
Abstract
This manuscript has not been published before and is not currently being considered for publication elsewhere. Increased septal convexity of left ventricle has been described in subjects with hypertrophic cardiomyopathy (HCM) -causing mutations without left ventricular hypertrophy (LVH). Our objective was to study septal convexity by cardiac magnetic resonance (CMR) in subjects with the Finnish founder mutation Q1016X in the myosin-binding protein C gene (MYBPC3). Septal convexity was measured in end-diastolic 4-chamber CMR image in 67 study subjects (47 subjects with the MYBPC3-Q1061X mutation and 20 healthy relatives without the mutation). Septal convexity was significantly increased in subjects with the MYBPC3-Q1061X mutation and LVH (n = 32) compared to controls (11.4 ± 4.3 vs 2.7 ± 3.2 mm, P < 0.001). In mutation carriers without LVH, there was a trend for increased septal convexity compared to controls (4.9 ± 2.5 vs 2.7 ± 3.2 mm, P = 0.074). When indexed for BSA, septal convexity in mutation carriers without LVH was 2.8 ± 1.4 mm/m2 and 1.5 ± 1.6 mm/m2 in controls (P = 0.036). In all mutation carriers, septal convexity correlated significantly with body surface area, age, maximal LV wall thickness, LV mass, and late gadolinium enhancement. Subjects with the MYBPC3–Q10961X mutation have increased septal convexity irrespective of the presence of LVH. Septal convexity appears to reflect septal remodeling, and could be useful in recognizing LVH negative mutation carriers.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disease and one of the most common causes of sudden cardiac death in the young1. HCM is caused by autosomal dominant mutations in genes encoding mainly sarcomeric proteins. More than 1400 mutations causing HCM have been identified in over 13 genes2. In familial HCM, there is a 50% chance that the first-degree relative will inherit the gene mutation leading to the disease. HCM is a heterogenous disease with variable clinical phenotype even in patients carrying the same causal mutation3. Clinical presentation may vary from asymptomatic to arrhythmia, heart failure and sudden death. Clinical manifestations of HCM usually develop during adolescence but myosin-binding protein C gene (MYBPC3) mutation carriers often have no LVH or develop LVH later in life4,5,6.
More than 40% of the mutations causing HCM are found in the MYBPC3 gene7. The founder mutation Q1061X in MYBPC3 (MYBPC3-Q1061X) is the most common single mutation causing HCM in Finland and is found in about 11% of Finnish HCM cases5,8,9. To our knowledge, this mutation is extremely rare outside Finland, with one reported mutation in non-Finnish populations according to the Genome Aggregation Database (gnomAD). This autosomal dominant mutation has a relatively low penetrance and often, but not always, mild phenotype6,8. Of the subjects with the Finnish founder mutation MYBPC3-Q1016X, 20 to 30% do not have LVH and are considered phenotype negative mutation carriers6,8,10.
Hypertrophic cardiomyopathy is characterized by asymmetric LV hypertrophy, most often located in the LV anterior septum or free wall. Of HCM patients with reverse septal curvature on echocardiography, nearly 80% have a positive genetic test for myofilament HCM, contrasting those with sigmoidal septal morphology, who have positive genetic test in fewer than 10% of cases11. Recently, CMR derived septal convexity into the LV has been suggested to be an additional, previously undescribed feature of subclinical HCM12. Compared to controls, abnormal convexity was significantly increased in phenotype negative carriers of HCM-causing mutations in multiple sarcomere genes. To our knowledge, there are no other studies regarding this topic. Therefore, the purpose of our study was to further investigate septal convexity by CMR in subjects carrying the single MYBPC3-Q1016X mutation and their healthy relatives.
Methods
Study population
All available subjects with the MYBPC3-Q1061X mutation and their healthy relatives from Kuopio and Helsinki University Hospital areas were prospectively recruited. Altogether, 74 adult subjects representing 25 families with the Finnish founder mutation MYBPC3-Q1061X were screened. The Genome Center of the University of Eastern Finland performed all the genetic studies as previously described8. All index patients of the present study were screened for 59 cardiomyopathy associated genes, and no other pathogenic or likely pathogenic mutations in addition to MYBPC3-Q1061X were found in any of the patients9. Seven subjects had implanted pacemaker precluding CMR imaging and were excluded from the study. The final study population consisted of 67 individuals (47 with the MYBPC3-Q1061X mutation with or without LVH, and 20 healthy relatives without the MYBPC3-Q1061X mutation).
The G+/LVH+ group consisted of 32 subjects with the MYBPC3-Q1061X mutation and no other cause for significant hypertrophy (LV maximal wall thickness [LVMWT] ≥ 13 mm in CMR), and 15 subjects with the same mutation but no hypertrophy (LVMWT < 13 mm in CMR) consisted the G+/LVH− group.
The study conforms to the principles outlined in the Declaration of Helsinki and the ethics committees at the Kuopio and Helsinki University Hospitals approved the study protocol. All subjects gave informed written consent.
Echocardiography
All subjects underwent echocardiographic studies which were carried out by experienced cardiologists (J.K., P.J., M.J.) with GE Vivid 7 ultrasound equipment (GE Vingmed, Norway). Conventional measurements were performed according to AHA/ASE guidelines. A resting gradient >30 mmHg through the left ventricular outflow tract (LVOT) was considered significant13.
CMR
CMR images were obtained with the same study protocol in both participating hospitals (Kuopio and Helsinki University Hospitals) as described previously14. Delayed-enhancement images were obtained 10 minutes after injection of gadoterate meglumine (Dotarem®) 0.2 mmol/kg contrast agent using segmented trueFISP IR sequence in short axis orientation and in two long axis orientations (4- and 2-chamber views). The imaging parameters were 700 msec temporal resolution, 1.08 msec echo time, flip angle 50°, acquisition matrix 192 × 144 and 340 × 340 field of view. Slice thickness was 8 mm and intersection gap 20%. Inversion time was optimized to null the signal intensity of normal myocardium.
Image Analyses
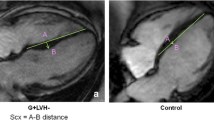
All of the anatomical measurements on CMR images were performed by using Sectra IDS7 workstation. Image analysis was performed by experienced analyzers with no data of the study subjects’ clinical or genetic findings. Septal convexity into the LV was measured in end-diastolic 4-chamber images as the maximal distance between LV septal endocardial border and a line connecting septal mid-wall points at the level of tricuspid valve insertion and at the level of apical right ventricular insertion on the LV as described previously12 [Fig. 1]. M.T. (with 5 years experience in CMR) performed all septal convexity measurements. All the other anatomical measurements of the CMR images were performed as described previously14. Areas of late gadolinium enhancement (LGE) were traced by QMass® late-enhancement analysis software (QMass® 7.2, Medis Medical Imaging Systems, Netherlands) in short axis images and quantified segmentally as absolute and relative mass in a standard 16-segment model. A threshold of +6 SD was used for detection of LGE15.

Measurement of septal convexity in end-diastolic 4-chamber CMR image. The red line is a reference line between the mid-wall at the level of tricuspid valve and the insertion point of the right ventricle into the left ventricle at the apex. The yellow line is the measured septal convexity. This particular subject has the measured septal convexity of 9 mm and left ventricular maximal wall thickness of 12 mm.
Measurement repeatability was evaluated by measuring septal convexity twice in 20 randomly selected subjects. M.T. tested intraobserver variability by re-measuring septal convexity more than 6 months after the initial measurement. Interobserver variability was calculated from the same 20 measurements by another radiologist (K.K.).
Statistical Analyses
Data is presented as mean ± standard deviation. Many of the variables had skewed distribution and were compared with Kruskal-Wallis one-way analysis of variance and independent samples with the Mann-Whitney U test. Receiver operating characteristic (ROC) curve was constructed to test the ability of septal convexity to differentiate between mutation carriers without LVH and control subjects. Paired samples T-test and Pearson correlation coefficient was used in repeatability testing. All statistical analyses were performed with IBM SPSS Statistics V22.0. and were considered significant with a P < 0.05.
Results
Characteristics of the study groups
Table 1 shows baseline, echocardiographic and CMR characteristics of the three study groups. G+/LVH− subjects were younger and had lower BSA compared to other groups. There were more males were in the G+/LVH− group compared to other groups. G+/LVH− subjects were all in NYHA 1 class14. They did not have cardiac symptoms or clinical findings (data not shown).
No septal interventions (myectomy or ablation) had been performed in any of the study subjects. There was one subject with significant LVOT obstruction (gradient >30 mmHg on echocardiography) in the G+/LVH+ group. However, the mean LVOT gradients did not differ significantly between the study groups.
CMR derived LVMWT and left ventricular mass index (LVMI) differed between study groups, as expected. Ventricular volume indices (LVEDVI and LVESVI) and LVEF did not differ between study groups. G+/LVH+ subjects had mostly moderate hypertrophy (LVMWT 22.1 ± 5.7 mm) located mainly in septum (72%) or anterior wall (25%). True apical hypertrophy was not found. No significant difference was found in LVMWT between G+/LVH− subjects and controls (9.5 ± 1.6 vs 9.6 ± 1.7 mm, P = 0.882). LVMI was significantly increased in G+/LVH+ group compared to other groups (68 ± 21 g/m2, P < 0.001), but not in G+/LVH− subjects compared to controls (49 ± 13 vs 45 ± 9 g/m2, P = 0.438). In the G+/LVH+ group, 23 subjects had LGE, as only one in the G+/LVH− group had LGE. In the subjects with LGE, the mean amount of LGE was 18 ± 12% of the LV myocardium (range 1–45%).
Septal convexity into the LV
Septal convexity measurement was feasible in all study subjects. Table 1 and Fig. 2a,b show the results of septal convexity measurements in the three study groups. There was a significant difference in septal convexity between G+/LVH+ group and controls (11.4 ± 4.3 vs 2.7 ± 3.2 mm, P < 0.001), and between G+/LVH+ and G+/LVH− groups, respectively (11.4 ± 4.3 vs 4.9 ± 2.5, P < 0.001) (Table 1 and Fig. 2a) (Fig. 3). The difference between the G+/LVH− group and the controls was not quite significant (4.9 ± 2.5 vs 2.7 ± 3.2 mm, P = 0.074; CI 95% 3.5–6.3 mm vs 1.2–4.2 mm). However, when indexed for BSA, the difference in SC values between all groups, including the G+/LVH− group and the control group, was statistically significant (2.8 ± 1.4 vs 1.5 ± 1.6 mm/m2, P = 0.036). Also the difference between G+/LVH+ group and controls (5.9 ± 2.2 vs 1.5 ± 1.6 mm/m2, P < 0.001) and G+/LVH+ group and G+/LVH− group (5.9 ± 2.2 vs 2.8 ± 1.4 mm/m2, P < 0.001) was significant (Fig. 2b).

(a) Septal convexity into the LV in study groups. There is a significant difference between the G+/LVH+ group and other groups. (b) Septal convexity into the LV indexed for BSA. There is a significant difference between the G+/LVH+ group and other groups. Also the difference between G+/LVH− and control group is significant.

Ventricular end-diastolic CMR images in 4-chamber orientation (a) a control subject, (b) a subject with MYBPC3-Q1061X mutation but no LVH and (c) a subject with MYBPC3-Q1061X mutation and LVH.
In MYBPC3 mutation carriers, increased septal convexity correlated significantly with BSA (r = 0.332, P = 0.006), age (r = 0.508, P < 0.001), LVMWT (r = 0.742 P < 0.001), LVM (r = 0.590, P < 0.001) and LGE (r = 0.683, P < 0.001). There was no correlation between septal convexity and LVEF (r = −0.083, P = 0.578) or LV volumes (LVEDV, r = 0.131, P = 0.379 and LVESV, r = −0.111, P = 0.459).
Figure 4 shows receiver operating characteristic (ROC) curve of septal convexity as a predictor of mutation presence but no LVH. Area under the curve (AUC) was 0.68 and a cutoff value of 3.85 mm performed best with sensitivity 67% and specificity 65%. When indexed for BSA, AUC was 0.71 and a cutoff value 2.34 mm/m2 performed best with sensitivity 60% and specificity 75%.

(a) ROC curve for septal convexity into the LV to differentiate between MYBPC3-Q1061X mutation carriers without LVH and controls. AUC is 0.68 (95% Confidence Interval (CI) 0.5–0.86). (b) ROC curve for BSA indexed septal convexity into the LV to differentiate between MYBPC3-Q1061X mutation carriers without LVH and controls. AUC is 0.71 (95% Confidence Interval (CI) 0.54–0.89).
Reproducibility of septal convexity measurements
There was no significant difference between the first and second measurement of the septal convexity in intraobserver analysis measurements (10.3 ± 4.6 vs 10.5 ± 4.7 mm, P = 0.66) and the correlation between measurements was excellent (r = 0.961, P < 0.001). Interobserver measurements showed no significant difference between measurements (10.3 ± 4.6 vs 10.4 ± 4.2 mm, P = 0.65) and an excellent correlation between measurements (r = 0.973, P < 0.001).
Discussion
Subjects with the MYBPC3–Q10961X mutation have increased left ventricular septal convexity irrespective of the presence of LVH. In mutation carriers, septal convexity correlates with BSA, age and CMR derived LV maximal thickness, mass and late gadolinium enhancement.
To our knowledge, there is only one previous study on CMR derived LV septal convexity in HCM-causing mutation carriers11. In aforementioned study, abnormal convexity was significantly increased in 36 phenotype negative carriers of HCM-causing mutations in multiple sarcomere genes, including MYBPC3, beta-myosin heavy chain (MYH7), troponin t (TNNT2), troponin I (TNNI3), myosin regulatory light chain (MYL2), myosin essential light chain (MYL3), tropomyosin (TPM1), and cardiac alpha-actin (ACTC1)11. In the present study, all mutation carriers had a single founder mutation of MYBPC3, and the septal convexity in LVH negative mutation carriers differed significantly from that of healthy controls only after indexing for BSA. In the present study, LV septal convexity with a cut point of >3.85 mm (AUC 68%, sensitivity 67% and specificity 65%) predicted MYBPC3-Q1061X mutation presence without LVH. Correspondingly, in the previous study, septal convexity cut point of >3.55 mm had best balance between sensitivity (77%) and specificity (89%) to predict mutation presence without LVH11. When indexed for BSA, LV septal convexity sensitivity was somewhat lower but specificity was higher (AUC 0.71 with cut point of >2.34 m/m2, sensitivity 60% and specificity 75%).
We found that septal convexity correlates significantly with BSA, age, and CMR derived LV maximal thickness, mass and late gadolinium enhancement in subjects with MYBPC3-Q1061X. To our knowledge, there are no previous correlation analyses on the topic.
Hypertrophic cardiomyopathy is characterized by asymmetric LV hypertrophy, most often located in the LV anterior septum or free wall. Bulging of septum, resulting in septal convexity, may precede increase in maximal wall thickness in the development of HCM, as suggested by the previous and present study. LV septal convexity correlated with BSA, age, LVMWT, LVM, and LGE in subjects with MYBPC3-Q1061X in our study. We propose that septal convexity reflects early-onset, body size and age -related septal remodeling in carriers of HCM-causing sarcomere mutations.
Measuring LV septal convexity in CMR is simple and highly reproducible, as shown by the present and the previous study. Septal convexity is measured in 4-chamber CMR images, which are usually included in CMR protocols used in clinical setting, and are therefore, readily available for measurement in subjects with HCM or LVH of unknown origin, and suspect carriers of HCM-causing mutations. Septal convexity, like most CMR parameters, correlates significantly with BSA. Consequently, indexing for BSA probably increases utility of septal convexity as shown by our study.
Our study population is relatively small. On the other hand, it is a cohort with a single HCM-causing mutation and, consequently, bias caused by multiple diverse mutations is avoided. MYBPC3 mutations are the most common cause of HCM accounting for about 40% of all HCM cases globally. Furthermore, MYBPC3-Q1016X might be regarded to represent most MYBPC3 mutations, as like two thirds of all MYBPC3 mutations, it leads to the production of a truncated protein and, consequently, to haploinsufficiency, characterized by absence of mutant protein in myocytes16,17,18.
Non-indexed measurements showed a non-significant trend for septal convexity abnormality in mutation carriers without LVH compared to control subjects. However, the G+/LVH− subjects in the present study were younger and had a smaller BSA than control subjects, and both age and BSA correlated with septal convexity. BSA indexed values showed significant difference in septal convexity between G+/LVH− subjects and controls.
Even in the present era of targeted sequencing of large cardiomyopathy gene panels, the pathogenic or likely pathogenic mutation is found only in about 60% of HCM cases9. Age-related increase in penetrance of HCM implies that a proportion of clinically unaffected mutation carriers will develop overt cardiomyopathy later in life, and thus, precautionary long-term evaluation of normal healthy mutation carriers and relatives of patients without disease-causing mutation is recommended19. Hence, we need imaging methods to identify the relatives at risk of developing HCM. Imaging is especially important in families with mutations which may represent LVH in the middle-ages, as in the case of MYBPC3 mutations2,20. One previous study has suggested that CMR derived septal convexity is a potential tool in finding phenotype negative HCM -causing mutation carriers12. In the present study, we showed that CMR derived septal convexity is significantly increased in carriers of the single mutation in MYBPC3. Septal convexity was related to BSA, and particularly if indexed for BSA, differed between LVH-negative mutation carriers and controls. Consequently, CMR derived septal convexity may have value in the evaluation of family members of patients with HCM to find relatives at risk of developing clinical HCM, particularly when genetic testing in the proband is not available or is negative. As septal convexity correlated with age and LVH and myocardial fibrosis in the whole mutation carrier group, it might be useful measure of severity of the disease also in the follow-up of patients with overt HCM. However, as septal derived convexity has been investigated only in two small studies, its utility requires further large-scale studies.
As a conclusion, subjects with the MYBPC3–Q10961X mutation have increased septal convexity irrespective of the presence of LVH. Septal convexity appears to reflect early onset, body size and age-related septal remodeling and could be useful in identifying phenotype-negative mutation carriers.
Ethical approval and informed consent
Institutional Review Board approval was obtained for the study and study methods were carried out in accordance with the relevant guidelines and regulations. Written informed consent was obtained from all subjects in this study.
References
Maron, B. J. Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320 (2002).
Maron, B. J. & Maron, M. S. Hypertrophic cardiomyopathy. Lancet 381(9862), 242–55 (2013).
Sipola, P. et al. Cine MR imaging of myocardial contractile impairment in patients with hypertrophic cardiomyopathy attributable to Asp175Asn mutation in the alpha-tropomyosin gene. Radiology 236(3), 815–824 (2005).
Niimura, H. et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 105(4), 446–51 (2002).
Jääskeläinen, P. et al. Mutations in the cardiac myosin-binding protein C gene are the predominant cause of familial hypertrophic cardiomyopathy in eastern Finland. J. Mol. Med. 80, 412–422 (2002).
Jääskeläinen, P., Miettinen, R., Kärkkäinen, P., Toivonen, L. & Kuusisto, J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann. Med. 36, 23–32 (2004).
Zhang, X. L., De, S., McIntosh, L. P. & Paetzel, M. Structural characterization of the C3 domain of cardiac myosin binding protein c and ita hypertrophic cardiomyopathy-related R502W mutant. Biochemistry (2014).
Jääskeläinen, P. et al. Two founder mutations in the alpha-tropomyosin and the cardiac myosin-binding protein C genes are common causes of hypertrophic cardiomyopathy in the Finnish population. Ann. Med. 45, 85–90 (2013).
Jääskeläinen, P. et al. Genetic basis and outcome in a nationwide study of Finnish patients with hypertrophic cardiomyopathy. ESC Heart Fail, https://doi.org/10.1002/ehf2.12420 (2019).
Kuusisto, J., Sipola, P., Jääskeläinen, P. & Naukkarinen, A. Current perspectives in hypertrophic cardiomyopathy with the focus on patients in the Finnish population: a review. Ann. Med. 48(7), 496–508 (2016 Nov).
Binder, J. et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc. 81, 459–67 (2006).
Reant, P. et al. Abnormal septal convexity into left ventricle occurs in subclinical hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 17(1), 64 (2015).
Jalanko, M. et al. Left ventricular mechanical dispersion is associated with nonsustained ventricular tachycardia in hypertrophic cardiomyopathy. Ann Med. 48(6), 417–427 (2016).
Tarkiainen, M. et al. Cardiovascular magnetic resonance of mitral valve length in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 18(1), 33 (2016).
Iles, L. M. et al. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur Heart J Cardiovasc Imaging. 16(1), 14–22 (2015).
Carrier, L., Mearini, G., Stathopoulou, K. & Cuello, F. Cardiac myosin binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 573(2), 188–97 (2015).
van Dijk, S. J. et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119(11), 1473–83 (2009).
Marston, S. et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circulation Research 105(3), 219–22 (2009).
Elliott, P. M. et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 35(39), 2733–79 (2014).
Kubo, T. et al. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac Myosin-binding protein C gene among Japanese. J Am Coll Cardiol. 46(9), 1737–43 (2005).
Acknowledgements
We thank research nurses Sini Weckström and Satu Nenonen for their assistance in data collection.
Author information
Authors and Affiliations
Contributions
M.T. conceived and planned the study, performed the CMR study, performed the statistics, and drafted the manuscript. P.S. conceived and planned the study, performed the CMR study and drafted the manuscript. M.J. conceived and planned the study and performed the echocardiographic study. T.H. conceived and planned the study. P.J. conceived and planned the study and performed the echocardiographic study. K.K. conceived and planned the study and performed the reproducibility study with M.T. M.L. conceived and planned the study. K.L. conceived and planned the study and performed the CMR study. J.K. conceived and designed the study and was in charge of overall direction and planning, performed the echocardiographic study, performed the statistics and drafted the manuscript. All authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tarkiainen, M., Sipola, P., Jalanko, M. et al. CMR derived left ventricular septal convexity in carriers of the hypertrophic cardiomyopathy-causing MYBPC3-Q1061X mutation. Sci Rep 9, 5960 (2019). https://doi.org/10.1038/s41598-019-42376-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42376-7
- Springer Nature Limited