
Abstract
In bacteria, the twin-arginine translocation (Tat) pathway allows the export of folded proteins through the inner membrane. Proteins targeted to this system are synthesized with N-terminal signal peptides bearing a conserved twin-arginine motif. The Tat pathway is critical for many bacterial processes including pathogenesis and virulence. However, the full set of Tat substrates is unknown in many bacteria, and the reliability of in silico prediction methods largely uncertain. In this work, we performed a combination of in silico analysis and experimental validation to identify a core set of Tat substrates in the opportunistic pathogen Pseudomonas aeruginosa. In silico analysis predicted 44 putative Tat signal peptides in the P. aeruginosa PA14 proteome. We developed an improved amidase-based Tat reporter assay to show that 33 of these are real Tat signal peptides. In addition, in silico analysis of the full translated genome revealed a Tat candidate with a missassigned start codon. We showed that it is a new periplasmic protein in P. aeruginosa. Altogether we discovered and validated 34 Tat substrates. These show little overlap with Escherichia coli Tat substrates, and functional analysis points to a general role for the P. aeruginosa Tat system in the colonization of environmental niches and pathogenicity.
Similar content being viewed by others


Introduction
In Gram negative bacteria, at least 30% of proteins are localized outside the cytoplasm, where they are produced1. Bacteria have evolved complex machineries to export these extracytoplasmic proteins across the cellular envelope (inner membrane, periplasm and outer membrane) and secrete them into the extracellular medium or into neighboring target cells2. The bacterial cytoplasmic membrane contains two export systems for the translocation of proteins, the Sec and Tat pathways. The Sec pathway is essential and constitutes the major route in most bacteria. Sec-dependent proteins are exported in an unfolded conformation and fold upon delivery to the periplasm. In contrast, the Tat pathway exports proteins in a folded conformation and is used more moderately in most organisms. Tat-dependent substrates are however essential for a wide range of bacterial processes such as adaptation and growth in particular environments, cell division and virulence3,4.
Proteins destined to either the Sec or the Tat pathway are synthesized as precursors with N-terminal signal peptides that allow their targeting and recognition by machinery components5. Signal peptides have a tripartite organization with a positively charged N-terminal region (N-region), a central hydrophobic core (H-region) and a polar C-terminal region (C-region)6. Despite their similar overall organization, Sec and Tat signal peptides have characteristics that allow specific recognition by their respective export pathway. In particular, Tat-dependent signal peptides differ from Sec-dependent signal peptides in the presence of an extended N-region that contains a conserved S/TRRXFLK motif. The two consecutive arginines in this motif are almost always invariant, and gave the system its name7. The other amino acids in this motif appear at a frequency greater than 50%7. Tat signal peptides are also longer and less hydrophobic than their Sec counterparts8. Finally, the C-region of Tat signal peptides contains conserved motifs which are recognition sequences for signal peptidases I and II8,9 as well as basic residues that impede targeting to the Sec pathway10.
Prediction algorithms that recognize the features of signal peptides based on their amino acid sequence have been developed over the years11. SignalP predicts classical Sec signal peptides cleaved by type I signal peptidase. For Tat signal peptides three prediction algorithms are available, TATFIND12, TatP13 and PRED-TAT14. TATFIND searches in the first 35 residues of a protein for the conserved twin-arginines within a specific hydrophobicity context, followed by an uncharged region of a certain position, length and hydrophobicity. TATFIND does not predict cleavage sites. TatP is also based on the search for a specific motif (RR.[FGAVML][LITMVF]), and integrates two machine learning algorithms (Artificial Neural Networks) for cleavage site recognition and hydrophobicity discrimination. Finally PRED-TAT uses a Hidden Markov Model (HMM) method with sub-modules for the distinguishing between Sec, Tat and TM (transmembrane) regions. Comparisons of these prediction algorithms on test sets tend to show that TatP generates fewer false positives than TATFIND and slightly more false negatives13. PRED-TAT outperforms both TATFIND and TatP in sensitivity, specificity and in prediction for the precise location of the cleavage site14. Overall these algorithms have both strengths and weaknesses, and using them in combination can improve the accuracy of signal peptide prediction. However, parallel experimental confirmation is still required to validate the export pathway and to determine the final localization of a protein. This is particularly true for Tat signal peptides, because the presence of a predicted Tat signal peptide does not always guarantee export via the Tat pathway15,16.
One technique used to identify the export pathway of a protein is proteome comparison after cell fractionation of wild type and export defective mutants. Defect in the final localization of a candidate protein in an export mutant can be used to assign the protein to the corresponding export pathway17. However the use of such approaches relies on the growth conditions that allow production of the candidate proteins, and on the availability of reliable fractionation procedures. Another means to identify transport pathway dependency is to use transport reporter assays. For Tat dependent transport, assays have been developed that consist in fusing a putative Tat signal peptide to a reporter protein whose Tat-dependent activity can be easily followed upon translocation. Reporter proteins such as the green fluorescent protein18, colicin V19, maltose binding protein20, β-lactamase21, agarase22 and the N-acetyl muramoyl-L-alanine amidase AmiA23 have been used. Of these only the Streptomyces coelicolor agarase and Escherichia coli AmiA are natural Tat substrates whose export is Tat-specific. Over the years the amidase reporter assay has been used to study the mechanism and function of the Tat system because it offers a facile and sensitive in vivo assay for very low levels of Tat activity24,25. This assay has also been used in a limited number of cases to confirm the Tat-dependent export of putative Tat substrates26,27,28,29. However the amidase assay has never been used to screen large numbers of candidate Tat substrates.
Pseudomonas aeruginosa is an opportunistic human pathogen, responsible for a variety of chronic and acute infections in susceptible hosts, including immunocompromised patients, those with injury to the epithelial barrier (burns or damages to the cornea) and patients with cystic fibrosis. It is one of the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa and Enterobacter spp) described as clinically relevant and highly multidrug resistant, and is a major menace to public health worldwide30. P. aeruginosa virulence is multifactorial and relies on numerous determinants that facilitate bacterial colonization, invasion and dissemination31. P. aeruginosa has a Tat system with the same TatABC core components as found in E. coli where TatC is essential32. We and others have shown that the Tat system is essential for full virulence of P. aeruginosa and that it plays a broad role in various processes such as the secretion of exoproteins, iron acquisition, anaerobic growth, osmotolerance, motility, biofilm formation, carbon metabolism and detoxification29,32,33,34,35,36. Except for a few exceptions, the molecular links between these defective processes and the tat mutation are still uncertain. This is mainly because the full repertoire of Tat proteins in P. aeruginosa is unknown.
In this study, we used a genome-wide in silico approach to identify putative Tat signal peptides in the P. aeruginosa PA14 strain. After development of the E. coli amidase reporter assay and its validation for P. aeruginosa signal peptides, we used it to probe all P. aeruginosa candidate Tat signal peptides identified in silico and reveal bona fide Tat signal peptides. This approach allowed us to confirm the Tat-dependent export of 8 previously known P. aeruginosa Tat substrates and to uncover 25 new P. aeruginosa proteins with a Tat signal peptide. Our approach also highlighted how the missassignement of one start codon on the PA14 genome impaired the subsequent prediction of a Tat signal peptide on the SphC protein. We then confirmed in vivo in P. aeruginosa that this signal peptide allows the periplasmic localization of SphC in a Tat-dependent manner. Altogether, our data reveal that P. aeruginosa possesses at least 34 Tat substrates and that the Tat system is involved in a wide range of processes allowing adaptation of this opportunistic pathogen to its niche and establishment of host infection.
Results
In silico screening of the P. aeruginosa genome sequence for N-terminal regions encoding twin-arginine signal peptides
Genomic and experimental approaches indicate that the Tat system is used to various extents in organisms. While the Tat system is utilized for more than 100 substrates in Streptomyces species, it is used for 27 substrates in E. coli, and only for 5 to 7 substrates in Bacillus subtilis despite the presence of two independent Tat machineries37. The P. aeruginosa proteome has been manually screened for the presence of Tat signal peptides in two studies either looking for the motif RRXFLK/R33 or the two conserved arginines followed by at least two of the FLK/R residues37,38. These studies respectively led to the prediction of 18 and 27 putative Tat signal peptides with 10 signal peptides common between them. Here, we screened the P. aeruginosa PA14 proteome for Tat signal peptides using two Tat prediction programs, TATFIND and TatP. Although the twin-arginine motif is highly conserved in Tat substrates, natural non canonical Tat signal peptides exist, where one of the consensus arginine residues is replaced by a lysine39,40. To take into account exceptions that might also exist in P. aeruginosa we modified the TATFIND and TatP prediction algorithm to consider RK or KR the same as RR. This analysis led to the prediction of 72 putative Tat signal peptides by TATFIND and 794 by TatP respectively (Table S1). As expected from the conceptual differences between these algorithms the candidate Tat signal peptides found show partial overlap and TatP predicted considerably more candidates than TATFIND. We selected the 39 Tat signal peptides predicted by both algorithms for further analysis (Table 1A and Fig. 1). To avoid dismissing real Tat substrates predicted only by TATFIND or TatP as occurred in Streptomyces coelicolor22, we next compared our list of candidates with the candidates obtained with the most recent program, PRED-TAT. This allowed us to select another five candidate signal peptides that were positive with this program (and either TatP or TATFIND) for further analysis (Table 1B and Fig. 1). The final set contained 44 putative Tat signal peptides. Of these, seven out of the eight already known P. aeruginosa Tat signal peptides (PA14_53360/PlcH, PA14_21110/PlcN, PA14_13330/EddA, PA14_33740/PvdP, PA14_33720/PvdN, PA14_48450/Agu2A’and PA14_10370/PA4140) were present (Fig. 1). Indeed, up to now 8 Tat substrates have been experimentally identified in P. aeruginosa: PlcH and PlcN32, PvdN34, PvdP35, Agu2A’36 and EddA, PA2699/PA14_29230 and PA414029. It was not surprising that the PA2699 Tat signal peptide was not identified in our analysis, because its start codon is missassigned on the P. aeruginosa genome29. While the Tat signal peptides of PlcH, PlcN, EddA, PvdP, PvdN, and PA4140 were identified by the three prediction programs, the Agu2A’ signal peptide was identified only by TatP and PRED-TAT. Among the final set 40 putative Tat signal peptides contained the typical twin-arginine repeat, while two possessed an RK (PA14_46750 and PA14_64720) and two a KR (PA14_44100 and PA14_09900). Altogether our in silico analysis identified 44 putative Tat signal peptides in P. aeruginosa PA14. 37 of these have never been experimentally tested for their dependence on the Tat system.

VENN diagram illustrating the comparison between the three Tat prediction program outputs. TATFIND (orange), TatP (green), and PRED-TAT (blue) output of P. aeruginosa proteome analysis. The numbers present inside the circles represent the total number of proteins identified with one, two or three prediction programs. Arrows from each overlapping output point to lists of P. aeruginosa candidate signal peptides tested in the study. These include 7 already known Tat substrates (underlined): with PA14_53360 (PlcH), PA14_21110 (PlcN), PA14_13330 (EddA), PA14_33740 (PvdP), PA14_33720 (PvdN), PA14_48450 (Agu2A’) and PA14_10370 (PA4140). Signal peptides shown later to target efficiently AmiAH to the Tat pathway (Fig. 6) are indicated in bold.
Development of the amidase Tat reporter assay
To experimentally validate selected putative P. aeruginosa Tat signal peptides, we next used the in vivo amidase reporter assay that we participated in developing for E. coli Tat candidates (ref.23 and Fig. 2). This assay indicates whether signal peptides of candidate Tat substrates can engage with the Tat pathway and permit translocation. It uses an E. coli strain where the Tat signal sequences of AmiA and AmiC, two Tat-dependent N-acetylmuramoyl-L-alanine amidases, have been deleted (ΔssamiAC41). AmiA and AmiC hydrolyse the peptide moiety from N-acetylmuramic acid in peptidoglycan during cell wall biogenesis. ΔssamiAC forms chains of cells and cannot grow in the presence of SDS because the peptidoglycan barrier is damaged. Notably these two phenotypes can be restored in a strictly Tat-dependent manner by AmiA sole production in trans. In the amidase-reporter assay a candidate signal peptide is fused to the mature AmiA coding sequence in place of the cognate Tat signal peptide and the chimeric protein is produced in the ΔssamiAC mutant. True Tat signal peptides allow the Tat-dependent translocation of AmiA and restore the ability of ΔssamiAC to form single cells and to grow in the presence of SDS. If the Tat signal peptide candidate is not functional AmiA is not exported and the ΔssamiAC mutant still forms chains of cells and does not grow in the presence of SDS (Fig. 2). This system has been used to show the Tat-dependence of single substrates with signal sequences from Salmonella enterica serovar Typhimurium28, Archaeoglobus fulgidus26 and Streptomyces coelicolor27. More recently we used this system to confirm proteomic data indicating that PA2699 was a genuine extracellular Tat substrate in P. aeruginosa29. However, this assay has never been used to systemically screen candidate Tat substrates.

Schematic representation of the amidase reporter assay or SDS sensitivity: an easy genetic screen to probe Tat signal peptides. In E. coli two Tat-dependent amidases, AmiA and AmiC allow the correct cleavage of the peptidoglycan (PG) layer in the periplasm. In the ΔtatC mutant (B1Lk0), the absence of export of AmiA and AmiC leads to a defect in cell division and envelope integrity (schematized here by a grey outer membrane). Consequently the ΔtatC mutant forms chains of cells and is sensitive to detergent. A ΔssamiAC strain (MCDSSAC), where AmiA and AmiC signal sequences have been deleted shows identical phenotypes to the tatC mutant41. Only providing AmiA in trans in this strain (ΔssamiAC pssAmiA-AmiA) allows the restoration of both cell division and SDS resistance in a Tat dependent manner. The amidase assay has been designed around the E. coli MCDSSAC strain and around the fact that AmiA alone is able to restore the envelope defect of the strain. In this assay, if the AmiA signal peptide of pssAmiA-AmiA is replaced by a signal peptide which can be recognized by the Tat system, SDS sensitivity and chain formation can be restored, unlike when AmiA signal peptide is swapped for a sequence not recognized by the Tat machinery. In microscopic images, the white bar represents 10 μm.
Absence or insufficient protein synthesis of signal peptide-AmiA constructs could prevent complementation of the ΔssamiAC mutant, and be interpreted as an absence of transport. Therefore, to monitor the production of protein fusions constructed later in this work we modified the original ssAmiA-AmiA fusion with the addition of a C-terminal hexa-histidine (His6) epitope-tag (ssAmiA-AmiAH). We showed that the new ssAmiA-AmiAH fusion is still functional because it allows the restoration of a wild type phenotype (SDS resistance and single cell formation) when produced in ΔssamiAC (Fig. 3A and B). Importantly ssAmiA-AmiAH did not complement the ΔtatC mutation (a tat null mutant) for SDS sensitivity and chain formation indicating that AmiAH activity is strictly Tat dependent (Fig. 3A). The ssAmiA-AmiAH protein fusion was identified by immunoblot analysis using anti-His6 antibodies in wild type (WT), ΔtatC and ΔssamiAC strains (Fig. 3C and D). Whereas the WT and ΔssamiAC strains showed the precursor and the mature forms of ssAmiA-AmiAH in similar amounts (annotated p and m), the ΔtatC cells showed mostly the precursor form indicating a role for the Tat pathway in AmiAH export to the periplasm. This interpretation was confirmed after fractionation of WT cells since the precursor form remained associated with spheroplasts while the mature form was released into the periplasmic fraction (Fig. S1 lanes 3 and 4). In contrast in ΔtatC cells both forms remained associated with the spheroplast fraction indicating that the faster migrating band (indicated with an asterisk) is not an exported form of AmiAH but rather a cytoplasmic degradation product that migrates to the same position as the mature AmiAH (Fig. S1 lanes 5 and 6). This was confirmed by Proteinase K accessibility experiment: both protein bands were intact after Proteinase K treatment of spheroplasts but were degraded once cells were lyzed with Triton X-100 (Fig. S1 lanes 8 and 10). This indicates that both forms are located in the cytoplasm. Altogether, these results indicate that AmiA carrying a C-terminal histidine tag, AmiAH, is exported by the Tat system and that its activity can be used as an efficient reporter of Tat translocation.

The ssAmiA-AmiAH protein fusion is functional. (A) SDS viability assay of wild type (MC4100), ΔtatC (B1LK0) and ΔssamiAC (MCDSSAC) strains carrying the empty vector (pUNIPROM) or pssAmiA-AmiAH (producing E. coli AmiA with an His6 epitope-tag). Strains were grown aerobically and 5 μl were spotted on LB agar plate containing 2% (w/v) SDS as indicated in the material and methods section (B) Microscopic observation of ΔssamiAC strains carrying the empty vector or pssAmiA-AmiAH. Scale: the white bar represents 10 μm (C) Immunoblot analysis of whole cells prepared from wild type, ΔtatC and ΔssamiAC strains carrying the empty vector or pssAmiA-AmiAH. The predicted sizes of AmiAH precursor (p) and mature (m) forms are 34 kDa and 28.9 kDa respectively. A form corresponding to AmiAH degradation product is indicated with an asterisk in the ΔtatC strain. The molecular masses (in kilodaltons) are indicated on the left of the gels. Full-length blots are presented in Supplementary Fig. 4 (D) The immunoblot bands from panel C were quantified from 6 experiments by densitometry analysis using the ImageQuant software.
Experimental validation of the amidase Tat reporter assay for P. aeruginosa signal peptides
Next we tested whether the amidase reporter system can recognize P. aeruginosa Tat signal peptides and robustly differentiate between Sec and Tat signal peptides. Fusions to all known P. aeruginosa Tat substrate signal peptides were constructed and tested for rescue of the ΔssamiAC mutant SDS sensitivity and chain formation phenotypes (Fig. 4A and B). All constructs were made by fusion of each signal peptide to the AmiAH mature domain (without signal peptide) thus creating protein fusions bearing the native E. coli tatABC promoter and Shine-Dalgarno (SD) sequence with identical distance between the SD sequence and initiation codon23. The production of each protein fusion was monitored by immunoblot analysis of whole cell fractions (Fig. 4C). Notably we found that Tat signal peptides from all known P. aeruginosa Tat substrates allowed the export of the AmiAH reporter protein and rescued the ΔssamiAC mutant phenotypes, even in the case of the least produced ones (PA2699 and EddA). To assess the reporter assay specificity we next tested its ability to discriminate between P. aeruginosa Sec and Tat signal peptides by constructing protein fusions between AmiAH and two Sec signal peptides (LasB and PA2377). We showed that neither of these Sec signal peptides rescued chain formation nor growth of the reporter strain on SDS (Fig. 4A and B) even if they were produced correctly (Fig. 4C). Finally, we studied the cellular localisation of AmiAH fused to the archetypal Tat (PlcH) and Sec (LasB) signal peptides after fractionation of a WT and a ΔtatC mutant (Fig. 5). Whereas AmiAH fused to PlcH Tat signal peptide was recovered in the periplasm as a mature form in WT cells, it accumulated clearly as a precursor in the spheroplasts of a ΔtatC mutant. This indicates that when fused to a Tat signal peptide AmiAH was exported to the periplasm in a strictly Tat-dependent manner (Fig. 5A). When AmiAH was fused to the Sec signal peptide of LasB, a single band was observed at the expected size of the precursor in WT and ΔtatC strains (Fig. 5B). After cell fractionation, this band stayed associated with the spheroplasts. This indicates that AmiAH was not exported to the periplasm when fused to a Sec signal peptide but was instead retained in the spheroplasts. We conclude from this set of experiments that P. aeruginosa Tat signal sequences can be efficiently and specifically recognized by the E. coli Tat machinery and that the E. coli amidase reporter assay can distinguish between the Sec and Tat-dependent nature of P. aeruginosa N-terminal regions.

The amidase assay discriminates between P. aeruginosa Tat- and Sec-dependent signal peptides. SDS viability assay (A), chain formation (B), and immunoblot analysis (C) of E. coli ΔssamiAC mutant (MCDSSAC) strain carrying pUNI-PROM (Empty vector), pssAmiA-AmiAH (producing E. coli AmiA with an His6 epitope-tag) or derivatives of pssAmiA-AmiAH where AmiA signal sequence has been replaced by the putative signal sequences of PA14 P. aeruginosa Tat-dependent and Sec-dependent known substrates. For each putative signal sequence the most likely twin-arginine motif is indicated in bold, the hydrophobic region is underlined and the predicted signal peptidase cleavage site is indicated by a star. For SDS-resistance assay, strains were grown aerobically and 5 μl were spotted on LB plate containing 2% (w/v) SDS where indicated. For microscopic observations the white bar represents 10 μm. For immunoblot analysis whole cell extracts were separated on a 12% SDS-PAGE gel and blots were probed with anti-his tag (His6). The predicted sizes of each signal peptide are indicated in amino acid at the bottom of the gel and the unprocessed (p) and processed (m) forms are indicated on the right of the gel. The molecular weight standards (in kilodaltons) are indicated on the left of the gels. The white line separates lanes from non-adjacent part of the same gel. Full-length blots are presented in Supplementary Fig. 4.

Localization of AmiAH fused to the archetypical Tat and Sec-dependent signal peptides. Immunoblot analysis of WT (MC4100) and ΔtatC (B1LK0) strains carrying pssPlcH-AmiAH (A) or pssLasB-AmiAH (B) after cell fractionation (where E. coli AmiA signal peptide has been replaced with P. aeruginosa PlcH or LasB signal peptide and where AmiA carries an His6 epitope-tag). Whole cell extracts (WC), spheroplasts (Sph) and periplasm (P) were separated on a 12% SDS-PAGE gel. Blots were probed with anti-his tag (His6), anti-elongation factor thermo unstable (EF-Tu) and anti-beta lactamase (Bla) antibodies. EF-Tu (43.3 kDa) is used as a cytoplasmic control and Bla (28.9 kDa) as a periplasmic control. The predicted sizes of ssPlcH-AmiAH unprocessed (precursor p) and processed (mature m) forms are 32.9 kDa and 28.6 kDa respectively while the predicted size of ssLasB-AmiAH unprocessed (precursor p) is 31.3 kDa and are indicated on the right of the gel. The molecular masses (in kilodaltons) are indicated on the left of the gels. A white line separates lanes from non-adjacent part of the same gel and with different exposures. Full-length blots are presented in Supplementary Fig. 4.
Systematic analysis of the P. aeruginosa candidate signal peptides by the amidase reporter assay
To determine the Tat-dependence of the 37 newly identified P. aeruginosa candidate Tat signal peptides we fused each candidate to the AmiAH mature domain and tested their capacity to engage the Tat pathway with the amidase reporter assay. The length of each candidate signal peptide was defined after prediction of signal peptidase cleavage site as described in Material and methods. For one of the candidates, PA14_15670, two versions of the region encoding the putative Tat signal peptide were cloned because two possible signal peptidase cleavage sites were predicted (AGG* and QVA*) (Table 1A). We systematically analyzed the SDS sensitivity and chain formation phenotypes of ΔssamiAC carrying each of the 38 fusion proteins (Fig. 6). Out of the 38 constructs tested, 26 clearly complemented the ΔssamiAC mutation indicating that they were real Tat signal peptides. Notably both versions of the PA14_15670 signal peptide (constructed with two different cleavage sites) restored the ΔssamiAC mutant phenotypes. Observation of the production and export of AmiAH fused to these two signal peptides indicate however that the export is slightly more efficient when AmiAH is fused after the AGG codon (Fig. S2). In contrast, 12 out of the 37 Tat candidates tested did not complement the ΔssamiAC mutation even if they were correctly produced (Figs 6 and S3). This indicates that these candidates are not authentic Tat signal peptides. Notably, these include four candidates that contain the unusual KR (PA14_44100 and PA14_09900) and RK (PA14_46750 and PA14_64720) motifs. In all, this approach led to the identification of 25 new Tat signal peptides in P. aeruginosa bringing the total number of P. aeruginosa Tat substrates to 33.

Screening of the putative Tat candidates by the amidase reporter assay. SDS viability assay of E. coli ΔssamiAC mutant (MCDSSAC) strain carrying derivatives of pssAmiA-AmiAH where AmiA signal sequence has been replaced by the 39 candidate Tat signal peptides identified in silico.
Identification of a new periplasmic Tat substrate with a missassigned start codon
Recently, a comparative proteomic approach allowed us to identify a new Tat substrate in P. aeruginosa with a missassigned start codon, PA269929. Our in silico approach misses such substrates because the screening was performed on the P. aeruginosa predicted proteome. Consequently, to identify other putative Tat substrates that may have missassigned start codons we ran a modified TATFIND prediction program on all PA14 open reading frames (instead of the P. aeruginosa protein database) bigger than 150 base pairs starting with an ATG and having two codons encoding RR, RK and KR within the first 120 nucleotides. This analysis led to the identification of 523 potential Tat peptides encoded by the 33416 putative ORFs tested (Table S1). As expected, amongst these we found all the putative Tat signal peptides starting with an ATG that were identified with the previous analysis. Next we manually curated the remaining hits to identify putative Tat signal peptides by removing (i) sequences in the opposite orientation to an annotated gene, (ii) sequences not directly in frame with an annotated gene and (iii) sequences further than 150 nucleotides from the start codon of an annotated gene. The remaining sequences were then analyzed for the presence of signal peptides using the SignalP, TatP and PRED-TAT algorithms and scrutinized for the presence of a putative SD sequence upstream of the putative missassigned start codon. This analysis led to the identification of one more Tat candidate, PA14_70330 or SphC. Of note PA2699, which we identified previously with a missassigned initiation codon, was not identified in this analysis because the TATFIND program does not recognize the PA2699 Tat signal peptide29.
Analysis of the nucleotide sequence of the annotated sphC gene showed the presence of another ATG codon 66 nucleotides upstream, with a typical SD sequence, and in frame with the annotated start codon of sphC (Fig. 7A). The utilization of an algorithm developed to identify bacterial start codons showed that the upstream ATG is better than the annotated ATG with a score of 28 compared with a score of 17 (a score of ≥26 is considered an initiation codon)42. Moreover, if the upstream ATG is used as a translation start (M−22) SphC is then predicted to possess a Tat signal peptide by the three prediction programs, with a clear Tat motif SRRQLL, a hydrophobic stretch and a typical leader peptidase cleavage site ALA (Fig. 7A).

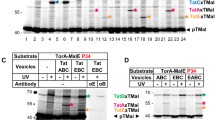
PA14_70330 is a new periplasmic Tat substrate with a missassigned start codon. (A) Nucleotide sequence of the PA14_70330/sphC upstream region containing the start codon annotated on the PA14 genome (M1) and the putative missassigned start codon (M-22) identified in this study. Deduced amino acid sequence of SphC is shown below the nucleotide sequence. A putative SD sequence based on the canonical AGGAGGU is underlined. The forward primer used to PCR amplifies sphC and its promoter region is underlined (dotted line). On the putative SphC protein sequence, the Tat motif is indicated in bold, the hydrophobic region is in italics, and the signal peptidase I cleavage site is indicated by an arrow. (B) SDS viability assay of E. coli ΔssamiAC mutant (MCDSSAC) strain carrying pUNI-PROM (empty vector) and pssSphCM-22-AmiAH (SphCM-22). (C) and (D) Immunoblot analysis of whole cells, extracellular fraction, spheroplasts (Sph) and periplasmic (P) fractions of PA14 (WT) and PA14Δtat (Δtat) carrying pJNSphCM-22H (RR) and pJNSphCM-22H-KK (KK), grown in phosphate depleted medium (C) or in rich medium (D) supplemented with arabinose as described in material and methods. The predicted sizes of SphCM-22H precursors (p) and mature (m) forms are respectively 52.6 kDa and 49.5 kDa. PlcH (precursor: 82.7 kDa, mature form: 78.3 kDa) is used as supernatant quality control. The protein disulfide isomerase DsbA (23.3 kDa) and EF-Tu (43.3 kDa) are shown as periplasmic and cytoplasmic controls respectively. The asterisk in panel D indicates an aspecific band. Molecular weight markers are indicated on the left of the blot. A white line separates lanes from different gels (C) or from non-adjacent part of the same gel (D). Full-length blots are presented in Supplementary Fig. 4.
First, to test the capacity of SphC putative N-terminal region (from M−22 to A8) to act as a Tat signal peptide, we fused it to AmiAH mature domain and introduced the construct into the ΔssamiAC reporter strain. Production of the SphCM-22 signal peptide-AmiAH fusion confered SDS resistance and rescued the chain formation phenotype of the reporter strain indicating that SphC putative N-terminal region contains an active Tat signal peptide (Fig. 7B). Next, we decided to confirm the dependence of SphC on the Tat system in vivo in P. aeruginosa. We constructed a C-terminal His6 epitope-tagged version of the protein (SphCM-22-H) and assessed its subcellular localization in the WT and ΔtatC strains. Immunobloting identified SphCM-22-H in both strains in whole cells rather than in the extracellular fraction indicating that the protein is not secreted (Fig. 7C). As expected for a Tat-dependent substrate, SphCM-22H was present as a slower migrating band (precursor) in the Δtat mutant in comparison to the WT strain where SphCM-22H migrated at the expected size for the mature form. In contrast when we replaced the two essential arginines of the Tat consensus sequence by two lysines (thus blocking Tat export) SphCM-22H-KK appeared as a precursor form in both WT and Δtat mutant strains confirming the Tat dependent export of SphCM-22H in P. aeruginosa (Fig. 7C). Finally, upon cellular fractionation, SphC was found in the periplasmic fraction of the WT strain and accumulated as a precursor form in a tatC background or when the twin-arginines where replaced by lysines (Fig. 7D). Altogether, our in silico approach and in vivo results reveal the existence of another Tat substrate with a missassigned start codon, SphC, and demonstrate that it is a periplasmic protein rather than a cytoplasmic protein. In conclusion, our previous data and the work presented here show that P. aeruginosa possesses 34 Tat substrates of which 2 have missassigned start codons.
Discussion
In this work we employed a combination of a stringent in silico analysis and in vivo reporter fusion assays to reveal that P. aeruginosa encodes 34 proteins with functional Tat signal peptides and by extrapolation strongly suggesting that P. aeruginosa contains 34 Tat substrates (Table 2). Of these 34 proteins, 8 were previously identified as Tat substrates, and the signal peptides of the remaining 26 were experimentally shown to allow Tat export for the first time in this study. Previously, manual screens of the P. aeruginosa genome identified 18 and 27 putative Tat signal peptides, with none or little experimental validation33,38. Here, we used a stringent in silico approach to identify potential Tat substrates: selecting only those substrates identified by at least two independent algorithms, relaxing the search criteria to allow identification of unusual Tat signal peptides, and additionally scanning the translated genome to identify substrates with missassigned start codons. Interestingly, compared to previous studies on P. aeruginosa, we found 15 new substrates, but we also did not identify all the predicted substrates previously reported. This is not surprising, considering that these studies were performed with a large manual component, and that even the expert algorithms that we used did not fully overlap in their predictions (Fig. 1). Therefore, the 34 experimentally validated Tat substrates that we identify represent the high quality core complement of P. aeruginosa Tat substrates. Our findings do not exclude that the P. aeruginosa proteome may harbor a limited number of other atypical Tat substrates. Interestingly, 24 of the Tat signal peptides we validated were predicted by the three Tat prediction programs, while four were predicted by TATFIND and TatP, one by TATFIND and PRED-TAT and three by TatP and PRED-TAT (Fig. 1). The signal peptides of SphC and PA2699 whose start codons are missassigned (Fig. 7 and ref.29) were predicted as Tat signal peptide respectively by the three prediction programs and by TatP and PRED-TAT. Altogether this shows the limitation of single prediction programs and highlights the need for experimental validation of Tat export.
Here we used an improved amidase reporter assay that we initially developed in E. coli23. We use it for the first time for the systematic screening of substrates identified in silico. We improved the assay with the addition of a C-terminal epitope tag to AmiA thus allowing detection and localization of the various protein fusions constructed. Importantly, the epitope tag allowed us to show that AmiA export is strictly Tat-dependent when fused to a Tat signal peptide and that the assay works even when the protein fusion is not strongly produced highlighting the sensitivity of the assay (Fig. 3). In addition we also showed that the amidase assay is robust, and can reliably discriminate Sec from Tat signal peptides (Fig. 4). Notably, while the assay was developed for E. coli signal peptides, we show here that it reliably recognizes P. aeruginosa signal peptides (Fig. 4). This is undoubtedly due to the fact that the substrates recognition sites on the E. coli and P. aeruginosa TatC protein (the receptor of the system) are perfectly conserved43. We therefore anticipate that the E. coli amidase reporter assay will be broadly applicable for the identification of Tat signal peptides from other organisms.
To visualize the sequence information content in P. aeruginosa Tat signal peptides we generated sequence logos with the 34 Tat signal peptides, from two amino acids before the twin-arginine dipeptide and from the signal peptidase I cleavage site (Table 2 and Fig. 8). Notably, the hallmark of Tat signal peptide can be visualized with the presence of the S/TRRXFLK motif where the twin-arginine dipeptide is strictly conserved. The FLK amino acids are also highly conserved despite the lysine not being present at a frequency in excess of 50% as defined previously7 but rather at a frequency around 32% (Fig. 8A). In addition the logo shows that the H-region, starting after the Tat motif (Fig. 8B residues −6 to −22), contains a significant number of glycine residues as is usually the case in Tat signal peptides. It also contains the typical leucine and alanine residues present in the hydrophobic region. Tat signal peptides are distinctly longer than Sec signal peptides with an average length of 38 amino acids compared to 24 amino acids44. Here the signal peptides vary between 23 and 60 amino acids with an average length of 34 amino acids. As previously noted8, the extended length arises mainly from a longer N-region (Table 2). Earlier studies suggested that Tat signal peptides contained positively charged amino acids in the C-region that could play the role of a “Sec avoidance signal”10. Notably, the sequence logo does not illustrate a high occurrence of lysine or arginine corresponding to such motif and only few P. aeruginosa Tat signal peptides contain positively charged amino acids in the C-region (Fig. 8B and Table 2). Finally, the sequence alignment shows that the canonical signal peptidase I cleavage site AXA is largely present in the C-region. Interestingly, we did not find any signal peptides with a signal peptidase II cleavage site as can be found in Tat signal peptides from Streptomyces and haloarchae9,22. Altogether we conclude from this analysis that P. aeruginosa Tat signal peptides share most of the characteristics of classical Tat signal peptides.

P. aeruginosa Tat signal peptide logo derived from signal peptides identified in this work and found in Table 2. Sequence logos of signal peptides aligned either (A) from two amino acids before the conserved twin-arginine or (B) from their cleavage sites. Amino acids are colored according to their chemical properties: polar amino acids (G, S, T, Y, C, Q, N) are green, basic (K, R, H) blue, acidic (D, E) red and hydrophobic (A, V, L, I, P, W, F, M) amino acids are black. The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each amino at that position.
The 34 Tat substrates identified previously and in this work are listed Table 2 along with their predicted function, the biological process they are involved in when known, their size and their predicted association with a metal cluster or a cofactor. For a long time the Tat system was thought to be mainly involved in the transport of proteins containing cofactors that needed to be folded in the cytoplasm before export, because the biosynthetic machinery for these cofactors was cytoplasmic. However, later studies showed that the Tat system was also involved in the export of cofactor-less proteins which for unknown reasons need to be folded before their export. Here half (17 out of 34) of the P. aeruginosa Tat substrates are predicted to contain cofactors. These cofactors can be classified into nucleotide based cofactors such as molybdopterin (7 substrates) and flavin (4 substrates), and metal clusters such as iron-sulfur clusters (3 substrates), copper centers (4 substrates) and zinc clusters (1 substrate). For comparison, in E. coli K12 where Tat substrates are the most characterized, 68% of Tat substrates (19 out of 28) bind cofactors of similar nature44. The Tat pathway is notorious for its versatility. Indeed, in addition to the export of folded soluble proteins, it can also export lipoprotein9,22, protein complexes where only one of the partners has a Tat signal peptide45, and proteins with N- or C-terminal anchors46,47. None of the P. aeruginosa Tat substrates are lipoproteins. One Tat substrate, FdnG, the major subunit of the formate dehydrogenase-O, is known to be exported as a complex with its partner FdnH (which is signal peptide-less) in other organisms. Once exported FdnH is probably attached to the inner membrane through a C-terminal transmembrane anchor48. Another of the P. aeruginosa Tat substrates is the Rieske iron sulfur protein, which is a typical Tat substrate whose N-terminal Tat signal peptide is not cleaved and is used as a transmembrane anchor instead46. The P. aeruginosa Tat system, in addition to transporting soluble proteins, therefore also appears capable of exporting protein complexes and proteins with N or C-terminal anchors.
As mentioned before Tat substrates have been best characterized in E. coli K12, as much for their identity as for their function44. Comparison of the P. aeruginosa Tat substrates with E. coli Tat substrates shows that they share the same range of sizes ranging from around 20 kDa (20.3 kDa for YbdK in E. coli and 20.8 kDa for PA14_57570 in P. aeruginosa) to around 110 kDa (FdnG for both organisms). Surprisingly only five Tat substrates are common between P. aeruginosa and E. coli: AmiC, CueO/CopA, FdnG, NapA and YedY. In addition to these E. coli orthologues, P. aeruginosa shares Tat substrates with known Tat substrates from other organisms. These include: the nitrous oxide reductase, NosZ, in Ralstonia eutropha49 and Pseudomonas stutzeri50, the Rieske protein in Paracoccus denitrificans46 and Legionella pneumophila51, the phosphodiesterase PhoD in Bacillus subtilis52, the phospholipases C PlcA and PlcB in Mycobacterium tuberculosis21,53. Analysis of the substrates with known function reveals that the P. aeruginosa Tat system is involved in a wide spectrum of functional classes centered on the adaptation of the bacteria to its niche and establishment of an infection. There are substrates involved in growth in the presence or absence of oxygen54, in survival during stress conditions (iron deficiency55, elevated levels of copper56…) and in pathogenesis57,58. Notably, a significant proportion of the Tat substrates we identified still have hypothetical or unknown functions. Future work will be devoted to understanding how these substrates promote P. aeruginosa survival and adaptation in a variety of ecological niches, including the eukaryotic host.
Material and Methods
In silico genome and sequence analysis
A modified version of TATFIND was used to screen the P. aeruginosa PA14 proteome for the presence of Tat signal peptides (File S1). For the identification of P. aeruginosa Tat substrates with TatP (http://www.cbs.dtu.dk/services/TatP) we specified the regular expression [RK][KR].[FGAVML][LITMVF] for output filtering. Identifiers with at least 1 “Yes” amongst the five scores represented in the output of TatP were selected as candidates Tat signal peptides.
For the identification of P. aeruginosa Tat substrates with PRED-TAT we ran the PRED-TAT (HMMER) program (available http://www.compgen.org/tools/PRED-TAT/supplement) on all PA14 predicted proteins then submitted the hits we obtained to the PRED-TAT webserver (http://www.compgen.org/tools/PRED-TAT/) using both the “original” and the “new” models. This analysis predicted 32 Tat substrates (Table S1).
For the identification of P. aeruginosa Tat substrates which might have a missassigned start codon, all PA14 ORFs of 50 amino acids starting with an ATG and having RR or RK or KR within the first 20 amino acids were extracted from a genbank file downloaded from https://www.ncbi.nlm.nih.gov/nuccore/NC_008463.1/ leading to 33416 sequences. The obtained amino acid sequences were then used as input to the modified TATFIND program.
For manual signal peptides prediction and analysis, SignalP3.0 (http://www.cbs.dtu.dk/services/SignalP-3.0/), TATFINDv1.4 (http://signalfind.org/TATFIND.html), TatP1.0 and PRED-TAT algorithms were used on the first 70 residues of P. aeruginosa PA14 proteins. Signal peptides domain prediction (N, H and C) was performed using SignalP 3.0 (HMM) and Phobius. Most likely signal peptide cleavage sites were predicted by SignalP3.0, TatP1.0 and PRED-TAT and after looking for sequence conservation of the cleavage site in orthologues if necessary. Identification of Shine-Dalgarno (SD) sequence was based on the AGGAGGT sequence. Identification of the most likely start codon was achieved using the Kolaskar and Reddy method that analyse the −18 to +18 nucleotides around the ATG/GTG42. Sequence logos were constructed using the web based application WebLogo and default settings http://weblogo.berkeley.edu/logo.cgi.
Bacterial strains, plasmids and growth conditions
E. coli strains were cultured in Lysogeny broth (LB) with antibiotics as required (50 μg ml−1 ampicillin (Ap), 25 μg ml−1 kanamycin (Kan), 15 μg ml−1 gentamicin (Gm). The E. coli DH5α strain was used to propagate plasmids. Recombinant plasmids were introduced in the reference strain P. aeruginosa PA14 using pRK201359 and transconjugants selected on Pseudomonas isolation agar (PIA, Difco Laboratories) supplemented with antibiotics as required (150 μg ml−1 Gm). P. aeruginosa strains were cultured in LB with antibiotics as required (50 μg ml−1 Gm). For E. coli whole cell analysis cells were grown in LB medium containing ampicillin overnight at 37 °C. For cell fractionation of P. aeruginosa strains carrying pJN105 cells were grown either in LB medium at 37 °C and arabinose (0.4%) was added after 4 hours of growth for 1 hour or in low inorganic phosphate medium containing 0.4% glucose at 30 °C until stationary phase (14 hours of growth) and arabinose (0.05%) was added at the beginning of the growth.
Plasmid and strain construction
The plasmids and strains constructed in this work are shown in Table S2, primers are shown in Table S3. All amplifications were carried out with Q5 High Fidelity DNA polymerase (New England Biolabs), using PA14 genomic DNA as template. All constructs were sequenced to verify the absence of any mutation (GATC-biotech).
Amidase plasmids are derivatives of pssAmiA-AmiAH (based on the pT7.5 vector60) and were generated by PCR amplification of DNA encoding the N-terminal region of candidates using P. aeruginosa PA14 chromosomal DNA as template. Primers used to amplify sequences encoding signal peptide from each protein were designed to include the sequence between the start codon and the most likely signal peptidase cleavage site.
In pssAmiA-AmiAH derivatives, AmiA signal sequence was replaced by each candidate N-terminal sequence by enzymatic restriction using BamHI and XbaI or one-step sequence- and ligation-independent cloning (SLIC) as described in61. Each construct allows the expression of N-terminal genetic fusion to mature (signal peptide lacking) AmiA carrying a C-terminal hexa-histidine tag under the control of the tatABC constitutive promoter. Each of these clones carries the tatA ribosome-binding site, with identical spacing between the ribosome-binding site and the start codon.
pJNSphCM-22H (encoding a C-terminal hexa-histidine tag on SphC) was constructed by PCR amplification of sphC and a 97 bp region upstream of the annotated start codon including the putative missassigned methionine (M−22) and Shine Dalgarno sequence from P. aeruginosa strain PA14 in two steps. First, sphC was amplified using sphCSDup/sphCHisXbaIdown and cloned into pCR2.1 plasmid (TA cloning, Invitrogen). Six histidine codons followed by the stop codon were included in the reverse primer. Secondly, this construct was cloned into the EcoRI site of pJN105 under the transcriptional control of an arabinose-inducible promoter62. pJNSphCM-22H-KK was constructed by mutating the CGA and CGC codons encoding putative arginines respectively 16 and 15 amino acid upstream of the annotated start codon of sphC (R−16 and R−15) in Lysine by QuickChange site-directed mutagenesis (Stratagene) using sphCKKup/sphCKKdown and pCR2.1-SphCM-22H as template then cloning this construct into the EcoRI site of pJN105 under the transcriptional control of an arabinose-inducible promoter.
Microscopy and outer membrane integrity assay
For phase-contrast microscopy, cells were grown to mid-exponential phase at 37 °C in LB medium and mounted on microscope slides covered with Poly-L-Lysine (Sigma-Aldrich) to immobilized cells. Fixed cells were imaged with a Zeiss AxioImager m2 equipped with a Hamamatsu OrcaR2 camera with a 100 × phase-contrast objective. The images were acquired with AxioVision software. For viability measurements, overnight cultures were adjusted to an OD600 of 1, serial dilutions from 10−1 to 10−6 were prepared in LB, and 5 µl of each dilution were spotted onto LB agar or LB agar + 2% (w/v) SDS. Plates were incubated overnight at 37 °C and photographed.
Preparation of extracts, cell fractionation, protease accessibility experiments and analysis by SDS-PAGE and immunobloting
For E. coli whole cell extracts, cells (1 ml) were pelleted by centrifugation for 2 min at 13,000 × g at room temperature and resuspended in denaturing buffer (63 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 0.0025% bromophenol blue, 10% beta-mercaptoethanol). Fractionation of E. coli cells into spheroplasts (cytoplasm and membranes) and periplasmic fractions were done as described previously63 with slight modifications. Briefly, cells (20 ml) were pelleted by centrifugation for 10 min at 2,000 × g, and resuspended in 0.2 ml of spheroplast buffer (0.2 M Tris-HCl pH 8.0, 0.5 M sucrose and 0.5 M EDTA). A freshly prepared solution (10 μl) of lysozyme (2 mg/ml in water) was added, followed by the addition of 0.4 ml of spheroplast buffer in 0.4 ml of water. The cells were gently mixed and then incubated for 20 minutes at 4 °C. After 10 minutes the suspension was brought to 250 mM NaCl to facilitate the release of AmiAH as described before64. The spheroplasts were pelleted by centrifugation at 2,000 × g for 10 minutes and resuspended in 0.5 ml of spheroplast buffer 0.5X. The supernatant was reserved as the periplasmic fraction.
For protease accessibility experiments, the spheroplasts were incubated for 20 minutes on ice with a solution (13 μl) of Proteinase K (PK) (250 μg/ml) in the presence or the absence of 1% (v/v) Triton X-100 (TX). Next, PMSF, a Proteinase K inhibitor (2 mM) is added to the suspension. Samples (whole cells, spheroplasts, periplasm, spheroplasts treated with PK+/−TX) were precipitated with 12% TCA overnight at 4 °C and centrifuged at 13,000 × g for 30 minutes. Pellets were washed twice with 90% acetone (1 ml), air dried and resuspended in denaturing buffer.
P. aeruginosa whole cells, supernatant, spheroplasts and periplasmic fractions were prepared as described before29,65.
Protein samples derived from equivalent amounts of culture (i.e. optical density equivalents) were loaded in each lane and analyzed by SDS-PAGE and immunoblotting as described before29 using primary polyclonal antibodies directed against DsbA (kindly gifted by K.E. Jaeger – university of Heinrich-Heine, dilution 1:25000) and PlcH (kindly gifted by M.L. Vasil – University of Colorado, dilution 1:500), or monoclonal antibodies directed against EF-Tu (Hycult-biotech, dilution 1:20000), His6 epitope-tag (Penta His, Qiagen, dilution 1:1000) and Bla (Millipore 1:5000). For anti His6 detection, manufacturer instructions were followed. Peroxidase-conjugated anti-Mouse or anti-Rabbit IgGs (Sigma, dilution 1:5000) were used as secondary antibodies. Nitrocellulose membranes were developed with homemade enhanced chemiluminescence and exposed to X-ray film for the appropriate time or were scanned using ImageQuant LAS4000 analysis software (GE Healthcare Life sciences). Protein samples equivalent to 0.1 OD600 units were loaded for whole cell and spheroplasts analysis while protein samples equivalent to 0.2 OD600 units (anti-DsbA and anti-EF-Tu immunoblots) or 1 OD600 units (anti-His6 immunoblot) were used for periplasm analysis and protein samples equivalent to 1 OD600 units were used for supernatant analysis.
Data availability
All data generated or analysed during this study are included in the published article (and its Supplementary Information files).
References
Orfanoudaki, G. & Economou, A. Proteome-wide Subcellular Topologies of E. coli Polypeptides Database (STEPdb). Mol. Cell. Proteomics 13, 3674–3687 (2014).
Dalbey, R. E. & Kuhn, A. Protein Traffic in Gram-negative bacteria - how exported and secreted proteins find their way. FEMS Microbiology Reviews 36, 1023–1045 (2012).
Berks, B. C., Palmer, T. & Sargent, F. Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Current Opinion in Microbiology 8, 174–181 (2005).
De Buck, E., Lammertyn, E. & Anné, J. The importance of the twin-arginine translocation pathway for bacterial virulence. Trends in Microbiology 16, 442–453 (2008).
Hegde, R. S. & Bernstein, H. D. The surprising complexity of signal sequences. Trends in Biochemical Sciences 31, 563–571 (2006).
von Heijne, G. Signal sequences. The limits of variation. J. Mol. Biol. 184, 99–105 (1985).
Berks, B. C. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22, 393–404 (1996).
Cristóbal, S., De Gier, J. W., Nielsen, H. & Von Heijne, G. Competition between Sec- and TAT-dependent protein translocation in Escherichia coli. EMBO J. 18, 2982–2990 (1999).
Giménez, M. I., Dilks, K. & Pohlschröder, M. Haloferax volcanii twin-arginine translocation substates include secreted soluble, C-terminally anchored and lipoproteins. Mol. Microbiol. 66, 1597–1606 (2007).
Bogsch, E., Brink, S. & Robinson, C. Pathway specificity for a ΔpH-dependent precursor thylakoid lumen protein is governed by a ‘Sec-avoidance’ motif in the transfer peptide and a ‘Sec-incompatible’ mature protein. EMBO J. 16, 3851–3859 (1997).
Nielsen, H. Protein sorting prediction in. Methods in Molecular Biology 1615, 23–57 (2017).
Rose, R. W., Brüser, T., Kissinger, J. C. & Pohlschröder, M. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol. Microbiol. 45, 943–950 (2002).
Bendtsen, J. D., Nielsen, H., Widdick, D., Palmer, T. & Brunak, S. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6 (2005).
Bagos, P. G., Nikolaou, E. P., Liakopoulos, T. D. & Tsirigos, K. D. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26, 2811–2817 (2010).
Jongbloed, J. D. H. et al. Selective contribution of the twin-arginine translocation pathway to protein secretion in Bacillus subtilis. J. Biol. Chem. 277, 44068–44078 (2002).
Pradel, N. et al. Contribution of the twin arginine translocation system to the virulence of enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 71, 4908–4916 (2003).
Maffei, B., Francetic, O. & Subtil, A. Tracking Proteins Secreted by Bacteria: What’s in the Toolbox? Front. Cell. Infect. Microbiol. 7 (2017).
Santini, C. L. et al. Translocation of Jellyfish Green Fluorescent Protein via the Tat System of Escherichia coli and Change of Its Periplasmic Localization in Response to Osmotic Up-shock. J. Biol. Chem. 276, 8159–8164 (2001).
Ize, B. et al. In vivo dissection of the Tat translocation pathway in Escherichia coli. J. Mol. Biol. 317, 327–335 (2002).
Blaudeck, N., Kreutzenbeck, P., Freudl, R. & Sprenger, G. A. Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J. Bacteriol. 185, 2811–2819 (2003).
McDonough, J. A. et al. The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J Bacteriol. 187, 7667–79 (2005).
Widdick, D. A. et al. The twin-arginine translocation pathway is a major route of protein export in Streptomyces coelicolor. Proc. Natl. Acad. Sci. USA 103, 17927–17932 (2006).
Ize, B. et al. Remnant signal peptides on non-exported enzymes: Implications for the evolution of prokaryotic respiratory chains. Microbiology 155, 3992–4004 (2009).
Huang, Q. et al. A signal sequence suppressor mutant that stabilizes an assembled state of the twin arginine translocase. Proc. Natl. Acad. Sci. 114, E1958–E1967 (2017).
Huang, Q. & Palmer, T. Signal peptide hydrophobicity modulates interaction with the Twin-Arginine translocase. MBio 8 (2017).
Coulthurst, S. J., Dawson, A., Hunter, W. N. & Sargent, F. Conserved signal peptide recognition systems across the prokaryotic domains. Biochemistry 51, 1678–1686 (2012).
Keller, R., de Keyzer, J., Driessen, A. J. M. & Palmer, T. Co-operation between different targeting pathways during integration of a membrane protein. J. Cell Biol. 199, 303–315 (2012).
James, M. J., Coulthurst, S. J., Palmer, T. & Sargent, F. Signal peptide etiquette during assembly of a complex respiratory enzyme. Mol. Microbiol. 90, 400–414 (2013).
Ball, G. et al. Contribution of the Twin Arginine Translocation system to the exoproteome of Pseudomonas aeruginosa. Sci. Rep. 6 (2016).
Pendleton, J. N., Gorman, S. P. & Gilmore, B. F. Clinical relevance of the ESKAPE pathogens. Expert Review of Anti-Infective Therapy 11, 297–308 (2013).
Kipnis, E., Sawa, T. & Wiener-Kronish, J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Medecine et Maladies Infectieuses 36, 78–91 (2006).
Voulhoux, R. et al. Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 20, 6735–6741 (2001).
Ochsner, Ua, Snyder, A., Vasil, A. I. & Vasil, M. L. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. USA 99, 8312–8317 (2002).
Voulhoux, R., Filloux, A. & Schalk, I. J. Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: The Tat system is required for PvdN but not for FpvA transport. J. Bacteriol. 188, 3317–3323 (2006).
Nadal-Jimenez, P. et al. PvdP is a tyrosinase that drives maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. J. Bacteriol. 196, 2681–2690 (2014).
Williams, B. J. et al. Discovery of an operon that participates in agmatine metabolism and regulates biofilm formation in Pseudomonas aeruginosa. Mol. Microbiol. 76, 104–119 (2010).
Palmer, T. & Berks, B. C. The twin-arginine translocation (Tat) protein export pathway. Nature Reviews Microbiology 10, 483–496 (2012).
Lewenza, S., Gardy, J. L., Brinkman, F. S. L. & Hancock, R. E. W. Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 15, 321–329 (2005).
Hinsley, A. P., Stanley, N. R., Palmer, T. & Berks, B. C. A naturally occurring bacterial Tat signal peptide lacking one of the ‘invariant’ arginine residues of the consensus targeting motif. FEBS Lett. 497, 45–49 (2001).
Ignatova, Z., Hörnle, C., Kasche, V. & Nurk, A. Unusual signal peptide directs penicillin amidase from Escherichia coli to the tat translocation machinery. Biochem. Biophys. Res. Commun. 291, 146–149 (2002).
Ize, B., Stanley, N. R., Buchanan, G. & Palmer, T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol. Microbiol. 48, 1183–1193 (2003).
Kolaskar, A. S. & Reddy, B. V. B. A method to locate protein coding sequences in DNA of prokaryotic systems. Nucleic Acids Res. 13, 185–194 (1985).
Rollauer, S. E. et al. Structure of the TatC core of the twin-arginine protein transport system. Nature 492, 210–214 (2012).
Sargent, F., Berks, B. C. & Palmer, T. The Tat Protein Export Pathway. EcoSal Plus 4, 260–274 (2010).
Rodrigue, A., Chanal, A., Beck, K., Müller, M. & Wu, L. F. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J. Biol. Chem. 274, 13223–13228 (1999).
Bachmann, J., Bauer, B., Zwicker, K., Ludwig, B. & Anderka, O. The Rieske protein from Paracoccus denitrificans is inserted into the cytoplasmic membrane by the twin-arginine translocase. FEBS J. 273, 4817–4830 (2006).
Hatzixanthis, K., Palmer, T. & Sargent, F. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol. Microbiol. 49, 1377–1390 (2003).
Sargent, F., Berks, B. C. & Palmer, T. Assembly of membrane-bound respiratory complexes by the Tat protein-transport system. Archives of Microbiology 178, 77–84 (2002).
Bernhard, M., Friedrich, B. & Siddiqui, R. A. Ralstonia eutropha TF93 is blocked in tat-mediated protein export. J. Bacteriol. 182, 581–588 (2000).
Dreusch, A., Bürgisser, D. M., Heizmann, C. W. & Zumft, W. G. Lack of copper insertion into unprocessed cytoplasmic nitrous oxide reductase generated by an R20D substitution in the arginine consensus motif of the signal peptide. Biochim. Biophys. Acta - Bioenerg. 1319, 311–318 (1997).
De Buck, E., Höper, D., Lammertyn, E., Hecker, M. & Anné, J. Differential 2-D protein gel electrophoresis analysis of Legionella pneumophila wild type and Tat secretion mutants. Int. J. Med. Microbiol. 298, 449–461 (2008).
Jongbloed, J. D. H. et al. TatC is a specificity determinant for protein secretion via the twin-arginine translocation pathway. J. Biol. Chem. 275, 41350–41357 (2000).
McDonough, J. A. et al. Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J. Bacteriol. 190, 6428–38 (2008).
Williams, H. D., Zlosnik, J. E. & Ryall, B. Oxygen, cyanide and energy generation in the cystic fibrosis pathogen Pseudomonas aeruginosa. Adv Microb Physiol. 52, 1–71 (2007).
Braud, A., Geoffroy, V., Hoegy, F., Mislin, G. L. & Schalk, I. J. Presence of the siderophores pyoverdine and pyochelin in the extracellular medium reduces toxic metal accumulation in Pseudomonas aeruginosa and increases bacterial metal tolerance. Environ Microbiol Rep. 2, 419–25 (2010).
Quintana, J., Novoa-Aponte, L. & Argüello, J. M. Copper homeostasis networks in the bacterium Pseudomonas aeruginosa. J Biol Chem. 292, 15691–15704 (2017).
Vasil, M. L. et al. A complex extracellular sphingomyelinase of Pseudomonas aeruginosa inhibits angiogenesis by selective cytotoxicity to endothelial cells. PLoS Pathog. 5, e1000420 (2009).
Minandri, F. et al. Role of iron uptake systems in Pseudomonas aeruginosa virulence and airway Infection. Infect Immun. 84, 2324–2335 (2016).
Figurski, D. H. & Helinski, D. R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. 76, 1648–1652 (1979).
Tabor, S. & Richardson, C. C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82, 1074–1078 (1985).
Jeong, J. Y. et al. One-step sequence-and ligation-independent cloning as a rapid and versatile cloning method for functional genomics Studies. Appl. Environ. Microbiol. 78, 5440–5443 (2012).
Newman, J. R. & Fuqua, C. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227, 197–203 (1999).
Petiti, M., Houot, L. & Duché, D. In Methods in Molecular Biology 1615, 59–64 (2017).
Bernhardt, T. G. & De Boer, P. A. J. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 48, 1171–1182 (2003).
Ize, B., Viarre, V. & Voulhoux, R. Cell fractionation. Methods Mol. Biol. 1149, 185–91 (2014).
Scheurwater, E. M., Pfeffer, J. M. & Clarke, A. J. Production and purification of the bacterial autolysin N-acetylmuramoyl-l-alanine amidase B from Pseudomonas aeruginosa. Protein Expr. Purif. 56, 128–137 (2007).
Acknowledgements
This work was supported by a joint grant from the Vaincre la Mucoviscidose and the Grégory Lemarchal associations (RF20140501138) and recurrent funding from the CNRS and Aix-Marseille Université. M.R.G. was supported by a PhD studentship and ATER position from Aix-Marseille Université. We thank J. Delisle and T. Doan for their help with microscopy, and M. Ba, I. Bringer, A. Brun and O. Uderso for technical assistance. We are grateful to S. Bigot, E. Cascales, B. Field, L. Houot, members of the Bleves/Voulhoux teams and all the members of the LISM for helpful discussion and support. We also thank B. Field for critical reading and comments about the manuscript.
Author information
Authors and Affiliations
Contributions
B.I. conceived, designed and supervised the project; M.G., B.I. performed experiments; P.V.O. made constructs for Fig. 7; G.C. performed in silico analysis; M.G., G.C., S.B. and B.I. analysed data; B.I. wrote the paper; M.G., R.V., B.I., S.B. discussed the data and read the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gimenez, M.R., Chandra, G., Van Overvelt, P. et al. Genome wide identification and experimental validation of Pseudomonas aeruginosa Tat substrates. Sci Rep 8, 11950 (2018). https://doi.org/10.1038/s41598-018-30393-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30393-x
- Springer Nature Limited