were often sterile, whereas colons from patients with CD contained increased bacterial numbers in the sub-mucosa, a relatively well oxygenated site, more than half of which were E. coli31. In parallel, we observed that the increase in E. coli population in the gut was correlated with C. glabrata overgrowth. Centeno et al. observed that piliated E. coli strains can enhance Candida attachment to epithelial cells32. E. coli also exhibits a synergistic effect with Candida by inducing high mouse mortality during experimental microbial peritonitis33. In biofilm studies, Hoarau et al. showed that the interaction of E. coli with C. tropicalis combined with Serratia marcescens enhanced fungal filamentation and biofilm maturation34.
In terms of anaerobic bacteria, the population of Bifidobacterium spp., B. thetaiotaomicron and other Bacteroides spp. decreased in response to intestinal inflammation and to C. glabrata overgrowth, while B. vulgatus was not affected by either DSS-induced colitis or C. glabrata overgrowth. Interestingly, C. glabrata overgrowth significantly decreased the L. johnsonii population during the development of colitis. This observation is consistent with previous reports, which indicate that Lactobacillus growth can antagonize colonization by Candida35.
Receptors and signaling pathways were selected according to our previous studies or other studies showing the involvement of these receptors in fungal recognition, in particular TLRs, MBL, or in the stimulation of signaling pathways in response to C. glabrata sensing7,36,37. In this study, C. glabrata WT was found to increase MBL-C and TLR-9 expression. This finding supports the findings of Choteau et al. who showed that the activation of MBL-C by Candida sensing in intestinal epithelial cells promoted the rapid elimination of C. glabrata from the gut37. Conversely, the C. glabrata WT strain decreased TLR-4 expression in the mouse colons as colitis developed when compared to colons of mice treated with DSS only, suggesting that stimulation of TLR-4 is driven by overgrowth of E. coli and E. faecalis, while the increase in MBL-C expression is related to Candida sensing35. Pro-inflammatory cytokine mRNA and protein expression (IL-6 and IL-1β) increased in the colons of DSS-treated mice, and a higher level of expression of these cytokines was observed in response to Candida sensing. This is consistent with different reports, which showed that DSS-induced colitis alone or combined with fungal colonization promotes overexpression of pro-inflammatory mediators that amplify the inflammatory cascade through NF-κB and Myd88 expression.
Previous studies have shown how the local environment affects morphogenesis, virulence gene expression, and stress resistance, but very little is known about how the inflammatory gut environment impacts on the fungal cell wall composition. In the present study, we found that C. glabrata undergoes cell wall remodeling during fungal colonization in the DSS-induced colitis model. Modification of the composition of the C. glabrata cell wall is related to intestinal inflammation and not to bacteria biodiversity changes, since the C. glabrata cell wall modification appears in the days corresponding to the onset of inflammation and not while the bacterial population changes. In the absence of colitis development, the level of α-mans increased in the C. glabrata cell wall on days 1 and 2 after fungal challenge and then this yeast was eliminated rapidly from the gut. The increase in C. glabrata α-man level enables C. glabrata to behave like S. cerevisiae transiting through the mouse gut. In contrast, in DSS-induced colitis, C. glabrata appears to alter towards a pathogenic form close to that of C. albicans, resulting in an increase in chitin and β-man, and a decrease in α-man levels. These data reveal that the inflammatory gut environment impacts on the C. glabrata cell wall leading to adaptation of the fungal cell within the host, which allows C. glabrata to persist in the gut environment.
In a previous study, we showed that a deficiency in β-mans reduced C. glabrata adherence to intestinal epithelial cells, favoring fungal elimination from the mouse gut, indicating that β-mans contribute to C. glabrata virulence. However, the role of chitin in modulation of intestinal inflammation, the gut microbiome and fungal colonization has not yet been thoroughly investigated. In the present study, orally administered chitin purified from C. glabrata decreased intestinal inflammation and C. glabrata overgrowth. Several studies have shown that chitin enhances the immune response and increases the clearance of pathogenic bacteria in animal models; this supports our observations that chitin from C. glabrata has beneficial activities for C. glabrata.
To assess the impact of chitin deficiency on C. glabrata in the inflammatory gut environment, we selected the two genes that encode chitin synthase involved in chitin biosynthesis, Chs1 and Chs338. Flow cytometry and confocal microscopy of the fungal cell wall revealed a significant reduction in chitin in C. glabrata Δchs3 while chitin levels in C. glabrata Δchs1 increased, indicating that in contrast to Chs1, chitin synthase Chs3 is a crucial enzyme for cell wall chitin synthesis (Supplementary data Fig. 8). This observation is consistent with other reports that show that chitin synthase Chs3 is involved in generating 80–90% of the total fungal chitin, while Chs1 is responsible for repairing the septa and the weakened cell walls of daughter cells after their separation from mother cells39. In the absence of DSS, mutant strains did not induce inflammation in the mice. In DSS-induced colitis, C. glabrata Δchs1, rich in chitin and β-glucan, is highly virulent in terms of colonization, fungal dissemination to the organs and inflammatory parameters. In contrast, a deficiency of chitin synthase Chs3 reduced the pathogenicity of C. glabrata in the inflammatory gut environment. Furthermore, C. glabrata Δ chs1, which has a high level of chitin, does not induce the expression of chitinase-3-like protein-1. Thus, this promotes C. glabrata overgrowth and persistence in the gut. In addition to the increase in chitin level in the C. glabrata Δchs1 cell wall, the β-man level, which is expressed in the outer fungal cell wall layer, was also increased. These data are consistent with other reports, which show that C. glabrata strains deficient in β-mans are less virulent than the WT strain suggesting the involvement of β-mans in the virulence and resistance of C. glabrata in the intestinal tract5.
Interestingly, the virulent C. glabrata Δchs1 strain reduced the expression of MBL-C, TLR-2, and TLR-9, while C. glabrata Δchs3 increased the expression of these receptors as colitis developed, indicating that the cell wall of C. glabrata Δchs3 is rich in α-mans, which are potential ligands for MBL-C and TLR4. In contrast, C. glabrata Δchs1, which has high levels of expression of β-man epitopes on its cell wall surface, is capable of reducing these intestinal epithelial cell receptors as an escape mechanism from the host defense.
Chitin acts as an anti-inflammatory agent and is involved in the process of wound healing40. Oral administration of chitin reduced all of the inflammatory parameters, and led to overgrowth of C. glabrata and reestablishment of the biodiversity of the gut microbiota. Chitin treatment increased chitinase-3-like protein-1, promoting chitin breakdown and the generation of small sized chitin particles that induce IL-10 production via PPARg, NOD-2, and TLR-8 sensing. These results are consistent with those of Wagner et al. who showed that chitin oligosaccharides have the potential to induce IL-10 secretion, via NOD-2 and TLR-9 signaling, promoting the attenuation of inflammation responses19. Surprisingly, the expression levels of TLR-4, TLR-5, TLR-6, TLR-7, TLR-9, and MBL-C were not upregulated by chitin treatment.
In conclusion, inflammation in the gut increased the aerobic bacteria population, in particular E. coli and E. faecalis, but decreased the population of anaerobic bacteria such as L. johnsonii, B. thetaiotaomicron, and Bifidobacterium spp. DSS-induced colitis led to cell wall remodeling through an increase in chitin production, which was involved in promoting C. glabrata overgrowth and persistence in the gut. C. glabrata colonization modulated the intestinal epithelial receptors, in particular MBL-C, TLR-4, and TLR-9, as well as expression of the signaling pathways, NF-κB and Myd88. In terms of fungal cell wall components, oral administration of chitin to mice reduced the overgrowth of aerobic bacteria and C. glabrata as well the production of inflammatory parameters through stimulation of intestinal receptors. Chitin deficiency affected the C. glabrata cell wall composition. This deficiency was compensated for by an increase in α-man levels in C. glabrata chs3, which induced fewer inflammatory parameters than the parental strain. Chitin treatment increased chitinase-3-like protein-1, enabling chitin digestion and the generation of small sized chitin particles that induced IL-10 production via PPARg, NOD-2, and TLR-8 sensing, promoting the attenuation of colitis and C. glabrata elimination. Finally, this study has increased our understanding of the nature of yeast molecular components that differentially affect inflammation and/or C. glabrata clearance.
Methods
Animals
Eight-to-10-week-old, female C57BL/6 mice were purchased from Charles River Laboratories, France. Mice were allocated to six experimental groups (Supplementary data, Table 1) and eight control groups, including assessment of the effect of DSS alone. Four complete experimental series were performed independently.
All experimental procedures were approved by the subcommittee for Research Animal Care, Regional Hospital Center, Lille, France (00550.05), and in accordance with institutional (86/609/CEE) and European guidelines for the care and use of laboratory animals.
Yeast strains
The C. glabrata strains used are shown in Supplementary data Table 2. For C. glabrata ΔChs3, CAGL0B04389g CHS3 was deleted (alias CHS3A)38. All C. glabrata strains were maintained at 4 °C in yeast peptone dextrose broth (YPD; 1% yeast extract, 2% peptone, 2% dextrose).
Extraction of chitin from C. glabrata
C. glabrata cell pellets were washed twice in phosphate-buffered saline (PBS). Chitin was extracted from C. glabrata yeast cells as described previously41. Briefly, the cell pellet was incubated twice in 20 ml of 10% KOH and autoclaved at 120 °C for 2 h. After washing several times with distilled water, the supernatant was removed and the pellet was oxidized with 50% hydrogen peroxide and 50% acetic acid, and then autoclaved at 120 °C for 2 h. After washing several times with distilled water, the chitin fraction was lyophilized. The nature of the chitin was confirmed by nuclear magnetic resonance (NMR) analysis. Intact chitin purified from C. glabrata was suspended in deuterated hexafluroisopropanol (Euriso-Top) at 70 °C until dissolved. All experiments were performed using a Bruker Avance 600 MHz (13.1 T) spectrometer with Bruker standard pulse programs. The chitin concentration was determined using a BiCinchoninic acid assay. The standard range for the N-acetyl-glucosamine (GlcNAc) control was 0.1–5 mg/mL.
Measurement of cell wall chitin content
The chitin content was measured as described previously42. C. glabrata HTL, C. glabrata ΔChs1, and C. glabrata ΔChs3 strains were grown in YPD broth. Yeast cells were suspended in 10 mL of PBS to a final concentration of 109 cells/mL and were disrupted with glass beads. The cells were then washed several times with 1 M NaCl and extracted in SDS-MerOH buffer (50 mM Tris, 2% sodium dodecyl sulfate (SDS), 0.3 M β-mercaptoethanol, 1 mM EDTA; pH 8.0) at 100 °C for 10 min, and then washed in distilled water. The chitin concentration was determined using a BiCinchoninic acid assay. The standard range for the N-acetyl-glucosamine (GlcNAc) control was 0.1–5 mg/mL.
Inoculum preparation and induction of colitis
The mice were inoculated on day 1 by oral gavage with 200 µL PBS containing 5 × 107 live C. glabrata cells. From day 1 to day 14, mice were also administered 2% DSS (36−50 kDa; MP Biomedicals, LLC, Germany) in drinking water in order to induce intestinal inflammation. For chitin treatment, mice were administered with chitin purified from C. glabrata (3 mg per mouse) orally and daily for 5 days, starting on day 1. The presence of C. glabrata in the intestinal tract was monitored daily by measuring the number of colony-forming units (CFUs) in feces (approximately 0.1 g/sample) collected from each animal4. Fecal samples were suspended in 1 mL saline, homogenized in a glass tissue homogenizer, and samples were then cultured on Candi-Select medium (Bio-Rad Laboratories, Marnes la Coquette, France)43. The number of C. glabrata colonies was counted after incubation of the plates at 37 °C for 48 h. The results are expressed as CFU/µg of feces. In order to determine the degree of C. glabrata colonization of the gut, the mice were sacrificed and the GI tract was removed and separated into the stomach, ileum, and colon. These portions of the GI tract were cut longitudinally and the intestinal contents were removed. The tissue samples were then washed several times in PBS in order to minimize contamination from yeasts present in the lumen44. Serial dilutions of the homogenates were prepared and plated onto Candi-Select medium. The number of colonies was noted after 48 h incubation at 37 °C and expressed as C. glabrata CFU/mg of tissue.
For the isolation of bacteria, we performed serial dilutions of the gut contents (stomach, colon, and caecum) or fecal samples collected from the mice. The samples were cultured on non-selective bacterial media (AC agar) focusing on the most representative cultivable anaerobic and aerobic bacteria that can undergo changes during intestinal inflammation45.
For the isolation of aerobic bacteria, the fecal samples and tissues were plated onto MacConkey agar (Sigma-Aldrich), bile esculin azide agar (BEA; Sigma-Aldrich), and MRS agar (Sigma-Aldrich) plates. Serial dilutions of the samples were prepared. Bacteroides bile esculin (BBE; Sigma-Aldrich), Columbia agar (Sigma-Aldrich), and Bifidus selective medium agar (BSM; Sigma-Aldrich) were used for the isolation of anaerobic bacteria. These plates were incubated in anaerobic jars containing an anaerobic atmosphere generator pack (AnaeroGenTM 2.5 L; Sigma-Aldrich) at 35 °C. All aerobic and anaerobic media contained 60 mg/L fluconazole (Fresenius Kabi) to inhibit the growth of fungal cells.
All plates were incubated at 37 °C and examined after 24 h and 48 h. To identify any bacteria on the plates, the colonies were mixed with 1.5 μl of matrix solution (α-cyano-4-hydroxycinnamic acid; Bruker Daltonics) dissolved in 50% acetonitrile, 47.5% water, and 2.5% trifluoroacetic acid, and analyzed by MALDI-TOF MS (Microflex-Bruker Daltonics).
Analysis of C. glabrata cell wall remodeling after passage through the GI tract
Fresh fungal cells obtained from fecal samples, and collected daily from each tagged animal, were diluted in PBS. 107 C. glabrata cells were prepared and washed several times with PBS. Washed yeast cells were incubated with PBS containing 2% fetal calf serum for 20 min, and were then incubated with fluorescein isothiocyanate-wheat germ agglutinin (FITC-WGA), rhodamine-labeled concanavalin A (Con A), or 5B2 antibody (FITC anti-rat IgM secondary antibody and the control isotype rat IgM). Analysis of the expression of glycan epitopes on the C. glabrata cell wall was performed by flow cytometry (Accuri® Sampler™). Mean fluorescence intensity (MFI) of each histogram was calculated as: labeled strain-unlabeled strain/mean fluorescence of labeled strain according to the number of days.
Assessment of clinical and histological scores
Body weight and mortality of the mice were recorded daily. The data were expressed as mean percent change from initial body weight. Clinical scores ranging from 0 to 12 and histologic scores ranging from 0 (no changes) to 6 (extensive cell infiltration and tissue damage) were calculated as described previously4,46.
Real-time mRNA quantification of pro-inflammatory cytokines and innate immune receptors
Total RNA was extracted from mouse colons using a Nucleospin RNA kit (Macherey-Nagel). RNA was quantified by spectrophotometry (Nanodrop; Nyxor Biotech, France). Reverse transcription of mRNA was carried out from 1 µg total RNA using a high capacity cDNA RT kit (Applied Biosystems) in a final volume of 20 µL. cDNA was amplified by PCR in the one-step system (Applied Biosystems) using Fast SYBR green (Applied Biosystems). The intensity of SYBR green dye was assessed using one-step software. The reference gene, POLR2A47, was used to normalize the results.
Quantification of cytokines and chitinase 3-like 1 by ELISA
Representative pro-inflammatory (IL-1β, IL-6) and anti-inflammatory (IL-10) cytokine profiles were selected in this study. Cytokine concentrations (IL-1β, IL-6, and IL-10) in the colons were measured using a commercial ELISA kit according to the manufacturer’s instructions (eBioscience, San Diego, CA) whereas the detection of murine chitinase 3-like 1 levels was done using an ELISA kit from R and D systems. The data are expressed as pg/mL.
Statistical analysis
All data are expressed as the mean ± standard deviation (SD) for each experimental group. Pairs of groups were compared using the Mann-Whitney U test. Differences were considered to be statistically significant when the P value was as follows: p < 0.05; p < 0.01; p < 0.001.
All statistical analyses were carried out using GraphPad Prism 4.0 and XLSTAT.
References
Baumgart, D. C. & Sandborn, W. J. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 369, 1641–1657, https://doi.org/10.1016/S0140-6736(07)60751-X (2007).
Standaert-Vitse, A. et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn’s disease. Gastroenterology 130, 1764–1775, https://doi.org/10.1053/j.gastro.2006.02.009 (2006).
Jawhara, S. & Poulain, D. Saccharomyces boulardii decreases inflammation and intestinal colonization by Candida albicans in a mouse model of chemically-induced colitis. Medical Mycology 45, 691–700, https://doi.org/10.1080/13693780701523013 (2007).
Jawhara, S. et al. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. Journal of Infectious Diseases 197, 972–980, https://doi.org/10.1086/528990 (2008).
Jawhara, S. et al. Murine model of dextran sulfate sodium-induced colitis reveals Candida glabrata virulence and contribution of beta-mannosyltransferases. Journal of Biological Chemistry 287, 11313–11324, https://doi.org/10.1074/jbc.M111.329300 (2012).
Lionakis, M. S. & Netea, M. G. Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathogens 9, e1003079, https://doi.org/10.1371/journal.ppat.1003079 (2013).
Gow, N. A., van de Veerdonk, F. L., Brown, A. J. & Netea, M. G. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nature Reviews. Microbiology 10, 112–122, https://doi.org/10.1038/nrmicro2711 (2012).
Poulain, D. Candida albicans, plasticity and pathogenesis. Critical Reviews in Microbiology, https://doi.org/10.3109/1040841X.2013.813904 (2013).
Miranda, L. N. et al. Candida colonisation as a source for candidaemia. Journal of Hospital Infection 72, 9–16, https://doi.org/10.1016/j.jhin.2009.02.009 (2009).
Poulain, D. et al. Yeasts: neglected pathogens. Digestive Diseases 27(Suppl 1), 104–110, https://doi.org/10.1159/000268129 (2009).
Shaw, J. A. et al. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. Journal of Cell Biology 114, 111–123 (1991).
Aufauvre-Brown, A., Mellado, E., Gow, N. A. & Holden, D. W. Aspergillus fumigatus chsE: a gene related to CHS3 of Saccharomyces cerevisiae and important for hyphal growth and conidiophore development but not pathogenicity. Fungal Genetics and Biology: FG & B 21, 141–152 (1997).
Bulawa, C. E., Miller, D. W., Henry, L. K. & Becker, J. M. Attenuated virulence of chitin-deficient mutants of Candida albicans. Proceedings of the National Academy of Sciences of the United States of America 92, 10570–10574 (1995).
Munro, C. A. & Gow, N. A. Chitin synthesis in human pathogenic fungi. Medical Mycology 39(Suppl 1), 41–53 (2001).
Nagahashi, S., Lussier, M. & Bussey, H. Isolation of Candida glabrata homologs of the Saccharomyces cerevisiae KRE9 and KNH1 genes and their involvement in cell wall beta-1,6-glucan synthesis. Journal of Bacteriology 180, 5020–5029 (1998).
Anandan, R., Nair, P. G. & Mathew, S. Anti-ulcerogenic effect of chitin and chitosan on mucosal antioxidant defence system in HCl-ethanol-induced ulcer in rats. Journal of Pharmacy and Pharmacology 56, 265–269, https://doi.org/10.1211/0022357023079 (2004).
Masuda, S. et al. Anti-tumor properties of orally administered glucosamine and N-acetyl-D-glucosamine oligomers in a mouse model. Carbohydrate Polymers 111, 783–787, https://doi.org/10.1016/j.carbpol.2014.04.102 (2014).
Fernandes, J. C. et al. Anti-inflammatory activity of chitooligosaccharides in vivo. Marine Drugs 8, 1763–1768, https://doi.org/10.3390/md8061763 (2010).
Wagener, J. et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathogens 10, e1004050, https://doi.org/10.1371/journal.ppat.1004050 (2014).
Seksik, P. et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 52, 237–242 (2003).
Sokol, H. et al. Fungal microbiota dysbiosis in IBD. Gut, https://doi.org/10.1136/gutjnl-2015-310746 (2016).
Bien, J., Palagani, V. & Bozko, P. The intestinal microbiota dysbiosis and Clostridium difficile infection: is there a relationship with inflammatory bowel disease? Therapeutic Advances in Gastroenterology 6, 53–68, https://doi.org/10.1177/1756283X12454590 (2013).
Standaert-Vitse, A. et al. Candida albicans colonization and ASCA in familial Crohn’s disease. American Journal of Gastroenterology 104, 1745–1753, https://doi.org/10.1038/ajg.2009.225 (2009).
Darfeuille-Michaud, A. et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004).
Liguori, G. et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. Journal of Crohns Colitis 10, 296–305, https://doi.org/10.1093/ecco-jcc/jjv209 (2016).
Kim, S. C., Tonkonogy, S. L., Karrasch, T., Jobin, C. & Sartor, R. B. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflammatory Bowel Diseases 13, 1457–1466, https://doi.org/10.1002/ibd.20246 (2007).
Heimesaat, M. M. et al. Exacerbation of murine ileitis by Toll-like receptor 4 mediated sensing of lipopolysaccharide from commensal Escherichia coli. Gut 56, 941–948, https://doi.org/10.1136/gut.2006.104497 (2007).
Lupp, C. et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host & Microbe 2, 204 (2007).
Bertin, Y. et al. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environmental Microbiology 13, 365–377, https://doi.org/10.1111/j.1462-2920.2010.02334.x (2011).
Garsin, D. A. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nature reviews. Microbiology 8, 290–295, https://doi.org/10.1038/nrmicro2334 (2010).
Saldena, T. A., Saravi, F. D., Hwang, H. J., Cincunegui, L. M. & Carra, G. E. Oxygen diffusive barriers of rat distal colon: role of subepithelial tissue, mucosa, and mucus gel layer. Digestive Diseases and Sciences 45, 2108–2114 (2000).
Centeno, A., Davis, C. P., Cohen, M. S. & Warren, M. M. Modulation of Candida albicans attachment to human epithelial cells by bacteria and carbohydrates. Infection and Immunity 39, 1354–1360 (1983).
Klaerner, H. G. et al. Candida albicans and Escherichia coli are synergistic pathogens during experimental microbial peritonitis. Journal of Surgical Research 70, 161–165, https://doi.org/10.1006/jsre.1997.5110 (1997).
Hoarau, G. et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. mBio 7, https://doi.org/10.1128/mBio.01250-16 (2016).
Erridge, C., Duncan, S. H., Bereswill, S. & Heimesaat, M. M. The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PloS One 5, e9125, https://doi.org/10.1371/journal.pone.0009125 (2010).
Choteau, L. et al. Role of TLR1, TLR2 and TLR6 in the modulation of intestinal inflammation and Candida albicans elimination. Gut Pathogens 9, 9, https://doi.org/10.1186/s13099-017-0158-0 (2017).
Choteau, L. et al. Role of mannose-binding lectin in intestinal homeostasis and fungal elimination. Mucosal Immunology 9, 767–776, https://doi.org/10.1038/mi.2015.100 (2016).
Schwarzmuller, T. et al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathogens 10, e1004211, https://doi.org/10.1371/journal.ppat.1004211 (2014).
Ueno, K., Namiki, Y., Mitani, H., Yamaguchi, M. & Chibana, H. Differential cell wall remodeling of two chitin synthase deletants Deltachs3A and Deltachs3B in the pathogenic yeast Candida glabrata. FEMS Yeast Research 11, 398–407, https://doi.org/10.1111/j.1567-1364.2011.00728.x (2011).
Azuma, K. et al. Chitin, chitosan, and its derivatives for wound healing: old and new materials. Journal of Functional Biomaterials 6, 104–142, https://doi.org/10.3390/jfb6010104 (2015).
Bulawa, C. E. et al. The S. cerevisiae structural gene for chitin synthase is not required for chitin synthesis in vivo. Cell 46, 213–225 (1986).
Munro, C. A. et al. CHS8-a fourth chitin synthase gene of Candida albicans contributes to in vitro chitin synthase activity, but is dispensable for growth. Fungal Genetics and Biology: FG & B 40, 146–158 (2003).
Sendid, B. et al. Prospective evaluation of the new chromogenic medium CandiSelect 4 for differentiation and presumptive identification of the major pathogenic Candida species. Journal of Medical Microbiology 56, 495–499 (2007).
Edwards-Ingram, L. et al. Genotypic and physiological characterization of Saccharomyces boulardii, the probiotic strain of Saccharomyces cerevisiae. Applied and Environmental Microbiology 73, 2458–2467 (2007).
Karrout, Y. et al. Colon targeting with bacteria-sensitive films adapted to the disease state. European Journal of Pharmaceutics and Biopharmaceutics 73, 74–81, https://doi.org/10.1016/j.ejpb.2009.04.003 (2009).
Siegmund, B. et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. Jouranl of Pharmacology and Experimental Therapy 296, 99–105 (2001).
Saviozzi, S. et al. Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer 6, 200, https://doi.org/10.1186/1471-2407-6-200 (2006).
Acknowledgements
The authors would like to thank Ms. Nadine François for her technical help. This work was supported by the digestScience Foundation, and was funded by the FP7 Health 260338 ‘ALLFUN’ project ‘Fungi in the setting of inflammation, allergy and auto-immune diseases: translating basic science into clinical practices’.
Author information
Authors and Affiliations
Contributions
R.C., Y.P., G.T., F.I., and S.J. performed the experiments. R.C., Y.P., G.T., F.I., B.S., and S.J. analyzed the data. R.C., Y.P., G.T., F.I., D.P., K.K., B.S., and S.J. interpreted the results of experiments. F.I., and K.K. contributed reagents/materials/analysis tools. S.J. designed the experiments and drafted the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Charlet, R., Pruvost, Y., Tumba, G. et al. Remodeling of the Candida glabrata cell wall in the gastrointestinal tract affects the gut microbiota and the immune response. Sci Rep 8, 3316 (2018). https://doi.org/10.1038/s41598-018-21422-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21422-w
- Springer Nature Limited
This article is cited by
-
The primate gut mycobiome-bacteriome interface is impacted by environmental and subsistence factors
npj Biofilms and Microbiomes (2022)
-
The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet
Communications Biology (2021)
-
First Study of Naturally Formed Fungal Biofilms on the Surface of Intragastric Balloons
Obesity Surgery (2021)
-
Bacteroides thetaiotaomicron and Lactobacillus johnsonii modulate intestinal inflammation and eliminate fungi via enzymatic hydrolysis of the fungal cell wall
Scientific Reports (2020)
-
Microbial-Based and Microbial-Targeted Therapies for Inflammatory Bowel Diseases
Digestive Diseases and Sciences (2020)