
Abstract
To explore the association between methylation of antisense non-coding RNA in the INK4 locus (ANRIL) and coronary artery disease (CAD) development. Methylation levels of ANRIL in 100 subjects with CAD and 100 controls were quantitatively analyzed using Sequenom MassARRAY. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was used to identify novel pathways. Our analyses indicated that 7 to 8 CpG sites within the 2nd CpG island located upstream of ANRIL, also known as cyclin-dependent kinase inhibitor 2B – antisense 1 (CDKN2B-AS1), are hyper-methylated in CAD subjects compared to controls (p = 0.034). The 40th CpG site within the 2nd CpG island located upstream of CDKN2B-AS1 was methylated to a lesser extent in CAD subjects compared to controls (p = 0.045). Both Pearson and Spearman analyses indicated that methylation levels were significantly associated with total cholesterol (r = 0.204, p = 0.004), fasting high-density lipoprotein cholesterol (r = 0.165, p = 0.020), and fasting low-density lipoprotein cholesterol (r = 0.265, p = 0.000). KEGG pathway analysis revealed a significant enrichment of genes associated with the tumor necrosis factor (TNF) signaling pathway. Among them, CCAAT/enhancer binding protein (C/EBPβ) was identified as a key transcription factor that promotes expression of CDKN2B-AS1 through promotor interaction. DNA methylation of the ANRIL promoter was significantly associated with CAD development in our study. Our analyses suggest that C/EBPβ is a key transcription factor that promotes CDKN2B-AS1 expression by directly interacting with the gene promotor mediated by TNF signaling.
Similar content being viewed by others


Introduction
Cardiovascular disease is the leading cause of mortality worldwide1. Atherosclerosis is a primary risk factor for coronary artery disease (CAD), which is the most common cause of cardiovascular disease. CAD susceptibility is mediated by gene-environment interactions, as well as changes in gene expression via epigenetic regulation2. Therefore, future investigation into epigenetic biomarkers and diagnostic markers for CAD is necessary.
Antisense non-coding RNA in the INK4 locus (ANRIL), also known as cyclin-dependent kinase inhibitor 2B antisense 1 (CDKN2B-AS1), resides on chromosome 9p21, which has been reported to be closely associated with CAD3. As an important epigenetic mechanism, DNA methylation provides a molecular basis for understanding how the environment impacts the genome to modify lifelong CAD risk4. It has been reported that epigenetic changes in p15 (INK4b) methylation and ANRIL expression account for chromosome 9p21 mediated CAD development5. However, the potential role of DNA methylation of ANRIL in CAD development has not been reported. In this case-control study in China, we evaluated the relationship between DNA methylation of ANRIL and CAD.
Subjects and Methods
Study subjects
The study was performed using the case-control principle in accordance with the protocol approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University and the Friendship Hospital of Ili Kazakh Autonomous Prefecture in China. All enrolled subjects provided informed consent prior to the start of the study. This case-control study included 100 subjects with CAD and 100 controls without CAD from the Friendship Hospital of Ili Kazakh Autonomous Prefecture between 2011–2014. The CAD study population included 67 males and 33 females (mean age: 59.93 ± 6.42) and 67 male and 33 female age-matched control subjects (mean age: 58.59 ± 6.18). All subjects underwent coronary angiography to estimate the extent of CAD. All angiograms were evaluated by a cardiologist. Significant CAD was defined as at least one major epicardial vessel with >50% stenosis; control subjects presented with <50% stenosis according to the criteria defined by an ad hoc committee of the American Heart Association6. The severity of coronary atherosclerosis was determined using the Gensini scoring system7.
Subjects with spastic angina pectoris, heart failure, hepatic or renal disease, cardiomyopathy, adrenal dysfunction, and thyroid dysfunction were excluded from the study. Medical history and demographic data of the study population were collected using a questionnaire. Blood pressure was also recorded.
Laboratory measurements
Venous blood (4 mL) was drawn after 12 hours of fasting on the second day of hospitalization for use in biochemical assays. Total cholesterol (TC, mmol/L), triglyceride (TG, mmol/L), fasting blood glucose (FBG, mmol/L), high-density lipoprotein cholesterol (HDL-C, mmol/L), low-density lipoprotein cholesterol (LDL-C, mmol/L), and creatinine (CR, μmol/L) were determined using an automated autoanalyzer (AU 2700 Olympus, 1st Chemical Ltd, Japan).
Cigarette smoking and alcohol use
A standardized questionnaire was used to assess cigarette smoking and alcohol use of the study subjects. Smoking status was classified as either “never smoking” (referred to those who never smoked) or “smoking” (including both former and current smokers). Subjects who reported consuming at least 50 g/week of alcohol were considered current drinkers. Alcohol intake status was classified as either “never drinking” or “drinking” (including both former and current drinkers)8.
DNA extraction and methylation analysis
Genomic DNA was extracted from whole blood samples using the Bioteke Corporation whole blood genomic DNA purification minikit (Beijing, China, #AU18016, lot number B016007017) following the manufacturer’s instructions. Purified DNA was free of protein, nucleases, and other contaminants or inhibitors. DNA purity and concentration were estimated using the NanoDropND-2000 spectrophotometer (Thermo, Wilmington, DE, USA).
The presence of CpG islands spanning the upstream region through the downstream region (−5,000 to 1,000 bp) of the transcription start site (TSS) of the CDKN2B-AS1 gene (chr9: 21989791 ~ 21995790) was analyzed using EMBOSS (European Molecular Biology of open software http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/). We found two CpG islands that overlapped with the CDKN2B-AS1 promoter 6,000 bp upstream in the gDNA CDKN2B-AS1 gene sequence, a CpG island spanning from 4,149 to 5,103 bp (length 955 bp), and another CpG island spanning from 5,244 to 5,764 bp (length 521 bp) (Fig. 1). The predicted sequences of the CpG islands overlapped with the sequences of the CDKN2B-AS1 promoter.

Overlapping CpG islands with the CDKN2B-AS1 promoter identified using EMBOSS software. CpG islands of unusual CG composition, EMBOSS_001 from 1 to 6,000; methylation island prediction rules: Observed/Expected ratio >0.60, Percent C + Percent G > 50.00, Length >200.
Methylation of the TSS of CDKN2B-AS1 was quantitatively analyzed using Sequenom MassARRAY (CapitalBio), which employs matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) and RNA base-specific cleavage. Using this system, the extracted genomic DNA was bisulfate-treated according to manufacturer’s protocol. The concentration of the bisulfate-converted DNA were determined by absorbance at 260 and 280 nm. Then, we used bisulfate-modified DNA for polymerase chain reaction (PCR) amplification. PCR primers were designed using the EpiDesigner tool (http://www.epidesigner.com). Each forward primer was tagged with a 10 mer (5′-aggaagagag-3′) to balance the PCR by adjusting for melting temperature differences, and each reverse primer had a T7-promoter tag (5′-cagtaatacgactcactatagggagaaggct-3′) for in vitro transcription. After PCR reaction, unincorporated deoxyribonucleotide triphosphates (dNTPs) were dephosphorylated by adding shrimp alkaline phosphatase (SAP), and SAP was then inactivated at 65 °C for 10 min9. The PCR reaction products were used as template in the in vitro transcription. After in vitro transcription, ribonuclease A (RNase A) was added to cleave the in vitro transcript. We used MALDI-TOF to analyze the products. The methylation level was expressed as the percentage of methylated cytosines over the total number of methylated and unmethylated cytosines10.
Two CDKN2B-AS1 primer pairs were designed using the EpiDesigner tool from Sequenom: forward: 5′-aggaagagagTTTTTGTTTTTTAGTTGGAAAGGAG-3′ and reverse: 5′-agtaatacgactcactatagggagaaggctATCCTTTATATCTAACCCATTTTTATTT-3′ (product size: 594, number of CpG’s: 43, coverage: 30); forward: 5′-aggaagagagTTTTTGTTTTTTAGTTGGAAAGGAG-3′ and reverse: 5′- cagtaatacgactcactatagggagaaggctATCCTTTATATCTAACCCATTTTTATTT-3′ (product size: 355, number of CpG’s: 28, coverage: 23). Altogether, 38 CpG sites in this region were checked. Spectra methylation ratios were generated using Epityper 1.0 (Sequenom, San Diego, CA).
Functional annotation of CDKN2B-AS1 promoter CpG sites
We used PROMO (version 8.3 of TRANSFAC) to identify putative transcription factor binding sites (TFBS) in the DNA sequences of CDKN2B-AS1’s promoter CpG sites (Figs 2 and 3). TFBS defined in the TRANSFAC database were used to construct specific binding site weight matrices for TFBS prediction11,12. We used Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment analysis to identify pathways that were potentially affected by gene DNA methylation. The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was used to determine ease of score differences between the CAD and control samples13,14.

Prediction of TFBS in DNA sequences within CDKN2B-AS1 promoter CpG sites.

Prediction of TFBS in DNA sequences within CDKN2B-AS1 promoter CpG sites.
Statistical analysis
Data were statistically analyzed using the Statistics Package for Social Sciences (ver. 16.0; SPSS Incorporated, Chicago, IL, USA). Subjects were classified into two groups according to CAD status. Systolic blood pressure (SBP), diastolic blood pressure (DBP), TC, TG, HDL-C, LDL-C, FBG, CR, Gensini scores, and methylation levels of the CDKN2B-AS1 promoter CpG sites (except for the 7 to 8 CpG sites within the 2nd CpG island and the 40th CpG site within the 2nd CpG island) were skewed parameters and were therefore expressed as median and quartile ranges. The Mann-Whitney U test was used to compare data parameters. Normally distributed variables, including age, methylation levels of the CDKN2B-AS1 promoter CpG sites (7 to 8 CpG sites within the 2nd CpG island and the 40th CpG site within the 2nd CpG island) are presented as mean ± standard deviation (SD), and comparisons were analyzed using the independent-samples t test. Categorical variables, including gender, smoking status, and drinking status, were compared between groups using chi-squared analysis. The Spearman two-way test was used to assess the relationship between two quantitative variables, and we evaluated the prediction of methylation of the CDKN2B-AS1 promoter CpG sites and traditional risk factors for CAD using receiver operator curve (ROC) analysis15. Statistical significance was considered if the two-tailed p value was <0.05.
Ethics approval and consent to participate
The study was performed in accordance with the protocol approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University and the Friendship Hospital of Ili Kazakh Autonomous Prefecture in China.
Consent for publication
All authors have read and approved the manuscript.
Results
Characteristics of the study population
Table 1 presents the characteristics of the study population. A total of 100 subjects with CAD and 100 controls were enrolled in this study. As expected, significantly elevated SBP (p = 0.002), DBP (p = 0.002), and FBG levels (p = 0.001) were risk factors for CAD in this population. Additionally, Gensini scores (p = 0.000) were significantly different between CAD and controls.
Methylation of CDKN2B-AS1 in the study population
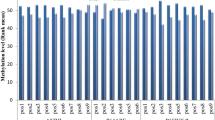
To further explore the possible molecular mechanism of CAD, we assessed DNA methylation of the CDKN2B-AS1 promoter region in CAD and control subjects. We identified 38 CpG sites upstream of the CDKN2B-AS1 gene that were methylated in the CAD and control samples using Sequenom EpiTYPER MassArra. As shown in Table 2 and Figs 4, 7 and 8 CpG sites within the 2nd CpG island located upstream of CDKN2B-AS1 were hyper-methylated in CAD subjects compared to the matched controls (p = 0.034). The 40th CpG site within the 2nd CpG island located upstream of CDKN2B-AS1 was methylated to a lesser, but still significant, extent in CAD subjects compared to the matched controls (p = 0.045).

Evaluation of promoter methylation of ANRIL.

Scatter plots of Spearman correlations between Gensini Scores and ANRIL methylation of 2CpG islands 7 to 8.

Scatter plots of Spearman correlations between Gensini Scores and ANRIL methylation of 2CpG islands 40.

The receiver operating characteristic curve for the predictive efficacy of 2CpG islands7to8 for CAD prevalence.

The receiver operating characteristic curve for the predictive efficacy of 2CpG islands40 for CAD prevalence.
Correlation of Gensini scores with methylation of CDKN2B-AS1
As shown in Table 3, Spearman correlation analysis indicated that Gensini scores were positively associated with methylation levels of 7 to 8 CpG sites within the 2nd CpG island located upstream of CDKN2B-AS1 (r = 0.137, p = 0.053), and that there was a significant negative association between Gensini scores and methylation levels of the 40th CpG site within the 2nd CpG island located upstream of CDKN2B-AS1 in the study population (r = −0.166, p = 0.019). Figures 5 and 6 show scatter plots of Spearman correlations between Gensini Scores and ANRIL methylation.
Predictors of CAD prevalence
To further explore the applicability of methylation levels of CDKN2B-AS1 as a potential diagnostic biomarker of CAD, subsequent ROC analyses were performed on the above data sets (Table 4). The AUC for predicting CAD prevalence was 0.577 for methylation of 7 to 8 CpG sites within the 2nd CpG island located upstream of CDKN2B-AS1 (95% confidence interval (CI): 0.497–0.657, p = 0.062)(Fig. 7), and 0.409 for methylation of the 40th CpG site within the 2nd CpG island located upstream of CDKN2B-AS1 (95% CI: 0.329–0.489, p = 0.028)(Fig. 8).
Pearson and Spearman correlations between CpG island methylation and clinical characteristics of the study population
Table 5 shows the results of both the Pearson and Spearman correlation analyses between CpG island methylation and clinical characteristics of the study population. Both the Pearson and Spearman analyses indicated that methylation of the 7 to 8 CpG sites within the 2nd CpG island was significantly associated with TC (r = 0.204, p = 0.004), HDL-C (r = 0.165, p = 0.020), and LDL-C (r = 0.265, p = 0.000), and methylation of the 40th CpG sites within the 2nd CpG island was significantly associated with age (r = −0.147, p = 0.037) and FBG (r = −0.178, p = 0.012).
KEGG pathway enrichment analyses of the CDKN2B-AS1 promoter region CpG sites
We predicted 23 major putative TFBS in DNA sequences in the CDKN2B-AS1 promoter CpG sites (within the upstream region to the downstream region spanning from −30 to 30 bp of the 7 to 8 CpG sites within the 2nd CpG island and the 40th CpG site within the 2nd CpG island). We then used KEGG pathway enrichment analyses to identify pathways possibly influenced by the genes nearest to the CpG sites in subjects with CAD compared to controls. KEGG pathway analysis revealed a significant enrichment of genes associated with the TNF signaling pathway (Fig. 9)16,17,18. Among them, CCAAT/enhancer binding protein β (C/EBPβ) was identified as a key transcription factor that could increase expression of CDKN2B-AS1 through interaction with its promoters.

Discussion
In this case-control study of a Chinese population, we found novel associations between CAD development and methylation levels of the CDKN2B-AS1 (ANRIL) promoter region CpG sites, which were significantly correlated with blood glucose and cholesterol levels, respectively. KEGG pathway enrichment analyses suggested that the TNF signaling pathway is involved in C/EBPβ-mediated increases in CDKN2B-AS1 expression. To the best of our knowledge, this is the first study focused on the relationship between DNA methylation of ANRIL and CAD.
Genome-wide association studies (GWAS) have found that multiple single nucleotide polymorphisms (SNPs) on chromosome 9p21 are highly associated with CAD19,20,21,22,23,24,25,26. However, the mechanism underlying this association remains elusive. One study suggested that epigenetic changes in p15 (INK4b) methylation and ANRIL expression are associated with chromosome 9p21 related risk of CAD development5. Recently, a few studies have explored the association between ANRIL promoter methylation and CAD risk factors. These studies demonstrated that epigenetic regulation of ANRIL promoter methylation is associated with childhood bone development27, adiposity28, and increased arterial pulse wave velocity (PWV, a measure of arterial stiffness)29. Therefore, it is necessary to explore the relationship between ANRIL promoter DNA methylation and CAD development.
In the present study, we found novel associations between CAD and methylation levels of the CDKN2B-AS1 promoter region CpG sites. Furthermore, we identified significant correlations between blood glucose and cholesterol levels with ANRIL promoter methylation. These findings may partially underlie the risk associated with chromosome 9p21 and CAD. However, the exact mechanism underlying ANRIL promoter DNA methylation and CAD remains unknown. Our KEGG pathway analysis demonstrated a significant enrichment of genes associated with the TNF signaling pathway, and C/EBPβ was identified as a key transcription factor that interacts with the ANRIL promotor. A previous study used loss-of-function and chromatin immunoprecipitation approaches to show that TNF-α induced ANRIL expression30. C/EBPβ is a transcription factor that belongs to a class of the basic-leucine zipper proteins that has phylogenetic, structural, and functional features. A related study confirmed that C/EBPβ regulates pro-inflammatory responses31. Therefore, C/EBPβ may be a potential target for treating CAD. It is important to note, however, that the results from our KEGG pathway analysis should be further confirmed in functional and mechanistic studies.
Our study has limitations that should be considered when interpreting the results. First, our study was a case-control, cross-sectional design with a small sample size. Moreover, our study only provides evidence for the potential association between ANRIL promoter DNA methylation and CAD; whether changes in DNA methylation are causative of CAD pathogenesis requires further evaluation. Finally, the mechanism underlying ANRIL promoter DNA methylation and CAD was predicted by KEGG pathway analysis rather than by experimental confirmation. Therefore, additional case studies with larger sample sizes are needed to confirm our results.
Conclusion
In summary, DNA methylation of the ANRIL promoter was significantly associated with CAD development in this Chinese study population. KEGG pathway enrichment analyses indicated C/EBPβ as a key transcription factor that promotes CDKN2B-AS1 expression mediated by TNF signaling.
Data availability
All data and materials have been made available.
References
Jankovic, N. et al. WHO guidelines for a healthy diet and mortality from cardiovascular disease in European and American elderly: the CHANCES project. Am J Clin Nutr 102(4), 745–56 (2015).
Yu, C. H., Pal, L. R. & Moult, J. Consensus Genome-Wide Expression Quantitative Trait Loci and Their Relationship with Human Complex Trait Disease. OMICS 20(7), 400–14 (2016).
Chi, J. S. et al. Long non-coding RNA ANRIL in gene regulation and its duality in atherosclerosis. J Huazhong Univ Sci Technolog Med Sci 37(6), 816–82 (2017).
Turgeon, P. J., Sukumar, A. N. & Marsden, P. A. Epigenetics of Cardiovascular Disease - A New “Beat” in. Coronary Artery Disease. Med Epigenet 2(1), 37–52 (2014).
Zhuang, J. et al. Methylation of p15INK4b and expression of ANRIL on chromosome 9p21 are associated with coronary artery disease. PLoS One 7(10), e47193 (2012).
Austen, W. G. et al. A reporting system on patients evaluated for coronary artery disease: report of the Ad Hoc Committee for Grading of Coronary Artery Disease, Council on Cardiovascular Surgery, American Heart Association. Circulation 5, 5–40 (1975).
Gensini, G. G. A more meaningful scoring system for determinating the severity of coronary heart disease. Am J Cardiol 51, 606 (1983).
Ge, P. C. et al. Synergistic Effect of Lipoprotein-Associated Phospholipase A2 with Classical Risk Factors on Coronary Heart Disease: A Multi-Ethnic Study in China. Cell Physiol Biochem 40(5), 953–968 (2017).
Li, Y. et al. Changes in DNA methylation of erythroid-specific genes in K562 cells exposed to phenol and hydroquinone. Toxicology 4(312), 108–14 (2013).
Cui, X. B. et al. Inactivation of miR-34a by aberrant CpG methylation in Kazakh patients with esophageal carcinoma. J Exp Clin Cancer Res 33(1), 20 (2014).
Xavier Messeguer et al. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18(2), 333–334 (2002).
Domènec Farré et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31(13), 3651–3653 (2003).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 4(1), 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1), 1–13 (2009).
Hanley, J. A. & McNeil, B. J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28, 27–30 (2000).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res 47(D1), D590–D595 (2019).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci (2019).
Chanock, S. J. et al. Replicating genotype-phenotype associations. Nature 447, 655–660 (2007).
McPherson, R. et al. A common allele on chromosome 9 associated with coronary heart disease. Science 316, 1488–1491 (2007).
Helgadottir, A. et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316, 1491–1493 (2007).
Welcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Preuss, M. et al. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet 3, 475–483 (2010).
Peng, W. H. et al. Chromosome 9p21 polymorphism is associated with myocardial infarction but not with clinical outcome in Han Chinese. Clin Chem Lab Med 47, 917–922 (2009).
Silander, K. et al. Worldwide patterns of haplotype diversity at 9p21.3, a locus associated with type 2 diabetes and coronary heart disease. Genome Med 1, 51 (2009).
Zhou, L. et al. Associations between single nucleotide polymorphisms on chromosome 9p21 and risk of coronary heart disease in Chinese Han population. Arterioscler Thromb Vasc Biol 28, 2085–2089 (2008).
Curtis, E. M. et al. Perinatal DNA Methylation at CDKN2A Is Associated With Offspring Bone Mass: Findings From the Southampton Women’s Survey. Bone Miner Res. 32(10), 2030–2040 (2017).
Lillycrop, K. et al. ANRIL Promoter DNA Methylation: A Perinatal Marker for Later Adiposity. EBioMedicine 19, 60–72 (2017).
Murray, R. et al. DNA methylation at birth within the promoter of ANRIL predicts markers of cardiovascular risk at 9 years. Clin Epigenetics 8, 90 (2016).
Zhou, X. 1 et al. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-κB pathway. RNA Biol 13(1), 98–108 (2016).
Pulido-Salgado, M., Vidal-Taboada, J. M. 1 & Saura, J. C/EBPβ and C/EBPδ transcription factors: Basic biology and roles in the CNS. Prog Neurobiol 132, 1–33 (2015).
Acknowledgements
We would like to thank Dr. En-Zhi Jia who is an Assistant Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine. Supported by the National Natural Science Foundation of China (Grants Nos 30400173, 30971257, and 81170180) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. This study received support from the National Natural Science Foundations of China (Nos 81170180, 30400173 and 30971257) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
As the guarantor, C.H.Z., J.Z. and E.Z.J. conceived the study. C.H.Z. and H.T.C. initially drafted the manuscript. Z.H.C. and L.H.L. enrolled participants and collected data under the supervision of F.H.A. Q.W.J., L.S.W., W.Z.M. and Z.J.Y. coordinated the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, CH., Cao, HT., Zhang, J. et al. DNA methylation of antisense noncoding RNA in the INK locus (ANRIL) is associated with coronary artery disease in a Chinese population. Sci Rep 9, 15340 (2019). https://doi.org/10.1038/s41598-019-51921-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51921-3
- Springer Nature Limited
This article is cited by
-
miR194 hypomethylation regulates coronary artery disease pathogenesis
BMC Medical Genomics (2022)
-
Genetic and epigenetic associations of ANRIL with coronary artery disease and risk factors
BMC Medical Genomics (2021)