
Abstract
Accumulation of amino acid (AA) insertions/substitutions are observed in the Gag-protein of HIV-1 variants resistant to HIV-1 protease inhibitors. Here, we found that HIV-1 carrying AA insertions in capsid protein (CA) undergoes aberrant CA degradation. When we generated recombinant HIV-1s (rHIV-1s) containing 19-AAs in Gag, such insertions caused significant CA degradation, which initiated in CA’s C-terminal. Such rHIV-1s had remarkable morphological abnormality, decreased infectivity, and no replicative ability, which correlated with levels of CA degradation. The CA degradation observed was energy-independent and had no association with cellular/viral proteolytic mechanisms, suggesting that the CA degradation occurs due to conformational/structural incompatibility caused by the 19-AA insertions. The incorporation of degradation-prone CA into the wild-type CA resulted in significant disruption of replication competence in “chimeric” virions. The data should allow better understanding of the dynamics and mechanisms of CA decomposition/degradation and retroviral uncoating, which may lead to new approach for antiretroviral modalities.
Similar content being viewed by others


Introduction
The currently available combination antiretroviral therapy (cART) for human immunodeficiency virus type 1 (HIV-1) infection has been shown to effectively suppress viral replication and significantly extend the life expectancy of infected individuals1,2. However, in the course of life-long treatment, drug-resistant HIV-1 often emerges, which has been a major contributor to treatment failure3,4,5. HIV-1 often develops high levels of cross-resistance against multiple drugs by acquiring a variety of amino acid (AA) substitutions near the active sites of targeted enzymes6,7,8,9,10. Such substitutions often compromise the enzymatic functions of the viral reverse transcriptase, protease (PR), and integrase11,12,13,14,15,16. During the development of resistance to certain protease inhibitors (PIs), HIV-1 often acquires, in addition to resistance-associated AA substitutions in PR, “compensatory” AA substitutions in Gag proteins. These compensatory AA substitutions do not directly confer drug-resistance but rather improve the otherwise compromised catalytic functions of the mutated PR17,18. Certain AA insertions in the proximity of Gag cleavage sites also restore the otherwise compromised enzymatic activity of PRs, enabling PI-resistant HIV-1 variants to remain replication-competent19. However, the replication competence of such HIV-1 variants does not get fully restored, viral replication of multi-PI-resistant variants remain poor compared with that of wild-type HIV-1.
In the present study, we introduced a 19-AA insertion at various positions in the Gag region of infectious HIV-1s using the Tn5-based transposon system and examined their effects on the processing and maturation of the Gag structural protein. Interestingly, we observed the emergence of enhanced CA degradation products, which had not been reported previously, in HIV-1 variants generated by transposon (HIVTPs)-containing the 19-AA insertion in the NTD region of CA. Our results also suggested that the observed CA degradation rapidly progressed in time-dependent manner, and CA degradation had no association with cellular/viral proteolytic mechanisms. CA degradation-inducing HIVTPs had drastic morphological abnormality, compromised infectivity, and no replicative ability. Furthermore, we show that upon the cleavage of the Gag between the CA and p2-NC, CA degradation occurs and that the CA degradation predominantly occurs in the CTD of the CA.
Results
CA degradation observed in HIV-1 variants with AA insertion in the Gag region
We have previously shown that certain AA insertions near the cleavage sites in Gag polyprotein result in the restoration of the otherwise compromised enzymatic activity of mutated PRs, enabling multi-PI-resistant HIV-1 variants to remain replication-competent19. We also found that drug-resistant HIV-1 variants carrying multiple (3~6) AA insertions in Gag undergoes aberrant CA degradation in Western blotting (WB) employing virion lysates of each HIV-1 with Gag AA insert produced by transfected COS-7 cells (19; Fig. S1a).
In order to further elucidate the effects of AA insertions on the infectivity and replication capacity of HIV-1, we generated a series of recombinant HIV-1 clones-containing a 19-AA insertion randomly introduced into the Gag region of HIV-1 using the Tn5-based transposon system (HIVTP: Figs 1a and S1b). HIVTP we generated included 5 HIVTP species-containing the 19-AA insertion in MA (HIVMAV35-W36, HIVMAG62-Q63, HIVMAV88-H89, HIVMAE105-E106, and HIVMAQ127-V128), 8 HIVTP species-containing the insertion in CA (HIVCAI2-V3, HIVCAR18-T19, HIVCAA64-A65, HIVCAM96-R97, HIVCAL111-Q112, HIVCAD152-I153, HIVCAG220-V221, and HIVCAK227-A228), one HIVTP-containing the insertion in p2 (HIVp2A1-E2), and one HIVTP-containing the insertion in p6 (HIVp6T8-A9)(Fig. S1b; AA sequences of inserts are described in Table S1). When the lysates of COS-7 cells producing the 19-AAs-containing HIVTP were subjected to WB using an HIV-1 CA-specific Gag polyclonal antibody (pAb)(Fig. 1b), notable levels of CA-specific signals, of which molecular-weights were less than that of CA, were observed. Such signals were predominantly and almost exclusively identified in HIVTP species that contained the insertions in CA. The signals (Fig. 1b, boxed) are identified as ladders consisting of different molecular-weight proteins but not of smear-like proteins. Thus, it is thought that those proteins represent orderly-processed CA degradation products. Moreover, the amounts of CA degradation products appeared to be greater when the inserts were introduced in the N-terminal domain of CA (HIVTP-CAINSNTD: HIVCAI2-V3, HIVCAR18-T19, HIVCAA64-A65, HIVCAM96-R97, HIVCAL111-Q112) than in the C-terminal domain of CA (HIVTP-CAINSCTD). In contrast, no or least degradation products were observed in the lysates of cells producing HIVWT or HIVTPs-containing the insert in MA, p2, or p6 proteins (Fig. 1b). These degradation products profiles strongly suggest that the insertion of the 19-exogenous-AA in CA caused the enhanced CA degradation. When we obtained condensed HIVTP particles with ultracentrifugation of the supernatants of COS-7 cells producing HIVCAI2-V3, immediately lysed the virions and subjected the virion lysates to WB, similarly large amounts of ladder-like degradation products were identified, confirming that the degradation products are also present within the virions (Fig. 1c). When HEK-293 cells were employed instead of COS-7 cells for transfection, similar ladder-like degradation products were observed in all the cell lysates producing HIVTP-containing the insert in CA (Fig. 1d). In order to determine whether other HIV-1-derived proteins were present in the degradation products, we subjected the cell lysates of COS-7 producing various HIVTP including HIVCAI2-V3 to WB using an MA-specific monoclonal antibody (mAb). No MA-derived degradation products were identified in any of the cell lysates of HIVTP-producing COS-7 cells (Fig. S2a). When an Env-specific pAb was used, no additional Env-derived products were identified (Fig. S2b). These data confirmed that the observed degradation occurred in CA proteins, not in MA proteins or Env glycoproteins.

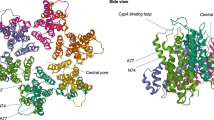
CA degradation observed in HIV-1 variants with the AA insertion in Gag region. (a) Locations of each 19-AA insertion in CA of HIVTP clones generated by using the Tn5-based transposon system are shown. The sequence of wild-type CA (CAWT) is shown with its secondary structure diagram. HIV-1 CA consists of the N-terminal domain (NTD; residues 1–145) and the C-terminal domain (CTD; residues 151–231) connected by 5 AAs that act as a flexible linker. Each insertion site is indicated by yellow arrow. The hexamer structure is stabilized by interactions formed by s hairpin and first 3 helices H1, H2, H3 as shown in blue. Two- and three-fold interactions take place between hexamer units are shown in green and orange, respectively. Cyclophilin A-binding site is indicated in purple. (b) CA degradation in the lysates of COS-7 cells producing various 19-AAs-containing HIV clones (HIVTPs). The lysates of COS-7 cells producing HIVWT or HIVTPs were examined for enhanced degradation. Each lysate sample was prepared 72 hrs after transfection and subjected to WB with anti-p24 polyclonal anti-serum. The boxes indicate lower-molecular-weight CA degradation products than the mature p24/p25 (CA). Results shown is representative of more than ten independent experiments. (c) The CA degradation products are identified in the lysates of HIVCAI2-V3. The supernatants of COS-7 cells producing HIVWT or HIVCAI2-V3 were collected 72 hrs after transfection, cleaned with 0.22 μm pore size filters, and subjected to ultracentrifugation (20,000 g, 4 °C, 24 hrs). The obtained virions were lysed and was subjected to WB. Results shown is representative of three independent experiments. (d) The lysates of HEK-293 cells producing HIVWT or HIVTPs showed the similar degradation products as seen in the COS-7 cell lysates as above. All samples in (b–d) were normalized by their protein concentrations (20 μg/well). Results shown is representative of seven independent experiments. Full-length blots/gels are presented in Fig. S9.
Time-dependent progression of the CA degradation in HIVTPs and recombinant CA-containing 19-AA insert, and morphological profiles of HIVTPs-containing 19-AA insertion in the CA region
We also examined if the observed CA degradation described above progressed when the lysates of COS-7 cells that produce various HIVTP were incubated over different periods of time at 37 °C using ELISA that employed two CA-specific mAbs (Lumipulse f). Figure 2a shows the changes in the amounts of CA in each cell lysate. The %CA at each time point was calculated as the percentage relative to the amounts of detectable CA in unincubated cell lysates. CA degradation generally occurred by 6 hours of incubation and relatively slowly progressed by up to 24-hours afterwards (Fig. 2a). The reduction of the amounts of CA was the greatest in the lysates of HIVTP-CAINSNTD-producing COS-7 cells (range: 96.8~68.1% reduction at 48-hours) compared to HIVTP-CAINSCTD-producing cells (range: 41.5~28.9% at 48-hours) (Fig. 2a). Virtually no reduction in the amounts of CA was seen in the cell lysates of COS-7 producing HIVWT or HIVTPs-containing the insert in MA, p2, or p6 proteins (Fig. 2a). These data are completely in line with our observation noted above (Fig. 1b,d) that the amounts of CA degradation products were greater in HIVTP-CAINSNTD-producing cells than in HIVTP-CAINSCTD-producing cells and that no CA degradation products were seen in the cell lysates of HIVWT or HIVTPs-containing the insert in MA, p2, or p6 proteins. These data also strongly suggest that as CA degradation occurred, the two antigenic epitopes, located in the amino-terminus of CA, were lost, which the two CA-specific mAbs used in the Lumiplulse f system (https://soled.co/pdf-docs/fujirebio-lumipulse-f-service-manual) should otherwise recognize. When we examined whether the CA degradation progressed time-dependently using virion lysates from the supernatants of COS-7 cell cultures, the greatest reduction was seen in the virion lysates with HIVCAI2-V3 (HIVTP-CAINSNTD), followed by the lysates with HIVCAD152-I153 (HIVTP-CAINSCTD)(Fig. S3a). No reduction was seen in the virion lysates with HIVWT, HIVMAQ127-V128, or HIVp2A1-E2. When an MA-specific mAb was employed in WB, no degradation product’s signals were seen even after the lysates were incubated over the 40-hours (Fig. S2c).

Time-dependent progression of the CA degradation in HIVTPs and recombinant CA-containing 19-AA insert. (a) Cell lysates of HIVWT- or various HIVTP-producing COS-7 cells were incubated at 37 °C for different periods of time, and the CA concentration of each sample was determined using ELISA. The sample at time 0 was stored at −80 °C immediately following the cell lysate preparation. “Percent p24 (CA)” was determined using equation described in the Methods section. Note that the lysates of COS-7 cells producing a panel of HIV-1-containing the 19-AAs in CA-NTD most rapidly lost CA, followed by the cells producing HIV-1 with the insert in CA-CTD. The data shown represent mean values derived from the results of two independent experiments. (b) Morphology of HIVCAI2-V3 and HIVCAR18-T19. HIVWT, HIVCAI2-V3 and HIVCAR18-T19 producing transfected COS-7 cells were subjected TEM study, as described in the Methods section. Note that HIVWT virions had the typically-condensed mature core, while the cores within HIVCAI2-V3 and HIVCAR18-T19 are much less condensed or absent. The frequency of mature and aberrant cores are 75.7% (56/74) and 24.3% (18/74) for HIVWT virions; 25.2% (27/107) and 74.8% (80/107) for HIVCAI2-V3 virions; 8.7% (2/23) and 91.3% (21/23) for HIVCAR18-T19 virions, respectively. Scale bars: 100 nm. (c) Various recombinant CAs such as wild-type HIV-1 CA (rCAWT) and the 19-AAs-containing CAs (rCAI2-V3, rCAR18-T19, rCAD152-I153, and rCAG220-V221) were expressed in COS-7 cells, the lysates of the cells incubated at 37 °C, subjected to WB using anti-p24 anti-serum, and the signal densities of rCAs quantified using the NIH Image J Program. Percent p24 (CA) was determined using equation described in the Methods section. The unincubated samples (0 hr) were normalized by their p24 concentrations (5 ng/well). 24 hrs and 48 hrs incubated samples were applied as same volumes as each unincubated (0 hr) sample. Results shown is representative of two independent experiments. (d) The same samples as in panel c were subjected to ELISA. Note that all the 19-AAs-containing CAs examined rapidly lost the immunogenicity as compared with rCAWT. Results shown is representative of two independent experiments. Full-length blots/gels are presented in Fig. S9.
Next, in order to determine if the CA degradation induced by the 19-exogenous-AA insertion had any effects on the morphology of virions, we conducted transmission electron microscopic (TEM) analysis using HIVTP-CAINSNTD-producing COS-7 cells. In the TEM images of HIVWT produced by COS-7 cells, the typically-condensed mature core morphology was seen (Fig. 2b, upper panels). In contrast, in the TEM of HIVTP-CAINSNTD-producing COS-7 cells, most virions are rather greater in their sizes and the cores within the virions are much less condensed or absent, strongly suggesting that the CA in such virions significantly degraded and failed to be assembled (Fig. 2b, lower panels).
We further constructed plasmids to express recombinant HIVWT CA (rCAWT) and recombinant CA-containing the 19-AA insert at different positions (two recombinant rCAINSNTD proteins: rCAI2-V3 and rCAR18-T19; two recombinant rCAINSCTD proteins: rCAD152-I153 and rCAG220-V221) and attempted to examine their structural integrity. As assessed with WB, rapid and drastic reduction in the signals of CA was seen within 24-hour incubation of the two rCAINSNTDs and moderate but yet drastic reduction was seen with the two rCAINSCTDs (Fig. 2c). The reduction levels became further greater by 48-hour incubation (Fig. 2c). The %signal-density reduction values in rCAWT, rCAI2-V3, rCAR18-T19, rCAD152-I153, and rCAG220-V221 were 23, 90, 87, 61, and 78% at 24-hours and 44, 97, 99, 85, and 75% at 48-hours, respectively, as determined using the NIH Image J Program. The ladder-like degradation products were also observed in unincubated COS-7 cell lysates expressing rCAINSNTDs and rCAINSCTDs (Fig. S3b). When the amounts of detectable CA were determined using ELISA, rapid reduction was seen within 24-hours with both rCAINSNTD and rCAINSCTD (Fig. 2d). However, the reduction of CA amounts was much less in rCAWT (Fig. 2c,d; the actual amounts determined using the Lumipulse f are illustrated in Table S2).
The CA degradation observed has no association with cellular/viral proteolytic mechanisms
We attempted to elucidate the mechanism of the CA degradation described above. Mammalian cells physiologically have multiple protein degradation pathways to regulate the levels of proteins. Such degradation pathways mainly include (i) the ubiquitin/proteasome20, (ii) autophagy21, and (iii) protease-associated proteolysis. Hence, we examined whether any of these degradation mechanism is responsible for the observed CA degradation. First, to examine whether the ubiquitin/proteasome system was in operation, we employed MG-132, a proteasome inhibitor, which caused the accumulation of ubiquitinated proteins at the concentration of 20 µM (Fig. S4a). However, the same concentration of MG-132 failed to block the CA degradation (Fig. 3a). 3-MeA, an autophagy inhibitor, blocked the formation of autophagosome (Fig. S4b); however, this inhibitor also failed to block the CA degradation (Fig. 3b). In order to ask whether cellular proteases were responsible for the CA degradation, we used a mixture of 7 cellular proteases inhibitors that fairly well blocked trypsin digestion of BSA (Fig. S4c), while the same mixture completely failed to block the CA degradation (Fig. 3c). It was also possible that HIV-1-derived aspartyl protease might have caused the CA degradation since precursors of CA proteins are digested by retrovirally-expressed protease. Thus, we used HIV-1 protease-inhibitor, saquinavir (SQV, 1 µM), which efficiently blocked HIV-1 protease activity (Fig. S4d). However, SQV also failed to block the CA degradation (Fig. 3d).

The CA degradation observed has no association with cellular/viral proteolytic mechanisms. (a–d) MG-132, an inhibitor of proteasome; 3-MeA, an autophagy inhibitor; a mixture of 7 potent cellular protease inhibitors; and SQV, a potent HIV-1-derived aspartyl protease failed to block CA degradation. (a) The lysates of COS-7 cells producing HIVWT or HIVTPs were prepared 72 hrs after transfection and subjected to WB with anti-p24 polyclonal antibody. MG-132 was added to transfection medium with 20 μM concentration at 6 hrs before harvest. Results shown is representative of four independent experiments. (b) The lysates of HEK-293 cells producing HIVWT or HIVTPCAI2-V3 were prepared 72 hrs after transfection and subjected to WB with anti-p24 polyclonal antibody. HEK-293 cells were incubated for 5 hrs with 5 mM of 3-MeA and were washed just before transfection (lane 2) or were incubated with 3-MeA for 72 hrs after transfection (lane 3). Results shown is representative of two independent experiments. All samples in (a) or (b) were normalized by their protein concentrations, 30 or 20 μg/well, respectively. (c,d) The lysates of COS-7 cells producing HIVWT or HIVTPCAI2-V3 were prepared 72 hrs after transfection with lysis buffer-containing the PI cocktail or 1 μM of SQV. Data are represented as mean ± S.D. of two independent experiments. (e) The lysates of COS-7 cells producing HIVWT or HIVCAI2-V3 were prepared with lysing buffer-containing 13.4 mM of sodium azide, incubated at 37 °C for different periods of time indicated and subjected to ELISA. Note that the presence of a high concentration of sodium azide did not block the CA degradation, suggesting that the degradation event is ATP-independent process. Data are represented as mean ± S.D. of two independent experiments. (f) rCAWT and rCAR18-T19 produced by E. coli staining with Coomassie Brilliant Blue (CBB) are shown. rCAWT and rCAR18-T19 were expressed in E. coli and each cultured cell preparation was lysed and ammonium-sulfate-precipitated. Subsequently, the lysed samples and ammonium-sulfate-precipitates were subjected to SDS-PAGE and CBB staining. (g) The ammonium-sulfate-precipitates of the rCAWT and rCAR18-T19 samples described above were run through size-exclusion-chromatography and the fractions-containing rCAWT and rCAR18-T19 were subjected to SDS-PAGE and CBB staining. Results shown is representative of two independent experiments. Full-length blots/gels are presented in Fig. S9.
A number of proteins in both prokaryotic and eukaryotic cells are subject to energy-dependent proteolysis. Since sodium azide is known to inhibit the production of ATP through inhibition of oxidative phosphorylation22, we examined whether the CA degradation observed is an ATP-dependent process. When we added >10 mM sodium azide that is known to cause ATP-depletion22 to the lysates of HIVCAI2-V3-producing cells, no inhibition of the CA degradation was observed (Fig. 3e), strongly suggesting that the CA degradation observed is not an energy-dependent process.
We also examined whether the degradation also occurred when rCAWT and rCAR18-T19 were expressed in E. coli and size-exclusion-chromatographically purified so that no human cellular components or HIV-1 viral components were involved in the degradation process. The lysates of E. coli were centrifuged to afford CA-containing supernatants, from which proteins were precipitated with 20% ammonium sulfate. In both the supernatants and precipitates obtained, p24 fractions-containing rCAWT or rCAR18-T19 were clearly identified as examined with SDS-PAGE and Coomassie-blue staining (Fig. 3f). When both rCAWT and rCAR18-T19 were fractionated with size-exclusion-chromatography and both samples were further examined with SDS-PAGE and Coomassie blue, rCAWT signals were readily identified; however, rCAR18-T19 signals were almost absent while ladder-like signals were identified (Fig. 3g) that were seen when COS-7 and HEK-293 cells producing HIVCAR18-T19 were examined (Fig. 1b,d). These data strongly suggest that degradation of rCAR18-T19 proceeded during the fractionation and SDS-PAGE procedures, while rCAWT remained intact.
Taken together, these results strongly suggest that no cellular/viral proteolytic mechanisms are involved in the CA degradation observed and that the degradation is not energy-dependent. Thus, we assumed that the CA degradation observed occurs most likely due to compromised structural integrity caused by the 19-AA insertion into CA.
The degradation of mutant CAs requires the cleavage of the C-terminus of CA and originates from the CA-CTD
We then investigated how the CA degradation is initiated and proceeds. CA, as a critically important viral protein, is under strong selective pressure to regulate its production, maturation, integral structure, and functions. In the replication cycle of HIV-1, Gag polyprotein undergoes cleavage and maturation to form the functional CA. Thus, we examined the relationships of the cleavage sites that flank the CA precursor in the polyprotein. We generated three HIVCAI2-V3-based constructs that lack the cleavage(s) of Gag polyprotein (Fig. 4a): (i) a construct, in which the MA-CA cleavage of HIVCAI2-V3 was blocked by changing the cleavage site from “NYPI” to “NAAI” (thus referred to NAAIHIVCAI2-V3), (ii) a construct, in which the CA-p2 and p2-NC cleavages were blocked by changing the two cleavage sites from “VLAE” to “VAAE” and “IMIQ” to “IAAQ” (VAAE/IAAQHIVCAI2-V3), and (iii) a construct, in which all the three cleavages were blocked (NAAI/VAAE/IAAQHIVCAI2-V3)(Fig. 4a). In COS-7 cells producing NAAIHIVCAI2-V3, the accumulation of the MA-CA precursor protein was seen as expected, while in COS-7 cells producing VAAE/IAAQHIVCAI2-V3 and NAAI/VAAE/IAAQHIVCAI2-V3, the accumulation of the CA-p2-NC precursor protein and MA-CA-p2-NC precursor protein were seen, respectively (Fig. 4b). Of note, the CAI2-V3 degradation products were identified only in COS-7 cells producing HIVCAI2-V3 and NAAIHIVCAI2-V3, but virtually absent in those producing VAAE/IAAQHIVCAI2-V3 and NAAI/VAAE/IAAQHIVCAI2-V3 (Fig. 4b, boxed). These data showed that the blockade of the CAI2-V3 cleavage at the C-terminal end resulted in the absence or notable decrease of CAI2-V3 degradation.

The degradation of mutant CAs requires the CA’s C-terminal cleavage and originates from the CA-CTD. (a) A genetic map of the three following HIVCAI2-V3-based constructs lacking the cleavage(s) of Gag polyproteins generated: (i) a construct, in which the MA-CA cleavage of HIVCAI2-V3 was blocked by changing the cleavage site “NYPI” to “NAAI” (thus referred to NAAIHIVCAI2-V3), (ii) a construct, in which the CA-p2 and p2-NC cleavages were blocked by changing the two cleavage sites “VLAE” and “IMIQ” to “VAAE” and “IAAQ”, respectively (VAAE/IAAQHIVCAI2-V3), and (iii) a construct, in which all the three cleavages were blocked (NAAI/VAAE/IAAQHIVCAI2-V3). (b) Reduced degradation in VAAE/IAAQHIVCAI2-V3 and NAAI/VAAE/IAAQHIVCAI2-V3. The lysates of COS-7 cells producing HIVCAI2-V3 (Lane 1), NAAIHIVCAI2-V3 (Lane 2), VAAE/IAAQHIVCAI2-V3 (Lane 3), or NAAI/VAAE/IAAQHIVCAI2-V3 (Lane 4) were subjected to WB using anti-p24 anti-serum. CA degradation was clearly seen in Lanes 1 and 2, while the degradation levels in Lanes 3 and 4 were drastically reduced, indicating that the blockade of cleavages in the CA-p2-NC cleavage sites resulted in reduction of the CA degradation. (c) The CAI2-V3 degradation products are derived from the CTD of CAI2-V3. The lysates of HEK-293 cells producing HIVCAI2-V3 were subjected to WB using two monoclonal antibodies specific to the CA-NTD (mAb-NTD1 and mAb-NTD2), and two monoclonal antibodies specific to the CA-CTD (mAb-CTD1 and mAb-CTD2). The locations of each immunogen recognized are shown in the top of Panel c. Note that the ladder-like signals were identified only in the lysates examined with the two CA-CTD-specific mAbs. All samples in (b,c) were normalized by their protein concentrations (20 μg/well). Results shown is representative of three to five independent experiments for each mAb. Full-length blots/gels are presented in Fig. S9.
We further attempted to determine the constituents of the CAI2-V3 degradation products in terms of NTD or CTD products using polyclonal anti-CA anti-serum, two mAbs specific to the CA-NTD [mAb-NTD1 and mAb-NTD2 recognizing immunogens at the N-terminus, aa1–104 and aa74–84, respectively], and two mAbs specific to the CA-CTD [mAb-CTD1 and mAb-CTD2 recognizing immunogens at the C-terminus, aa180–190 and aa165–231, respectively]. When the lysates of HEK-293 cells producing HIVCAI2-V3 were subjected to WB using anti-CA pAb, the ladder-like signals were seen in HIVCAI2-V3-producing cells but not in HIVWT-producing cells (the very left panel in Fig. 4c). However, as examined with mAbs, the ladder-like signals were identified only in the lysates examined with the two CA-CTD-specific mAbs (Fig. 4c). Of note, the signals seen with the pAb are those with mAb-CTD1 added with those with mAb-CTD2 (Fig. 4c). These findings were corroborated when the lysates of HEK-293 cells producing five other HIV-1 constructs (HIVCAR18-T19, HIVCAA64-A65, HIVCAM96-R97, HIVCAL111-Q112, and HIVCAD152-I153) were subjected to WB using mAb-NTD1 or mAb-CTD1. Significant amounts of CA degradation products were seen as examined with pAb and mAb-CTD1, while virtually no significant degradation products were identified as examined with mAb-NTD1 (Fig. S5).
Taken together with the data above that the CAI2-V3 cleavage blockade at the C-terminus end resulted in the absence or notable decrease of CAI2-V3 degradation (Fig. 4b), the results strongly suggest that CA degradation observed is initiated at the CTD and proceed in a time-dependent manner.
Degradation-prone HIVTP-CAINS variants are replication-incompetent
We next determined the single-round infectivity of HIVTP virions harvested from the culture supernatants of transfected COS-7 cells with the MAGI assay using U373-MagiCD4+CXCR4+ cells. All five HIVTP-CAINSNTDs virions examined (HIVCAI2-V3, HIVCAR18-T19, HIVCAA64-A65, HIVCAM96-R97, and HIVCAL111-Q112) that had high-level CA degradation (Figs 1b,d and 2a) showed drastically reduced infectivity compared to HIVWT virions (Table 1 and Fig. 5a). The three HIVTP-CAINSCTDs virions examined (HIVCAD152-I153, HIVCAG220-V221, and HIVCAK227-A228) that had moderate-level CA degradation (Figs 1 and 2) had rather moderately reduced infectivity compared to the five HIVTP-CAINSNTDs virions examined. When we determined the replication kinetics of HIVTP-CAINSNTDs and HIVTP-CAINSCTDs virions using the MT-4 cell-based replication assay, both the virion groups examined had completely lost their replication capacity (Table 1 and Fig. 5b). Interestingly, HIVTPs-containing the AA insertion in the NTD of MA (HIVMAV35-W36, HIVMAG62-Q63, and HIVMAV88-H89), none of which showed CA degradation (Figs 1b and 2a) but had moderately to drastically reduced infectivity (Fig. 5a), showed no replication capacity (Fig. 5b). In addition, one HIVTP-containing the AA insertion in the p2 protein, which showed no CA degradation (Figs 1b and 2a) or no drastic infectivity reduction, had also completely lost its replication capacity (Table 1 and Fig. 5).

Degradation-prone HIVTP-CAINS variants are replication-incompetent. (a) The infectivity of each of HIVTP-CAINSNTDs (shown in red) and HIVTP-CAINSCTDs (in blue) was determined with the single-round infectivity assay using U373-MagiCD4+CXCR4+ cells. Note that the infectivity of HIVTP-CAINSs was substantially reduced, while that of three HIVTP-MAINSs (HIVMAV35-W36, HIVMAG62-Q63, and HIVMAV88-H89) were also substantially reduced. Data are represented as mean ± S.D. (n = 3). (b) Loss of replication capacity of HIVTP-CAINSNTDs (in red) and HIVTP-CAINSCTDs (in blue). Replication kinetics of HIVTP-CAINSs was determined with the amounts of p24 production in cultures of MT-4 cells exposed to each HIVTP. The boxes denote that all HIVTP-CAINSs examined completely failed to replicate in MT-4 cells, while the three HIVTP-MAINS (in green) described above and HIVp2A1-E2 (in purple) also failed to replicate. HIVp6T8-A9 (in orange) showed slower replication in MT-4 cells compared to HIVWT. Data are represented as mean ± S.D. (n = 2).
Co-expression of pHIVWT and pHIVTP-CAINSNTD produces highly compromised or replication-incompetent HIV-1
We then attempted to examine the impact of CAINSNTD production onto the replication competence of HIVWT by co-transfection of HEK-293 cells with pHIVWT and pHIVTP-CAINSNTD. pHIVTP-CAINSNTD has all the HIVWT genetic components except the CA-encoding gene carrying the 19-AAs in the NTD. When cells were transfected with pHIVWT only, a large amount of p24 (516 ng/ml) was produced, while co-transfected with pHIVWT and either of 3 pHIVTP-CAINSNTD (pHIVCAI2-V3, pHIVCAR18-T19, and pHIVCAA64-A65), p24 production was drastically reduced by 71.1, 86.4, and 74.2%, respectively, strongly suggesting that the co-expression of rCAWT with rCAI2-V3, rCAR18-T19, and rCAA64-A65 caused drastically enhanced degradation of the presumably chimeric rCA molecules comprised of rCAWT and rCAINSNTD (Fig. 6a). In order to corroborate and extend these results, we performed co-transfection experiments using different ratios of pHIVWT and pHIVTP amounts. When HEK-293 cells were co-transfected with 1:1, 1:1/6, and 1:1/10 ratios of pHIVWT and pHIVMAQ127-V128 amounts, no drastic reduction of p24 amounts produced was seen (Fig. 6b). In contrast, co-transfection of 1:1/6, and 1:1/10 ratios of pHIVWT and pHIVCAI2-V3 amounts caused as much as 51.8 and 40.4% reduction in the amounts of detectable p24. Co-transfection of 1:1/6, and 1:1/10 ratios of pHIVWT and pHIVCAR18-T19 amounts also caused 78.8 and 72.1% reduction in the amounts of detectable p24 (Fig. 6b).

Co-expression of pHIVWT and pHIVTP-CAINSNTD produces highly compromised or replication-incompetent HIV-1. (a) HEK-293 cells were transfected with pHIVWT alone, pHIVWT plus pHIVWT, pHIVMAE105-E106, pHIVMAQ127-V128, pHIVCAI2-V3, pHIVCAR18-T19, pHIVCAA64-A65 or pHIVp2A1-E2, cultured for 72 hrs, and the p24 amounts of each culture supernatant were determined. Note that when co-transfected with pHIVWT and either of 3 pHIVTP-CAINSNTD (pHIVCAI2-V3, pHIVCAR18-T19, pHIVCAA64-A65) and pHIVp2A1-E2, p24 production was drastically reduced as compared when transfected with pHIVWT and pHIVWT plus pHIVWT. (b) HEK-293 cells were transfected with pHIVWT alone, co-transfected with 1:1, 1:1/6, and 1:1/10 ratios of pHIVWT and pHIVp17Q127-V128, pHIVCAI2-V3, or pHIVCAR18-T19, cultured for 72 hrs, and the p24 amounts of each culture supernatant were determined. Results shown is representative of three independent experiments. (c) The supernatants harvested from each of the cultures described in Panel a were subjected to 9-day HIV-1 replication assays using MT-4 cells as target cells, after all the supernatants were normalized to contain an equal amount of p24. Note that the supernatants from transfection with pHIVWT alone or pHIVWT plus pHIVWT, pHIVp2A1-E2, pHIVMAE105-E106, or pHIVMAQ127-V128 produced large amounts of p24 over the 9-day culture period, while those from transfection with pHIVWT plus either of the three pHIVTP-CAINSNTD (pHIVCAI2-V3, pHIVCAR18-T19, pHIVCAA64-A65) produced no or least p24 protein. All p24 values are single-point determinations.
The supernatants harvested from each of the transfectant cultures of HEK-293 cells (Fig. 6a) were subsequently subjected to HIV-1 replication assays. After all the supernatants were normalized to contain an equal amount of p24 (10 ng/ml), serving as a source of infectious virions. MT-4 cells were exposed to each supernatant, washed, and cultured over 9 days. The supernatants from HEK-293 cells transfected with pHIVWT alone produced large amounts of p24 proteins (~1,200 ng/ml) by the end of the 9-days-culture period. Of note, MT-4 cells exposed to the supernatants of HEK-293 cells transfected with a two-fold greater amount of pHIVWT (pHIVWT plus pHIVWT as a control for other co-transfections) produced comparable amounts of p24 (~1,200 ng/ml) as compared to the amounts with only pHIVWT transfection (Fig. 6c). However, MT-4 cells exposed to the supernatants from HEK-293 cells, which were co-transfected with pHIVWT plus either of the three pHIVTP-CAINSNTD, produced no or the least p24 proteins over the 9-days-culture period, indicating that those supernatants contained virions with substantially compromised replication capacity or replication-incompetent virions. These data also strongly suggest that the presence of the three rCAINSNTD caused the failure of the formation of functional CA, resulting in replication inhibition of the otherwise replication-competent HIVWT. HEK-293 cells co-transfected with pHIVWT and either of two pHIVTP-MAINS (pHIVMAE105-E106 and pHIVMAQ127-V128) produced moderately reduced p24 in the culture (Fig. 6a); however, MT-4 cells exposed to those culture supernatants produced comparable p24 amounts compared to the supernatants from transfection with pHIVWT alone and with pHIVWT plus pHIVWT (Fig. 6c). MT-4 cells exposed to the supernatants from HEK-293 cells co-transfected with pHIVWT plus pHIVp2A1-E2 also produced p24 proteins comparably to the supernatants from transfection with pHIVWT alone (Fig. 6c).
Modelling study of the mechanism of the CA degradation
In an attempt to delineate the possible mechanism of the CA degradation, we first generated 19-AA insertion models and examined how the insertions could pose structural and functional impacts to each CA monomer that is to form hexamers (Figs 7, S6, and Movie S1). In the case of CAI2-V3, the insertion I2-V3 is located at the very N-terminal region (Fig. 7a–d) and is highly likely to effect the beta-hairpin loop that has crucial function by stabilizing hexamer complex. The view of the hexamer comprised of six CAI2-V3 monomers (Fig. S6d) strongly suggests that the insert should disrupt the structure and integrity of the pore, whose function is reportedly required for nucleotide entry to fuel RT activity inside the CA shell23. In the case of CAR18-T19, the insert R18-T19 is also located in the proximity of the N-terminal region of CA monomer and is thought to do the same as does the insertion I2-V3. In fact, both of the two insertions (I2-V3 and R18-T19) resulted in a total loss of the replication capacity of HIV-1 carrying those inserts (HIVCAI2-V3 and HIVCAR18-T19), most likely resulting from significantly disturbed structural and functional integrity of CA. In the cases of CAA64-A65, CAM96-R97, and CAL111-Q112, where the 19-AA insertion occurred in the proximity of H4, H5, and H6, which, unlike the cases of CAI2-V3 and CAR18-T19, are rather distant from the N-terminal region. As seen in the Fig. S6a, the insert is located relatively distant form the center of the hexamer and seems to cause less drastic effects on the structure of the hexamer and the pore. Although the levels of CA degradation in and the incompetent replication capacity of HIVCAA64-A65, HIVCAM96-R97, and HIVCAL111-Q112 were similar to those in HIVCAI2-V3 and HIVCAR18-T19, when COS-7 cells was co-transfected with pHIVWT and pHIVCAA64-A65, the replication of the otherwise totally replication-incompetent pHIVCAA64-A65 was moderately rescued (Fig. 6c), in contrast to the co-transfection of COS-7 cells with pHIVWT plus pHIVCAI2-V3 and pHIVWT plus pHIVCAR18-T19, which resulted in no identifiable rescue-effect by pHIVWT was seen (Fig. 6c). These data should suggest that the insert, which occurred rather distant from the N-terminal region, posed relatively moderate detrimental impact to the structure and function of CA hexamers. When the insertion occurred in the C-terminal region of CA (HIVCAD152-I153, HIVCAG220-V221, and HIVCAK227-A228), the insert apparently does not interfere with essential intramolecular interactions and the flexible 19-AAs sequence could adopt different conformation without blocking hexamer formation (Fig. S6a) and the insert probably does not pose severe crippling effects on the structure or functions of the hexamers. In fact, the levels of CA degradation in the three infectious clones are only moderate (Figs 1b,d and 2a) and their infectivity was also only moderate (Fig. 5a). The structural examination of the locations of the insert and the phenotypic features of each clone with the insert strongly suggest that the impacts of the insert are drastically affected by the location of the insert, as were seen in the cases of inserts Tamiya et al. reported causing significant CA degradation (19; Fig. S1a). However, it should also be noted that, while the levels of CA degradation, the rescue-effects by co-transfection with pHIVWT, and the infectivity assessed in the single-round HIV-1 replication assay were moderate or not-too-debilitating, all the 19-AAs-containing clones were virtually equally replication-incompetent (Fig. 5b). In this regard, it is rather understandable that for the occurrence of successive HIV-1 replication, robust structural and functional integrity of CA is required24,25. Also, we considered that the 19-AA insertion at the CA-NTD induced conformational alternations that extended into the distant C-terminal region of CA resulting in an unstable CA-CTD and the induction of degradation initiation at the CTD as shown in Fig. 4, suggesting that the 19-AA insertion at the CA-NTD causes gross conformational changes, which extended to the CA-CTD, resulting in structural fragility in both CA domains. Taken together, the present results suggest that the CA-p2 and p2-NC cleavage sites should be intact or the structural integrity of the CA-p2-NC domains should also be kept intact for the infectivity and replication capacity of HIV-1 and that if either (or both) of these molecular events is affected, HIV-1 CA degradation is initiated in the CTD and the degradation of virtually entire CA ensues.

Modeling study of the mechanism of the CA degradation. The 19-AA inserts are indicated by red color for both CAI2-V3 and CAR18-T19. On the upper panel, the model of CAI2-V3 in orange are superimposed into the monomer (a) and the hexamer (b–d) forms of the CAWT structure (PDB ID: 5HGL). Similarly, lower panel dedicated to the structural comparison of the CAR18-T19 model colored in purple with the CAWT monomer (e) as well as the hexamer form (f–h) structures. Hexamers are shown from top views (b,f), 90°-rotated side views (c,g), and 180°-rotated views (d,h). Note that the presence of 19-AA inserts let to formation of extended loops, which wrap around the CA pore composed of 6 arginine residues (shown in cyan). It is assumed that the extended loops are likely to push each monomer apart and effectively block the formation of physiologic hexamer complexes.
Discussion
The class of HIV-1 PIs represents a major component of the currently available cART of HIV-1 infection and AIDS, which has been shown to potently suppress the replication of HIV-1 and significantly extend the life expectancy of HIV-1-infected individuals26. It has been well reported that in the course of cART including PIs, HIV-1 acquires AA substitutions in the PR-encoding gene and avoids the binding of PIs to PR, thus developing resistance to PIs. The introduction of such AA substitutions into PR, however, often results in compromised proteolytic activity of PR, causing impaired replication competence of the virus11,27,28,29. In compensation of the reduced catalytic activity of mutant PRs, HIV-1 is known to add AA substitutions/insertions in the cleavage sites of the Gag proteins, substrates for PR, to restore the otherwise compromised enzymatic cleavage activity of mutated PRs, enabling multi-PI-resistant HIV-1 variants to keep or restore replication-competence19,30,31,32. Moreover, certain AA substitutions in non-cleavage sites within the Gag proteins have been shown to contribute to the development of higher levels of HIV-1 resistance to PIs33,34.
In the present study, we first found that drug-resistant HIV-1 carrying multiple AA insertions in Gag undergoes aberrant CA degradation (19; Fig. S1a). To elucidate the mechanism of the aberrant degradation and to determine the role of AA insertions into CA in the infectivity and replication capacity of HIV-1, we generated a series of recombinant HIV-1 clones-containing a 19-AA insertion randomly introduced into the Gag region using the Tn5-based transposon system (HIVTP). Although 19-AA insertion is relatively long and can be conformationally bulkier and rather more complex than the inserts identified in HIV-1 Gag in the context of HIV-1’s acquisition of resistance to PIs19, but we chose the 19-AA insertion in the present study since it is technically efficient and less labor-intensive than the site-directed mutagenesis to generate various replication-competent HIV-1 variants with AA insertions in CA and elucidate the effects of such AA insertions on the behavior of the Gag polyproteins in drug-resistant HIV-1 variants. The 19-AA insertion per se probably does not have structural fragility or instability, in view that the same AA insertion in MA underwent no undue degradation (Fig. S2a,c).
If one carefully takes a look at our previous paper by Tamiya et al.19, no significant consecutive amino acid substitutions or repeats were observed or described except for one or two sporadic substitutions in the CA-encoding region. These data should strongly suggest that if such significant amino acid substitutions (i.e., 3 to 6 or longer consecutive amino acids insertions) occur in the CA-encoding region, such mutant HIV-1 viruses would lose their replicable fitness, presumably due to the CA degradation we have seen in the current study, and are not seen in the clinical setting. In this regard, the integrity of CA and its formation is more susceptible to the impact of 3 or longer consecutive amino-acid insertions in CA rather than in matrix or p6. We would assume that the magnitude of CA degradation and the loss of viral replicability should be associated with both “the size and location of the inserts”. The longer the insert, the greater the magnitude of degradation and compromised replication. At this time, we have not directly asked whether “smaller AA sequences” would promote the degradation and compromised replication, which we are afraid goes beyond the scope of the current study. However, Tamiya et al. generated a recombinant clone HIV-1NL4–3 containing the “SQVN” insert, located 8 amino-acid-upstream from the I2-V3 insert of HIVCAI2-V3 (HIVSQVN), which proved that HIVSQVN showed significant capsid degradation as well as compromised replicability19. Thus, such previous data by Tamiya et al. and our current data should strongly suggest that the significant CA degradation observed is caused by the inserts (3 or longer consecutive amino acids) and is associated with the loss of replicability.
In Tamiya’s report, the magnitude of the compromised replicative fitness of recombinant clones engineered to have 3 to 6 AA insertions was not that great (downed by ~20-fold compared to HIV-1NL4–3)19 compared to the recombinant clones engineered to have 19-AA inserts in CA presented in the current study. This difference should be, as we mentioned above, due to that the integrity of CA and its formation is more susceptible to the impact of AA insertions in CA rather than in matrix or p6 documented in Tamiya’s report19. In this regard, we have newly conducted an experiment, in which a recombinant clone engineered to contain two inserts, AQQA and PTAPPA inserts that did not cause “severe” reduction in viral replication in Tamiya’s report, designated HIVMAAQQAp6PTAPPA. As we expected, this HIVMAAQQAp6PTAPPA had significant CA degradation and “severe” reduction in replicability (Fig. S8).
The CA degradation identified also progressed in a time-dependent manner when incubated at 37 °C (Fig. 2). Moreover, the CA degradation was shown to begin at the CA-CTD as examined using multiple different CA-specific mAbs, which recognized different antigenic epitopes of CA (Fig. 4c). Also, the data strongly suggest that proteasome, autophagy, or cellular/viral proteases are not responsible for the CA degradation (Fig. 3a–d). In addition, the CA degradation does not seem to be an energy-dependent process (Fig. 3e). Moreover, CA degradation also occurred when rCAR18-T19 was expressed in E. coli (Fig. 3f,g). Altogether, it is assumed that the CA degradation is caused by the structural instability and/or compromised structural integrity (Fig. 3).
The mature CA mainly exists as a hexamer and the CA-NTD appears to play an important role in its formation in view of both geographical and functional perspectives. In this regard, Jacques and his colleagues have shown that each CA hexamer has a size-selective pore bound by a ring of six Arg-18 residues and a ‘molecular iris’ is formed by the amino-terminal β-hairpin23. In this respect, a study of >70,000 protein structures and complexes for unusual formation of arginine residues by Neves et al.35 has shown that arginines form various clusters that are exposed to interact with and potentially be controlled or switched by nucleic acids, charged metabolites, membrane lipids, and side chains of other proteins. Also, Mallery et al. reported that these pores bind the cellular polyanion IP6, transforming viral stability and allowing newly synthesized DNA to accumulate inside the CA36. These data, together with the data on CA hexamers23 strongly suggest that the control of the stability endowed by arginine clusters may play an important role in protein-protein oligomerization, molecular recognition, DNA accumulation, and ligand binding. Interestingly, the CA degradation and the loss of infectivity and replication of HIV-1 were most profound in the present study when 19-AA insertions were added to CA-NTD (residues 1–145) involving the arginine clusters, while the loss of infectivity was less extensive when the insertions were added to CA-CTD (residues 151–231), distant from the CA-NTD. The loss of replication capacity of HIV-1s that contained the 19-AA insertions in the CA-NTD was relatively more drastic than their infectivity was probably since the replication capacity represents the results of multiple cycles of HIV-1 replication and would show more significant compromised results.
It is of note that all HIVTP-CAINSNTDs and HIVTP-CAINSCTDs totally failed to replicate in MT-4 cells, although HIVWT and three other rHIVTPs (HIVMAE105-E106, HIVMAQ127-V128, and HIVp6T8-A9) vigorously replicated (Fig. 5b). The replication failure in all the recombinant HIV-1 carrying TP at the CA-NTD and CA-CTD should stem from the disturbed structural integrity of CA. In this regard, when the morphological abnormality is drastic as examined with TEM, no replication capacity is seen as easily expected; but with much less or even apparently normal morphology of the virions, the virus virtually completely loses replication capacity37. These observations suggest that the induction of even nominal CA abnormality could possibly completely abrogate the replication capacity of HIV-1. Indeed, when we assessed the morphology of two HIVTP-CAINSNTDs (HIVCAI2-V3 and HIVCAR18-T19), which totally failed to replicate in MT-4 cell culture (Fig. 5b), the cores of those virions were much less condensed or absent as compare to normal virions (Fig. 2b). These drastic morphological abnormalities are fairly in line with the failure of their replication loss. In the relevant context, the co-expression of HIVTP-CAINSNTD with HIVWT notably impaired the replication capacity of HIVWT (Fig. 6c), probably supports the “assembly defect” of the chimeric virions with physiologically robust CA plus structurally compromised CA due to the insert would diminish or nullyfy the replication capacity. Indeed, CAINSNTD expressed from co-transfected mutant HIVTPs probably incorporated into CA multimers within HIVWT virions, thereby disrupting the formation of normal CA shells and resulting in viral replication failure (Fig. 6c).
Also, it is of note that CA-CTDs contribute to hexamer-hexamer interactions during the formation of a conical shaped mature CA shell in HIV-1 virions38,39. Thus, we hypothesize that the 19-AA insertions in the CA-NTD directly interfere with CA hexamer formation and that the degradation initiation induced at CA-CTD additionally prevents CA-CTD-mediated hexamer-hexamer interactions. Our TEM studies also support this notion, revealing that normal CA cores are not present in HIVTP-CAINSNTD virions (Fig. 2b). It is possible that the two distinct structural effects mentioned above (the presence of insertion in the NTD and following CA degradation initiated from CTD) induce this drastic morphological abnormality in the HIVTP-CAINS virions.
With regard to our failure to determine the exact mechanism of the CA degradation we have observed other than the possible spontaneous degradation due to the disintegrity of the CA structure, the best study would be to determine the X-ray structure of 19-AA insertion-containing CA. However, it should be probably impractical to express and crystalize 19-AA insertion-containing CA due to its highly degradation-prone property. Indeed, CA has the most unique biological feature: a certain level of structural stability of CA conical shell is needed for the protection of viral RNA copies and other enzymes such as reverse transcriptase, while at the same time the CA shell should be “brittle” and “unstable” to disintegrate upon “uncoating” around the intracellular entry of HIV-1, eventually allowing the transport of the viral pre-integration complex into the cytoplasm, then to the nucleus. In fact, even “when, where, and how CA uncoating occurs” still remains hypothetic. It is not known as to (i) whether “uncoating” takes place immediately after the HIV-1 fusion with the cellular membrane; (ii) whether a partial uncoating occurs within the cytoplasm; or (iii) whether uncoating occurs at the nuclear pore. Whether viral/cellular factors are involved in the CA uncoating process is also presently unclear40. Nevertheless, physiological CA degradation is obviously initiated upon/through the uncoating process. We believe that the 19-AA insertion in CA induces conformational/structural disintegrity, significantly enhancing the CA degradation. In fact, as illustrated in Fig. 1d, WB results of HIVWT cell lysates show multiple signals although with less intensity compared to the degradation products of 19-AA insertion-containing CA, such as HIVCAI2-V3. It should be noted that the weak signals in the HIVWT cell lysates are of the same molecular weights as in the degradation products of 19-AA insertion-containing CA, strongly suggesting that the 19-AA insertion potentiated the otherwise slow and modest physiological CA degradation. Indeed, in recombinant CAWT expressed in E. coli, similarly weak degradation signals at the same mobilities as seen in recombinant CAR18-T19, were also identified (Fig. 3g).
The reason why cells co-transfected with pHIVWT plus pHIVp2A1-E2 produced a notably reduced amount of p24 protein is not readily understood, although the supernatants had contained robustly replication-competent virions (Fig. 6a,c). However, it is noted that the insert had occurred between alanine-1 and glutamic acid-2 of p2, that is located just next to the cleavage site CAL231-p2A1. Thus, it is possible that the structural conformation of the cleavage site CAL231-p2A1 was drastically altered and the cleavage efficiency was compromised. The notable restoration of the infectivity and replication capacity seen when co-expression of pHIVWT plus pHIVp2A1-E2 was made well explain since physiological p2 produced with pHIVWT rescued the otherwise compromised and non-replicative HIVWT (Fig. 6c).
The WB results showed that recombinant CA-containing the 19-AAs in both NTD and CTD underwent drastic degradation in time-dependent manner, although CA degradation in HIVTP-CAINSNTD and rCAINSNTD underwent substantially greater levels of degradation than in HIVTP-CAINSCTD and rCAINSCTD (Figs 1b,d and 2a,c). Of note, when the quantities of CA were determined as p24 antigen levels using a quantitative immunoassay employing two CA-specific mAbs, the CA signals were rapidly lost over the 48-hour incubation of various HIVTP-CAINSNTD (Fig. 2a). Although the CA signals were also lost when HIVTP-CAINSCTD were similarly placed at 37 °C, the intensities of the loss were moderate compared to those of HIVTP-CAINSNTD. It is noteworthy that the degradation of rCAWT was not drastic even after 48-hour incubation as assessed with WB using a pAb against CA, although in the cases of rCAI2-V3 and rCAR18-T19, both of which carry the insert in the CA-NTD, CA degradation rapidly progressed within 24-hours and virtually no drastic signals were detected in 48-hours. When the amounts of CA in two rCAINSNTDs (rCAI2-V3 and rCAR18-T19) and two rCAINSCTD (rCAD152-I153 and rCAG220-V221) were quantified using the automated quantitative immunoassay, the signals were also progressively lost over the incubation. When using another manufacturer’s ELISA kit to corroborate these results, similar degradation profiles were observed (Fig. S7). These data strongly suggest that CA degradation was so extensive when incubated over 24 to 48 hours at 37 °C that most of the antigenic epitopes on CA that would be detected by the anti-serum had been nearly entirely destroyed. It is worth noting that when we attempted to determine the AA-sequences of each of the CA degradation product ladders using the pieces of nitrocellulose membrane-containing the ladders with the Edman degradation method41; however, we failed to obtain the AA-sequences.
HIVTPs carrying the insert at the NTD of MA (HIVMAV35-W36, HIVMAG62-Q63, and HIVMAV88-H89) had reduced infectivity and replication capacity (Table 1 and Fig. 5); however, none of them had notable protein degradation (Figs 1b and 2a). In this regard, the positions V35 and W36 of HIVMAV35-W36 are located in the basic domain of MA, which plays an important role in Env incorporation over the assembly stage. The positions G62/Q63 and V88/H89 are located in the proximity of or within the “virus assembly domain” and “Gag-targeting domain”39, respectively, both of which are essential for HIV’s acquisition of infectivity and replicative ability. Thus, the AA insertion in the NTD of MA presumably disrupts these functional domains.
Most importantly, in the present study, when we examined the impact of CAINSNTD production onto the replication competence of HIVWT conducting co-transfection of cells with pHIVWT and pHIVTP-CAINSNTD, the resultant probably “chimeric” virions showed highly compromised replication capacity or replication-incompetence (Fig. 6). The p24 found in the supernatants of the co-transfected COS-7 cells shown in Fig. 6a should represent p24 from replicative HIV-1 (chimeric HIV containing CAWT plus mutated MA or mutated p2) and p24 from non-replicative HIV-1 (chimeric HIV containing CAWT plus mutated CA). Figure 6c shows that the chimeric HIV containing CAWT plus mutated MA or mutated p2 were capable of replicating; while the chimeric HIV containing CAWT plus mutated CA failed to replicate in the case of co-transfection with pHIVCAI2-V3 and pHIVCAR18-T19. The data shown in Fig. 6c strongly suggest that the mutant CA such as CAI2-V3 and CAR18-T19, if integrated into the capsid-shell formed with CAWT, induces degradation of the whole CA. In other words, these data strongly suggest that the incorporation of degradation-prone (unstable or fragile) CAINSNTD into the otherwise functional CA results in drastic disruption of the formation of normal CA shells, leading to the loss of replication competence in such resultant “chimeric” virions (Fig. 6c). It is possible that if certain mutated CA-encoding genetic materials could be introduced to express such degradation-prone CA species within the cells productively infected with HIVWT or even drug-resistant HIV, the resultant “chimeric virions” would lose their replication capacity, causing “intracellular killing of infectious virions” and eventual cellular apoptosis without production of infectious virions, a potentially new modality of reduction or elimination of the HIV-1-infected cells.
In conclusions, the present observations of CA degradation may allow us to understand how CA decomposes and possibly the dynamics and mechanism of the formation of CA hexamers and decomposition/degradation process upon the initial phases of HIV-1’s cellular entry. The present observation may also open a new avenue for discovery of new antiretroviral modalities to compromise the orderly CA degradation and the uncoating process, or new anti-HIV-1 agents inducing abnormal CA degradation.
Methods
Cells
MT-4 cells were grown in RPMI 1640-based culture medium supplemented with 10% fetal calf serum (FCS; PAA Laboratories GmbH, Linz, Austria) plus 50 U of penicillin and 100 μg of kanamycin per ml. COS-7 and HEK-293 cells (DS pharma biomedical, Osaka) were grown in DMEM-based culture medium supplemented with 10% FCS plus 50 U of penicillin and 100 μg of kanamycin per ml. U373-MAGICD4+CXCR4+ cells were obtained through the AIDS Reagent Program, NIAID, NIH, were maintained in DMEM with 10% FCS, 0.2 mg/ml G418, 0.1 mg/ml hygromycin B, and 1.0 µg/ml puromycin.
Inhibitors of cellular/viral protein degradation pathways/enzymes
To determine whether cellular/viral protein degradation pathway/enzyme have any effects on the CA degradation observed, we employed the following inhibitors: (i) a proteasome inhibitor, MG-132 (Sigma-Aldrich Japan, Tokyo, 20 µM); (ii) a cocktail of seven mammalian protease inhibitors (PIs; Sigma): AEBSF (final concentration, 1 mM), aprotinin (0.8 μM), bestatin (40 μM), E-64 (10 μM), leupeptin (100 μM), pepstatin (15 μM), and phosphoramidon (10 μM); (iii) an autophagy inhibitor, 3-methyladenine (3-MeA, Sigma, 5 mM); and (iv) an HIV-1 PI, saquinavir (SQV, 1 μM), which was kindly provided by Roche Products Ltd. (Welwyn Garden City, United Kingdom).
Generation of recombinant HIV-1 clones
We generated a series of HIV-1NL4-3-based recombinant clones containing a 19-AA insert randomly introduced into Gag proteins using the Tn5-based transposon system (EZ-Tn5™ In-Frame Linker Insertion Kit, Epicentre, Madison, WI) according to the manufacture’s protocol. In brief, a plasmid (TA cloning kitTM with pCRTM 2.1 vector, Thermo Fisher Scientific, Waltham, MA) containing the BssHII-SmaI fragment of the wild-type HIV-1HXB2 (nucleotides 712 to 2591, encoding Gag and PR) was exposed to EZ-Tn5™ transposon, which contains the kanamycin-resistance-associated gene, and transposase, followed by E. coli transformation and kanamycin selection. The resulting colonies with 19 AA-containing BssHII-SmaI fragment was picked up, kanamycin-resistance-associated gene was removed by NotI restriction enzyme, and HIV-1NL4-3 constructs with the 19 AA-containing BssHII-SmaI fragment were generated using pHIV-1NLSma, which had been generated to have a SmaI site by changing two nucleotides (2590 and 2593) of pHIV-1NL4-3, and expanded. Thus obtained 19 AA-containing Gag-encoding gene was subjected to DNA sequencing and the amino acid sequences and locations of the 19 AA within Gag were determined through deducing the nucleic acid sequences. Insert-containing CA-encoding plasmids were generated as follows. pCMV-Myc (Takara Bio, Kusatsu, Shiga) was digested with ApaI/XhoI to remove Myc-epitope and linearize the vector. CA fragment of each recombinant HIV-1 clone was amplified with PCR, and each of thus-generated fragments was introduced into the linearized pCMV-Myc using In-fusion enzyme (Clontech Laboratories, Takara bio, Kusatsu, Shiga).
Western blot analysis
To analyze whether HIV-1 polyproteins in each of molecular HIV-1 clones generated were cleaved by HIV-1 PR, Western blot (WB) analysis with the lysates of HIV-1-producing cells and cell-free virions was conducted. Briefly, at 72 hours after transfection using Lipofectamine LTX (Invitrogen, Thermo Fisher Scientific, Waltham, MA) with plasmid preparations, COS-7 or HEK-293 cells were washed with PBS and lysed in M-per solution (Thermo Fisher Scientific, Waltham, MA), and the cell lysates were subjected to WB. Culture supernatants containing virions were collected 72 hours after transfection, filtered through Steriflip Millipore Express PLUS membranes (0.22 μm pore-size, Millipore, Bedford, MA), and centrifuged at 20,000 g for 24 hours to pellet virions, were then lysed in M-per solution. In the WB assay, samples were normalized based on the protein concentration and subjected to electrophoresis on sodium dodecyl sulfate 4–12% polyacrylamide gel, followed by electroblotting onto nitrocellulose membranes.
The HIV-1 Gag proteins and Env glycoprotein were visualized with SuperSignal WestPico (Thermo Fisher Scientific, Waltham, MA) and Can Get Signal (Toyobo, Kitaku, Osaka) by using anti-HIV-1 p24 (CA) polyclonal antibody (pAb; Advanced Biotechnologies, Eldersburg, MD, Cat# 13-203-000, RRID:AB_1929205), 4 different anti-HIV-1 CA monoclonal antibodies (mAbs; United States biological, Salem, MA, Cat# H6005-08A, -08B, -08D, and -08F), anti-HIV-1 p17 (MA) mAb (Advanced Biotechnologies, Cat# 13-103-100, RRID:AB_1929216), and anti-HIV-1 gp120 pAb (D7324; Aalto Bio Reagents, Dublin). We also used following polyclonal antibodies (pAbs); anti-ubiquitin pAb (Calbiochem, San Diego, CA Cat# 662099, San Diego, CA), anti-LC3 pAb (Novus Biologicals, Littleton, CO, Cat# NB100-2220, RRID:AB_10003146), anti-BSA pAb (BioVision, Milpitas, CA, Cat# 5998-100, RRID:AB_2225924).
Determination of time-dependent CA degradation
The cell/virion lysates were prepared 72 hours after transfection and incubated at 37 °C for different periods of time, and CA concentrations of each cell/virion lysates at optional time points were measured using a fully-automated chemiluminescent enzyme immunoassay system (Lumipulse f: Fujirebio Inc., Tokyo). Graphs were generated based on the percentage of the CA concentration in each lysate sample incubated at 37 °C for different time periods, compared to the CA concentration of the control samples, which had been stored at −80 °C immediately after preparation and served as 100%. Percent p24 (CA) represents the percentage determined using the following equation: 100 × (CA conc. of cell lysates at each incubation time)/(CA conc. of the time 0 lysates)(%).
Determination of single-round infectivity and replication capacity of each recombinant infectious HIV-1 clone containing an insert in CA
To determine the single-round infectivity of each recombinant infectious clone containing an insert in CA, we conducted the Magi assay using U373-MAGICD4+CXCR4+ cells42. The U373-MAGICD4+CXCR4+ cells (6 × 104 cells/500 µL/well) were cultured in DMEM containing 10% FCS in 24-well plates for 24 hours, exposed to each infectious clone (10 ng/ml of p24-equivalent/300 µL/well) for 2 hours, replenished with 1 mL fresh culture medium, and further cultured for another 48 hours. Following removal of the supernatants, the cells were fixed with 1% formaldehyde and 0.2% glutaraldehyde in PBS for 5 minutes, the fixing solution was removed, and the cells were washed with PBS and visualized with staining solution (950 μl PBS, 20 μl 0.2 M potassium ferrocyanide, 20 μl 0.2 M potassium ferricyanide, 1.0 μl 2.0 M Mg2Cl, and 10 μl 40 mg/ml X-gal stock) over 2 hours. Finally, the stained cells were washed twice with PBS and the number of blue-stained infected cells was determined under microscopy.
To determine the replication capacity of each recombinant infectious clones, replication kinetics assay employing fresh MT-4 cells was performed as previously described43,44. In brief, COS-7 cells were transfected with pHIVWT or pHIVTPs and incubated for 3 days, then concentrations of p24 in culture supernatants were measured and stored at −80 °C until use. MT-4 cells (1 × 105) were exposed to the each HIVWT or HIVTP preparation containing 10 ng/ml of p24 in 6-well culture plates for 5 hours, and those MT-4 cells were washed and cultured with fresh culture medium. The amounts of p24 in supernatants were determined every two days for up to 9 days.
Transmission electron microscopy (TEM) analysis
COS-7 cells were transfected with pHIVWT, pHIVCAI2-V3, or pHIVCAR18-T19, centrifuged, fixed in phosphate-buffered 2% glutaraldehyde, post-fixed in 2% osmium tetra-oxide in ice, dehydrated in ethanol, and embedded in the epoxy resin. Ultrathin sections of the samples were stained with uranyl acetate for 10 min and with lead-staining solution for 5 minutes, and submitted to TEM (JEM-2000EX and JEM-1200, JEOL, Peabody, MA).
Expression and purification of recombinant CA in E.coli
For the generation of recombinant CA expression vectors, the entire HIV-1 CA-encoding gene from pHIVNL4-3 or pHIVCAR18-T19 was introduced to pET30a (Merck KGaA, Darmstadt, Germany) using the In-Fusion method. The CA-encoding gene-containing pET30a was transformed into RosettaTM (DE3) pLysS Competent Cells (Novagen, Merck Millipore, Kenilworth, NJ) grown in LB medium supplemented with kanamycin and chloramphenicol to an OD600 of 0.5 at 37 °C, and the expression of rCA was induced with 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3–4 hours at 37 °C. The cells were sonicated in lysis buffer (5 mM 2MetOH, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4), centrifuged for 15 min at 3,500 rpm, and the obtained supernatants were added with 20% of (NH4)2SO4, and the precipitates were collected with centrifugation for 15 min at 3,500 rpm. Thus obtained precipitates were resuspended in lysis buffer, centrfuged for 20 min at 15,000 rpm at 4 °C, and the supernatants were passed through 0.45 μm filter and loaded onto a GE Hi Load 16/600 Superdex (GE Healthcare, Chicago, IL) attached to a AKTA prime plus (GE Healthcare). rCA-containing fractions were pooled and concentrated using Amicon Ultra 10 K device (Merck Millipore, Kenilworth, NJ) in the lysis buffer. The concentration of protein was determined using BCA Protein Assay Reagent Kit (Thermo Fisher Scientific, Waltham, MA).
Computational modeling of AA insertions into CA
Computational models of AA insertions at various sites of CA were generated using the Structure Prediction Server (PS)2 version 3.045 using the complete sequence of the insertion along with the flanking sequences from the wild-type CA. The models generated were downloaded from the server and were analyzed using PyMol version 1.7.6.6 (Schrodinger, LLC). The flanking sequence of the obtained model was then superposed onto the corresponding matching sequence of the wild-type CA (PDB ID: 5HGL).
Data Availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information Files.
References
Department of Health and Human Services. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. (2106).
Yeni, P. G. et al. Antiretroviral treatment for adult HIV infection in 2002: updated recommendationsof the International AIDS Society-USA Panel. JAMA 288, 222–235 (2002).
Grabar, S. et al. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. AIDS 14, 141–149 (2000).
Mitsuya, H. & Erickson, J. W. In Textbook Of AIDS Medicine. (eds Merigan, T. C., Bartlet, J. G. & Bolognes, D.), 751–780 (The Williams & Wilkins Co., Baltimore, Md., 1999).
Paredes, R. et al. Predictors of Virological Success and Ensuing Failure in HIV-Positive Patients Starting Highly Active Antiretroviral Therapy in Europe. Arch. Intern. Med. 160, 1123–1132 (2000).
Condra, J. H. et al. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374, 569–571 (1995).
Whitcomb, J. M., Parkin, N. T., Chappey, C., Hellmann, N. S. & Petropoulos, C. J. Broad Nucleoside Reverse-Transcriptase Inhibitor Cross-Resistance in Human Immunodeficiency Virus Type 1 Clinical Isolates. J. Infect. Dis. 188, 992–1000 (2003).
Shafer, R. W. et al. Combination therapy with zidovudine and didanosine selects for drug-resistant human immunodeficiency virus type 1 strains with unique patterns of pol gene mutations. J. Infect. Dis. 169, 722–729 (1994).
Shirasaka, T. et al. Changes in drug sensitivity of human immunodeficiency virus type 1 during therapy with azidothymidine, dideoxycytidine, and dideoxyinosine: An in vitro comparative study. Proc. Natl. Acad. Sci. USA 90, 562–566 (1993).
Shirasaka, T. et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 92, 2398–2402 (1995).
Croteau, G. et al. Impaired Fitness of Human Immunodeficiency Virus Type 1 Variants with High-Level Resistance to Protease Inhibitors. J. Virol. 71, 1089–1096 (1997).
Gulnik, S. V. et al. Kinetic Characterization and Cross-Resistance Patterns of HIV-1 Protease Mutants Selected under Drug Pressure. Biochemistry 524, 9282–9287 (1995).
Mahalingam, B. et al. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur. J. Biochem. 245, 238–245 (1999).
Schock, H. B., Garsky, V. M. & Kuo, L. C. Mutational Anatomy of an HIV-1 Protease Variant Conferring Cross-resistance to Protease Inhibitors in Clinical Trials. J. Biol. Chem. 271, 31957–31963 (1996).
Ueno, T., Shirasaka, T. & Mitsuya, H. Enzymatic characterization of human immunodeficiency virus type 1 reverse transcriptase resistant to multiple 2′,3′-dideoxynucleoside 5′-triphosphates. J. Biol. Chem. 270, 23605–23611 (1995).
Andreatta, K. N., Goodman, D. D., Miller, M. D. & White, K. L. Reduced Viral Fitness and Lack of Cross-Class Resistance with Integrase Strand Transfer Inhibitor and Nucleoside Reverse. Antimicrob. Agents Chemother. 59, 3441–3449 (2015).
Borman, A. M., Paulous, S. & Clavel, F. Resistance of human immunodeficiency virus type 1 to protease inhibitors: selection of resistance mutations in the presence and absence of the drug. J. Gen. Virol. 77, 419–426 (1996).
Ho, D. D. et al. Characterization of Human Immunodeficiency Virus Type 1 Variants with Increased Resistance to a C2-Symmetric Protease Inhibitor. J. Virol. 68, 2016–2020 (1994).
Tamiya, S., Mardy, S., Kavlick, M. F., Yoshimura, K. & Mitsuya, H. Amino Acid Insertions near Gag Cleavage Sites Restore the Otherwise Compromised Replication of Human Immunodeficiency Virus Type 1 Variants Resistant to Protease Inhibitors. J. Virol. 78, 12030–12040 (2004).
Peters, J. M., Frankes, W. W. & Kleinschmidtfl, J. A. Distinct 19S and 20S Subcomplexes of the 26S Proteasome and Their Distribution in the Nucleus and the Cytoplasm. J. Biol. Chem. 269, 7709–7718 (1994).
Seglen, P. O. & Gordon, P. B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 79, 1889–1892 (1982).
Schwoebel, E. D., Ho, T. H. & Moore, M. S. The mechanism of inhibition of Ran-dependent nuclear transport by cellular ATP depletion. J. Cell Biol. 157, 963–974 (2002).
Jacques, D. A. et al. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353 (2017).
Rihn, S. J. et al. Extreme Genetic Fragility of the HIV-1 Capsid. PLoS Pathog. 9, e1003461 (2013).
Reicin, A. S. et al. Linker Insertion Mutations in the Human Immunodeficiency Virus Type 1 gag Gene: Effects on Virion Particle Assembly, Release, and Infectivity. J. Virol. 69, 642–650 (1995).
Palella, F. J. J. et al. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 338, 853–860 (1998).
Brenner, B. G. et al. Persistence and Fitness of Multidrug-Resistant Human Immunodeficiency Virus Type 1 Acquired in Primary Infection. J. Virol. 76, 1753–1761 (2002).
Kavlick, M. F. & Mitsuya, H. In In The Art Of Antiretroviral Therapy. (ed. De Clercq, E.), 279–312 (American Society for Microbiology, Washington, D.C., 2002).
Rose, R. E. et al. Human immunodeficiency virus type 1 viral background plays a major role in development of resistance to protease inhibitors. Proc. Natl. Acad. Sci. USA 93, 1648–1653 (1996).
Maguire, M. F. et al. Changes in Human Immunodeficiency Virus Type 1 Gag at Positions L449 and P453 Are Linked to I50V Protease Mutants In Vivo and Cause Reduction of Sensitivity to Amprenavir and Improved Viral Fitness In Vitro. J. Virol. 76, 7398–7406 (2002).
Prabu-jeyabalan, M., Nalivaika, E. A., King, N. M. & Schiffer, C. A. Structural Basis for Coevolution of a Human Immunodeficiency Virus Type 1 Nucleocapsid-p1 Cleavage Site with a V82A Drug-Resistant Mutation in Viral Protease. J. Virol. 78, 12446–12454 (2004).
van Maarseveen, N. M. et al. Modulation of HIV-1 Gag NC/p1 cleavage efficiency affects protease inhibitor resistance and viral replicative capacity. Retrovirology 9, 29 (2012).
Gatanaga, H. et al. Amino Acid Substitutions in Gag Protein at Non-cleavage Sites Are Indispensable for the Development of a High Multitude of HIV-1 Resistance against Protease Inhibitors. J. Biol. Chem. 277, 5952–5961 (2002).
Myint, L. et al. Gag Non-Cleavage Site Mutations Contribute to Full Recovery of Viral Fitness in Protease Inhibitor-Resistant Human Immunodeficiency Virus Type 1. Antimicrob. Agents Chemother. 48, 444–452 (2004).
Neves, M. A. C., Yeager, M. & Abagyan, R. Unusual Arginine Formations in Protein Function and Assembly: Rings, Strings and Stacks. J. Phys. Chem. B 116, 7006–7013 (2012).
Mallery, D. L. et al. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife. 7, e35335 (2018).
Kageyama, S. et al. A C2 Symmetry-Based HIV Protease Inhibitor, A77003, Irreversibly Inhibits Infectivity of HIV-1 In Vitro. AIDS Res. Hum. Retroviruses 10, 735–743 (1994).
Ganser-pornillos, B. K., Cheng, A. & Yeager, M. Structure of Full-Length HIV-1 CA: A Model for the Mature Capsid Lattice. Cell 131, 70–79 (2007).
Freed, E. O. HIV-1 Gag Proteins: Diverse Functions in the Virus Life Cycle. Virology 251, 1–15 (1998).
Campbell, E. M. & Hope, T. J. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 13, 471–483 (2015).
Edman, P. Method for determination of the amino acid sequence in peptides. Acta Chem. Scand. 4, 283–293 (1950).
Vodicka, M. A. et al. Indicator Cell Lines for Detection of Primary Strains of Human and Simian Immunodeficiency Viruses. Virology 198, 193–198 (1997).
Amano, M. et al. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 51, 2143–2155 (2007).
Amano, M. et al. A Modified P1 Moiety Enhances In Vitro Antiviral Activity against Various Multidrug-Resistant HIV-1 Variants and In Vitro Central Nervous System Penetration Properties of a Novel Nonpeptidic. Antimicrob. Agents Chemother. 60, 7046–7059 (2016).
Huang, T. T. et al. (PS)2: protein structure prediction server version 3.0. Nucleic Acids Res. 43, 338–342 (2015).
Acknowledgements
This work was supported in part by the Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re-emerging Infectious Diseases (Renkei Jigyo: No. 78, Kumamoto University) of Monbu-Kagakusho (H.M.), the Grant from the National Center for Global Health & Medicine (H.M.), the Intramural Research Program of Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.), and by grants from the JSPS KAKENHI Grant Numbers JP23791140 and JP15K09574 (M.A.). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, M.D. (http://biowulf.nih.gov). We also thank Drs. Masao Matsuoka, Kenji Maeda, Toshikazu Miyakawa, Hirotomo Nakata, Yasushi Tojo, Tahei Nakamura, Nobutoki Takamune, Ravikiran S. Yedidi, Debananda Das, Matthew L. Danish, Hiromi Aoki-Ogata, and Pedro Miguel Salcedo-Gómez for their discussions and technical supports.
Author information
Authors and Affiliations
Contributions
M.A. designed and performed almost all the experiments and analyzed the data. S.T. generated all the transposon-related AA-insertion-containing HIV-1 variants. H.B. conducted structural analyses regarding AA-insertion-containing CAs. T.N. was involved in the preparation of recombinant CAs. Y.K. supported to design the experiments. H.M. supervised all the experiments. M.A. and H.M. wrote and edited the MS. H.B. and T.N. supported preparation of the MS. All authors read, commented on, and approved the MS.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amano, M., Bulut, H., Tamiya, S. et al. Amino-acid inserts of HIV-1 capsid (CA) induce CA degradation and abrogate viral infectivity: Insights for the dynamics and mechanisms of HIV-1 CA decomposition. Sci Rep 9, 9806 (2019). https://doi.org/10.1038/s41598-019-46082-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46082-2
- Springer Nature Limited
This article is cited by
-
Rotten to the core: antivirals targeting the HIV-1 capsid core
Retrovirology (2021)