
Abstract
Bats are reservoirs of numerous zoonotic viruses. The Picornaviridae family comprises important pathogens which may infect both humans and animals. In this study, a bat-related picornavirus was detected from Algerian Minioptreus schreibersii bats for the first time in the country. Molecular analyses revealed the new virus originates to the Mischivirus genus. In the operational use of the acquired sequence and all available data regarding bat picornaviruses, we performed a co-evolutionary analysis of mischiviruses and their hosts, to authentically reveal evolutionary patterns within this genus. Based on this analysis, we enlarged the dataset, and examined the co-evolutionary history of all bat-related picornaviruses including their hosts, to effectively compile all possible species jumping events during their evolution. Furthermore, we explored the phylogeny association with geographical location, host-genus and host-species in both data sets.
Similar content being viewed by others


Introduction
In the last several decades, bat-related virological studies revealed an increase in the major virus groups highlighting outstanding diversity and prevalence among bats (e.g., Astroviridae, Coronaviridae and Picornaviridae)1,2,3. Although several novel viruses were discovered in these animals worldwide, fewer studies examined the evolutionary patterns regarding these pathogens. Among bat-harbored viruses, members of the Picornaviridae family remains neglected with limited available sequence data4.
The virus family consists of nearly 80 species grouped into 35 genera, and includes several well-known human and animal pathogens, causing various symptoms ranging from mild febrile illness to severe diseases of heart, liver or even the central nervous system5. The family members are small, spherical, non-enveloped viruses, with icosahedral symmetry. The viral genome is a monopartite, linear, polyadenylated positive ssRNA of 7.1–8.9 kb in length, including a single ORF encoding a large polyprotein6,7. To date, bat picornaviruses (BtPVs) discovered are associated to the Mischivirus, Hepatovirus, Crohivirus, Kunsagivirus, Kobuvirus and Shanbavirus genus or remain unassigned8,9. To the best of our knowledge, M. schreibersii bats are the primary hosts regarding mischiviruses, which are classified in three distinct species, namely Mischivirus A10, Mischivirus B11, and Mischivirus C. However, new putative mischivirus sequences were described from both Romanian Myotis myotis and Myotis oxygnathus. Additionally, other detected BtPVs clustered together with canine and feline picornaviruses12 suggests host-jumping events during their evolution.
By understanding virus-host co-evolution history and patterns, disease prediction efforts become more reliable13. Coevolutionary studies of major RNA virus groups were performed on coronaviruses and flaviviruses14,15. However, BtPVs is a rapidly growing group comprising 8 genera in 200816, and 35 genera in 20179, yet coevolution related studies are still missing.
Bats and viral zoonoses are both neglected and not well researched in Algeria. Therefore, the aim of the current study was drawing the different possible coevolutionary scenarios and assess the degree of association between phenotypic traits and phylogeny, in order to understand more about the virus-host co-evolution within the bat Picornavirus family.
Materials and Methods
Sample collection and laboratory procedures
Guano samples were collected in the Jiri Gaisler cave, both the Aoukas and the Melbou caves located in the city of Bejaia, Algeria during 2016 and 2017. Thirty-five fresh bat guano samples were collected from the terrain directly under roosting bats then stored in 2 ml cryo-tubes containing 1 ml of 1x PBS, using sterile dissecting forceps. Samples were next transported in liquid nitrogen and stored at −80 °C until laboratory processes. Nucleic extractions were performed using the Gene JET Viral DNA/RNA Purification Kit (Thermo Scientific), in full accordance the manufacturer’s recommended protocol. Library preparation and sequencing regarding Ion Torrent viral metagenomic analysis were conducted as previously published, including bioinformatics processes, and de novo assembly of sequence readouts17. Genome end sequences were amplified with 5′/3′ RACE protocol as described elsewhere11, bat DNA barcoding was performed in accordance to Walker and colleagues18.
Sequence data selection
All known bat picornavirus sequences, representing either complete or partial coding sequences were retrieved from GenBank, including the novel mischivirus sequence presented in this study. A total of 70 sequences were analysed (3 kobuvirus, 9 mischivirus, 1 crohivirus, 1 kunsagivirus, 1 sapelovirus, 4 hepatovirus, 1 shanbavirus, 50 unassigned viruses), plus 1 amphibian ampivirus which was represented as an outgroup. The sampling location, collection date, and host genus were listed as indicated in GenBank sequence annotation, and/or the literature19 (Supplementary Table S1). The BtPVs sequences were sampled from 26 bat species belonging to 9 genera (Table 1). Sequences provenance hail nearly entirely from Europe n = 35, Asia n = 25, Africa n = 9 and America n = 1. Sequences with unknown hosts were discarded. In order to represent mammalian host evolution, we downloaded both complete and partial mitochondrial cytochrome b gene (CYTB)20. In reference to the M. schreibersii bat, we obtained additional CYTB sequences from the Hungarian Natural History Museum in Budapest.
Sequence data editing and phylogenetic analysis
Both BtPVs and mischiviruses, and their respective hosts’ sequences were aligned using the MAFFT alignment tool21. Sequence length adjustment was acquired in the use of GeneDoc22. The size ranged between 1419–1838 bp regarding the P1 region, 1724–2082 bp representative of the P2 region and 343–1404 bp in reference to the RdRp. Host sequences were not modified.
Prior to applying the datasets for phylogenetic reconstruction, we implemented the finest substitution model selection using Mega v623. The GTR + G substitution model was applied for phylogenetic construction based on 3Dpol gene from Mischiviruses and their hosts. Additionally, the same substitution model was used to create a tree from P1 region of all BtPVs. Likewise, the GTR + G + I substation model was used to implement phylogeny based on P2 region and 3Dpol gene of all BtPVs and their hosts. An amphibian picornavirus Ampivirus A sequence was used to root the viral phylogenies9, while Furipterus horrens CYTB sequence was used as a representative of the outgroup in the host tree. Non-clock Bayesian phylogenetic trees were constructed using MrBayes v3.2.4 software24. Each analysis operated for 10 million generations (25% were discarded as burn-in) and sampled every 1000 generations, and the resultant trees were then edited using iTOL25.
Pairwise genetic distances were calculated between RdRp nucleotide and amino acid sequences, using the MegAlign pro program (DNASTAR v15.2.0) with uncorrected pairwise distances as a metric, and P distance in MEGA v623.
To assess the temporal signal in the viral data above, a regression method of root-to-tip distances against dates of sampling was implemented regarding the RdRp Bayesian trees, of which, TempEst was used26.
Phylogeny-trait association analysis
Phylogeny-trait statistics were performed, using the association index (AI), parsimony score (PS), and maximum monophyletic clade (MC) index statistics available in the BaTS package27. Mischiviruses and all bat picornaviruses (based on the 3Dpol gene) were inspected using BaTS software. Both of these exhibited significant bunching by the following character states of interest: bat host genus, species, or sampling location. The values obtained were interpreted according to Parker and colleagues27. This analysis compared the posterior distribution of trees regarding our data formerly mentioned, to a null distribution of 1000 trait-randomized trees. The results were interpreted in accordance with Parker and colleagues27. Prior, the trace files generated by MrBayes were analyzed in Tracer v1.626, with the aim of discarding the burn-in trees.
Co-evolution analysis
In order to estimate the virus-host co-divergence scope, we simultaneously analysed picornaviruses (RdRp) and their hosts’ phylogenies along with mischiviruses and their hosts’ phylogenies, all in the operational use of Jane v4.028. It deduces the nature and the frequency of different evolutionary events, by determining the congruence with the least costly reconstructions of the host-parasite connection, using the tree topologies. Thus, the parameters for the entire event costs (co-speciation, duplication, duplication and host switch, loss and failure to diverge) were set to 0, then 1, and after co-speciation, equal to 0 and other events equal to 1 with a population size equal to 100 and 100 generations for both datasets mentioned above.
Tanglegrams for all bat PVs and their hosts, in addition to mishiviruses and their hosts were created using Dendroscope v3-9-529.
Recombination analysis
To effectively detect recombination, phylogenetic trees generated from various regions of BtPVs genomes (P1, P2 and 3Dpol) were examined regarding tree structure incongruities. Subsequently, all BtPVs aligned nucleotide sequences were imported in the Recombination Detection Program (RDP 4). Recombination events, parental and recombinant sequences as well as putative breakpoints, all underwent analysis using, GENECONV, BOOTSCAN, GENCONV, SISCAN and MAXCHI methods aligned to default settings30, and RDP with internal references only as a parameter.
Statistical analysis
The strength of the correlation among picornavirus diversity (the number of detected PV clusters) and the number of PV species for each host genus was estimated using the Spearman coefficient (r). The value interpretation was as follows: 0.00–0.39 “weak” correlation, 0.40–0.59 “moderate”, 0.60–0.79 “strong” and 0.80–1.0 “robust”.
Results
Detection and genome organization of the novel mischivirus from bats
Out of the 35 sequenced metagenomic libraries regarding viral discovery, picornavirus (PV) was found in one library with 1179 reads. Bat DNA barcoding revealed the virus was detected from a M. schreibersii bat. Based on genome sequence identity level, the novel sequence (MG888045) described in this study is grouped within the Mischivirus genus. Nearly the entire genome (6961 nt) was obtained (some 1400 bp are missing, 5′ UTR and the beginning of the L protein). This demonstrates the typical PV characteristic genome organization of UTR [L-P1(VP0, VP3, VP1)-P2(2 A, 2B, 2Chel)-P3(3 A, 3BVPg, 3Cpro, 3Dpol)] UTR-poly(A); and the conservative motifs were very similar to the Hungarian Mischivirus B described by Kemenesi and colleagues11. Genome organization pattern, hypothetical cleavage sites and conserved motifs according to the first start codon in the obtained sequence are indicated in Fig. 1.

Schematic representation of the novel Algerian BatPV genome organization. 3′ UTR, P1, P2 and P3 regions are included. Also, the putative cleavage sites and the conserved motifs are depicted.
Phylogeny and PVs bunching by host and sampling location
According to the Blast results, the novel Algerian sequence shared 85% of nucleotide identity with the Hungarian virus and 73% identity with the Chinese strain. Moreover, it shared between 91–94% identity with shorter sequences available from the Bulgarian tentative mischiviruses.
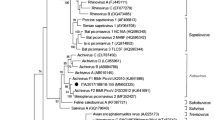
The phylogenetic analysis predicated on RNA-dependent RNA polymerase gene (RdRp) of mischivirus sequences (Fig. 2) revealed how the novel Algerian BtPV formed a monophyletic clade together with the Hungarian Mischivirus B sequences11; in addition, the Bulgarian and Romanian tentative Mischivirus B12, including the Chinese Mischivirus A10. The phylogenetic relationship is supported with high posterior probability values (>90%).

Bayesian interference phylogenetic tree of mischiviruses. The tree exhibits the relationship among the new Algerian mischivirus and other described mischiviruses. The analysis was performed using MrBayes. 3.2.4. ten million generations were performed. Posterior probabilities are indicated at nodes. Branch symbols indicate Mischivirus species. Yellow color: Mischivirus C species, green: Mischivirus A species, red: Mischivirus B species. Solid circles indicate ICTV classified viruses, empty circles indicate the unclassified viruses, and the new Algerian Mischivirus is represented in the use of a star.
An extended phylogenetic analysis to all bat picornaviruses (Fig. 3, Supplementary Fig. S1) exhibited a grouping, related in some areas of the tree with both host genus and large-scale sampling location (continent), while in other areas it is interspersed. Furthermore, bat mischiviruses clustered according to host genus, and sampling location for each virus species (Fig. 2).

A phylogenetic overview of PVs sequences analyzed. A Bayesian analysis of 70 RdRp sequences, rooted using ampivirus A sequence (NC027214). Branch lengths represent the number of substitutions per site. Genus-specific clusters are colored, based on bat genus. Solid circles represent Large-scale sampling locations, red for Europe, purple for Asia, yellow for Africa, and chartreuse for America. The bar encircling the tree represents the RdRp length range, sequences <500 bp are colored in light grey, sequences between 700 bp and 900 bp in dark grey, and black for sequences >1,000 bp. ICTV virus classification is indicated, if and when available.
Regression analyses (Supplementary Fig. S2) exhibited no association between sampling times and root-to-tip genetic distances, neither for bat mischivirus dataset R2 = 0.0829 (Supplementary Fig. 2a) nor for all BtPVs R2 = 0.0218 (Supplementary Fig. 2b), and due to this, any molecular clock dating was excluded.
Phylogeny-trait association analysis tests (AI = 0.028, PS = 2) offered statistical support (p < 0.05) regarding the clustering of bat mischivirus (n = 16) when considering host genus. The null hypothesis of no association between phylogeny and host species character trait was accepted based on the association index test (AI = 0.38, p = 0.065), while it was rejected based on the parsimony score test (PS = 3, p = 0). The MC statistic supported the association for both Miniopterus (MC = 12, p = 0.001) and Hipposideros (MC = 3, p = 0.001) genera, in addition to the M. schreibersii species (MC = 12, p = 0.001). Regarding the geographical macro area of sampling, the association index (AI = 0.029, p = 0.003) suggested a strong phylogeny-trait association, once the parsimony score (PS = 3, p = 0.23) exhibited no significant relationship. Furthermore, the MC permitted an inspection in each geographic region alone and provided connecting proof for character-trait Europe (MC = 10, p = 0.009). Note how individual traits (single countries, species) consistently provided non-significant results (see Supplementary Table S2).
Furthermore, the same analyses were performed regarding all BtPVs (n = 69), AI and PS provided support (p < 0.05) for associations, when taking into account host genus (AI = 2.25, PS = 21), host species (AI = 3.47, PS = 32), and large-scale sampling area (AI = 0.60, PS = 15). Additionally, the MC statistics representative of each geographical location, host genus, and host species revealed the continents Europe (MC = 10), Africa (MC = 4), and Asia (MC = 5), in addition to the host genera Miniopterus (MC = 12), Myotis (MC = 4), and Rhinolophus (MC = 9), also the species M. schreibersii, M. myotis, R. euryale, M. magnate, R. sinicus, M. fuliginosus were not randomly distributed on the tips of the matching phylogenetic tree (p < 0.05) (Supplementary Table S3). Based on the phylogenetic analyses (Fig. 3) executed using the 69 RdRp gene sequences, 28 different bat genus-specific clusters were identified: three in Kubovirus, three in Mischivirus, one in Crohivirus, one in Shanbavirus, one in Kunsagivirus, one in Sapelovirus, four in Hepatovirus, and fourteen clusters in different unassigned viruses. Likewise, we tallied six PV phylogenetic clusters for Miniopterus genus, five for Myotis, five for Rhinolophus, five for Eidolon, two for Hipposideros, two for Nyctalus, one for Coleura, and one for Ia. Since these BtPVs are related to different host species and sampled from varying locations, the number of RdRp sequences and/or host species within each cluster are different. Strikingly, several of the clusters are composed of a single BtPV RdRp sequence. A very strong correlation was observed among the number of BtPV specific clusters and both its species richness (r = 0.94; p = 0.0001), or geographical sampling area (r = 0.84; p = 0.002). The accurate classification of BtPVs is related to the length of the viral sequences, nonetheless, even short RdRp sequences permitted us to acquire a primary classification of the unassigned sequences.
BtPV sequences are very diverse and pairwise distances calculation resulted in relevant data. The mean nucleotide divergence between sequences from cluster C5 (Myotis) and C4 (Hipposideros) suggest they are closely related, thus C5 (unassigned) may belong to the genus Mischivirus (Table 2 and Fig. 3). The same bat genus can host different virus species which explain the percentage of differences observed among sequences belonging to the same host genus. Mean nucleotide and amino-acid divergence within the same genus were 36% and 54.8% for Miniopterus; 45.6% and 60.3% for Myotis; 40% and 45.7% for Rhinolophus; 55.3% and 78.5% for Hipposideros; 44% and 52.3% for Nyctalus; 58.7% and 75.5% for Eidolon.
Coevolutionary analyses
In a dataset comprising ICTV classified mischiviruses and our sequence, topological similarities were observed mischiviruses and their hosts’ phylogenies (Fig. 4) suggesting first, a co-divergence evolutionary scenario, which interestingly, was not supported by reconciliation analysis (using Jane), when considering the least costly event. When taking into account both all putative and classified mischiviruses, incongruence topology was observed, in addition to reconciliation analysis which revealed how host-jumping events were involved in the evolution of mischiviruses (Fig. 5).

Tanglegram and Jane results of ICTV classified mischiviruses plus the novel Algerian sequence and their hosts. The least costly result is the best evolutionary scenario.

Tanglegram and Jane results of all mischiviruses and their hosts.
When extending the analysis to all BtPVs and their hosts’ phylogenies, the history of their evolution was explained by more host-jumping events than co-speciation events, generating incongruent tree topologies (Supplementary Fig. S3). Jane analysis displayed more duplication and host switch events than co-divergence events, independently of co-divergence costs (Table 3). The least costly events represented the best evolutionary scenario, Jane results helped us to determine hypothetic donors and receptors in cross-species transmission events.
Recombination analysis
The phylogenetic trees built from P1, P2 and 3Dpol regions of BtPVs genomes displayed discordances in their structures indicating potential recombination events (Fig. 6a–c). Out of the 69 recombination events detected with the RDP 4 program, only 11 unique putative recombination events identified with two or more methods were retained (Table 4).

(a) Bayesian phylogenetic reconstruction of P1 region for all BtPVs genomes included in this study. (b) Bayesian phylogenetic reconstruction of P2 region for all BtPVs genomes included in this study. (c) Bayesian phylogenetic reconstruction of RdRp region for all BtPVs genomes included in this study. Ampivirus A sequence (NC027214) used as an outgroup for the three viral phylogenetic trees. Genus-specific clusters are colored, based on bat genus. Recombination may be reflected in tree structure incongruities between phylogenetic trees (a–c).
Discussion
Numerous factors (such as geographical, demographical or ecological) likely influence the occurrence of spill-over events and act as driver factors in viral evolution31,32. Destruction of the natural habitat of bats worldwide facilitated the urbanization of several dedicated species or simply established more opportunities for human-bat contact events33. This constitutes novel possible factors regarding viral emergence as already seen with coronaviruses34 and rabies35. Understanding the evolutionary mechanisms of BtPVs is a prominent direction of research.
In addition, the zoonotic potential of all documented BtPVs is clearly unidentified yet and fairly misunderstood36. Although animal originated PVs were previously exemplified as potential zoonotic agents, as in the case of the encephalomyocarditis virus, which was clearly revealed through experimental infections on human tissues and primary cell cultures37.
Understanding viruses and their hosts’ coevolution is crucial to show up the evolutionary character and understand potential disease emergence factors38,39. Thus far, several phylogenetic and systematic evolutionary studies were performed with regards to the Picornaviridae family, as the virus members of this family are increasingly discovered40,41. However, little is still relatively unknown in reference to the virus-host coevolution patterns for this group. In this study, we describe the first picornavirus from Algerian bats, and we include this novel sequence in detailed coevolutionary analysis, focusing on BtPVs.
Picornaviruses exhibit a high genetic diversity (quasi-species) similar to other RNA viruses42,43, and a broad geographical spectrum in Chiroptera order. Moreover, a positive relationship was previously described between the viral richness and geographical distribution of bats44,45. Similarly, in the present study, a positive correlation between PV diversity, species richness, and geographical distribution was revealed. Furthermore, more than one virus cluster and larger viral clusters were obtained for genera in which more species have been sampled in a large geographical range.
BtPVs originating from the same host genus were very diverse and not closely related, hence, both BtPVs and bat mischiviruses did not demonstrate any specificity, whether on the species level nor on the genus level. Likewise, for tortoise picornavirus40 and contrariwise for coronaviruses46.
Several BtPVs clusters possess a single viral sequence whereas several sequences were considerably short. For instance, mischiviruses displayed three host genus related clusters. Miniopterus genus virus related cluster comprising the Asian mischivirus sequence (8468 bp), the new Algerian sequence (6,961 pb), the six Hungarian sequences (6,855 bp), and four partial 3Dpol Bulgarian sequences (three sequences 343 bp, one 983 bp). Myotis genus cluster composed of three Romanian partial 3D pol sequences (993 bp), while the Hipposideros genus cluster consisting of a single Mischivirus C sequence from the Congo (8096 bp). Both the number and the length of sequences often limit the analyses. By way of illustration through the use of BaTS software if and when the number of sequences regarding a character trait is lower than three, the result is declared insignificant (case of America). Therefore, short sequences may likely be misclassified.
According to Lewis-Rogers and Crandall43, PVs and their hosts evolve through host-jumping events and not via co-speciation. Chiropteran order was not included in the previous study. The main finding of the present study was the frequency regarding host-jumping events occurring in the evolutionary history of BtPVs, and reflected as incongruence between the virus and the host phylogeny. Furthermore, we could observe nearly all bat genera host phylogenetically divergent viruses and an absence of species specificity. According to studies performed on coronaviruses14 it may likely be due to multiple introductions of PVs, this supposition is coherent with the detection of highly related PVs in humans and different animal species, such as the aichivirus 1 in humans47 canine kobuvirus 148,49, murine kobuvirus 150, feline kobuvirus51,52, all belonging to the virus species Aichivirus A in the genus Kobuvirus. Moreover, the Mischivirus genus was supposedly restricted to the M. schreibersii bat, however, later it was associated to the H. gigas, M. oxygnathus, M. myotis and surprisingly, to the foxhound53.
Cross-species transmission event occurrence is multifactorial, nevertheless, we concluded in some cases of BtPVs, sympatry may increase host-jumping events. Co-roosting of Myotis and Miniopterus bats may likely explain the detection of closely related kobuvirus associated among these bats14. The migratory ability of Miniopterus bats with the longest distance of 883 km recorded in Europe, which possibly eases the spread of viruses among bat populations spread out in geographically distant areas. For example, M. schreibersii, originating from Europe and Algeria, which possess different geographical distribution and share the same virus species Mischivirus B54.
Despite the fact in which no interaction is known regarding M. oxygnathus and H. gigas in accordance with their ecologies, hypothetical cross-species transmission events were detected among these two species. The length of the viral sequence obtained from M. oxygnathus (993 bp) is a limitation regarding accurate classification. Based on the phylogenetic analyses (Fig. 2), the putative mischivirus sequence obtained from M. oxygnathus and M. myotis were closely related to Mischivirus C, identified from H. gigas. Although, they revealed an identical branching pattern upon the structure of the phylogenetic tree as Mischivirus A from M. schreibersii sampled in China and Mischvirus B from Algeria and both Bulgaria and Hungary, indicating BtPVs from Romania may not belong to Mischivirus C. Furthermore, it has been demonstrated in which M. schreibersii bats from Hainan were actually M. fuliginosus10,55. This is due to the cryptic nature of several Miniopterus bat species. Distinctly, the identification based on their morphology is not sufficient, and typically requires the use of DNA barcoding or echolocation studies56.
Our results emphasized the evolution among PVs within bats horizontally, through host jumping mechanism, rather than co-speciation. Additionally, host specificity was not observed, suggesting it is not involved in the evolutionary history regarding BtPVs. Moreover, we found the occurrence of cross-species transmission events may likely increase with sympatry. Notwithstanding the exploitation of all available information in reference to BtPVs, admittedly, this study bears limitations, primarily related either to the quality of the available data, or the lack thereof. To cite an example, the precise classification of the Romanian putative mischiviruses was not possible due to the short length of the viral sequences. Additionally, we could not use several BtPVs sequences originating from America since the hosts’ species were unknown. Undeniably, the impact of the misclassification of several host species should not be entirely ignored.
Recombination occurrence was highlighted in this research and believed to play a part in both the diversity and the evolution regarding BtPVs, as aptly demonstrated in previous studies57,58. The phylogenetic resolution was affected by a lack of data reflected in polytomies observed in the virus phylogeny. Moreover, we noticed how the BtPVs data panel is small (n = 69) and unbalanced, since most studies were undertaken primarily throughout Europe and Asia, while other continents are characteristically, under-represented (America n = 1, Australia = 0). Furthermore, the sampled bats included in this study represent just 4.8% of the total currently recognized bat genera, and 2.2% of the total bat species known so far, leaving the greater majority yet unexplored.
Frankly speaking, our study is a starting point regarding further investigations in pursuit of the evolution of the PV family within these important flying mammals. Specifically, for this very purpose, we support additional sampling, detection and the acquisition of more BtPVs with longer sequences, including accurate host species identification.
References
Fischer, K., Pinho dos Reis, V. & Balkema-Buschmann, A. Bat Astroviruses: Towards Understanding the Transmission Dynamics of a Neglected Virus Family. Viruses 9, 34 (2017).
Lau, S. K. P. et al. Complete Genome Analysis of Three Novel Picornaviruses from Diverse Bat Species. Journal of Virology 85, 8819–8828 (2011).
Memish, Z. A. et al. Middle East Respiratory Syndrome Coronavirus in Bats, Saudi Arabia. Emerging Infectious Diseases 19 (2013).
Hayman, D. T. S. Bats as Viral Reservoirs. Annual Review of Virology 3, 77–99 (2016).
Yu, J. et al. Identification of a Novel Picornavirus in Healthy Piglets and Seroepidemiological Evidence of Its Presence in Humans. PLoS ONE 8, e70137 (2013).
Hulo, C. et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Research 39, D576–D582 (2011).
Jiang, P., Liu, Y., Ma, H.-C., Paul, A. V. & Wimmer, E. Picornavirus Morphogenesis. Microbiology and Molecular Biology Reviews 78, 418–437 (2014).
King, A. M. Q. et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Archives of Virology 163, 2601–2631 (2018).
Zell, R. et al. ICTV virus taxonomy profile: Picornaviridae. Journal of General Virology 98, 2421–2422 (2017).
Wu, Z. et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces. Journal of Virology 86, 10999–11012 (2012).
Kemenesi, G. et al. Genetic characterization of a novel picornavirus detected in Miniopterus schreibersii bats. Journal of General Virology 96, 815–821 (2015).
Lukashev, A. N. et al. Close genetic relatedness of picornaviruses from European and Asian bats. Journal of General Virology 98, 955–961 (2017).
Olival, K. J. et al. Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650 (2017).
Leopardi, S. et al. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infection, Genetics and Evolution 58, 279–289 (2018).
Lobo, F. P. et al. Virus-Host Coevolution: Common Patterns of Nucleotide Motif Usage in Flaviviridae and Their Hosts. PLoS ONE 4, e6282 (2009).
Knowles, N. J. et al. Taxonomy of Picornaviridae: Current Situation and Future Proposals. Group 2008–2008, https://doi.org/10.1371/journal.pone.0001847.Oberste (2008).
Bányai, K. et al. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infection, Genetics and Evolution 48, 19–26 (2017).
Walker, F. M., Williamson, C. H. D., Sanchez, D. E., Sobek, C. J. & Chambers, C. L. Species From Feces: Order-Wide Identification of Chiroptera From Guano and Other Non-Invasive Genetic Samples. PLOS ONE 11, e0162342 (2016).
Wu, Z. et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. The ISME Journal 10, 609–620 (2016).
Agnarsson, I., Zambrana-Torrelio, C. M., Flores-Saldana, N. P. & May-Collado, L. J. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Currents 3, RRN1212 (2011).
Katoh, K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30, 3059–3066 (2002).
Nicholas, K. B. et al. GeneDoc: a tool for editing and annotating multiple sequence alignments. (1997).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution 30, 2725–2729 (2013).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 61, 539–542 (2012).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research 44, W242–W245 (2016).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evolution 2, vew007 (2016).
Parker, J., Rambaut, A. & Pybus, O. G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infection, Genetics and Evolution 8, 239–246 (2008).
Conow, C., Fielder, D., Ovadia, Y. & Libeskind-Hadas, R. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms for Molecular Biology 5, 16 (2010).
Huson, D. H. & Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Systematic Biology 61, 1061–1067 (2012).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution 1 (2015).
Cortes, M. C. et al. Characterization of the Spatial and Temporal Distribution of Nipah Virus Spillover Events in Bangladesh, 2007–2013. The Journal of Infectious Diseases 217, 1390–1394 (2018).
Wang, L.-F. & Crameri, G. Emerging zoonotic viral diseases: -EN–FR- Les maladies zoonotiques virales émergentes -ES- Enfermedades zoonóticas emergentes de origen vírico. Revue Scientifique et Technique de l’OIE 33, 569–581 (2014).
Russo, D. & Ancillotto, L. Sensitivity of bats to urbanization: a review. Mammalian Biology 80, 205–212 (2015).
Hu, B., Ge, X., Wang, L.-F. & Shi, Z. Bat origin of human coronaviruses. Virology Journal 12 (2015).
Pape, W. J., Fitzsimmons, T. D. & Hoffman, R. E. Risk for Rabies Transmission from Encounters with Bats, Colorado, 1977–1996. Emerging Infectious Diseases 5, 433–437 (1999).
Geldenhuys, M. et al. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 13 (2018).
Carocci, M. & Bakkali-Kassimi, L. The encephalomyocarditis virus. Virulence 3, 351–367 (2012).
Lukashov, V. V. & Goudsmit, J. Evolutionary Relationships among Parvoviruses: Virus-Host Coevolution among Autonomous Primate Parvoviruses and Links between Adeno-Associated and Avian Parvoviruses. Journal of Virology 75, 2729–2740 (2001).
Sharp, P. M. & Simmonds, P. Evaluating the evidence for virus/host co-evolution. Current Opinion in Virology 1, 436–441 (2011).
Farkas, S. L. et al. Sequencing and phylogenetic analysis identifies candidate members of a new picornavirus genus in terrestrial tortoise species. Archives of Virology 160, 811–816 (2015).
Hughes, A. Phylogeny of the Picornaviridae and differential evolutionary divergence of picornavirus proteins. Infection, Genetics and Evolution 4, 143–152 (2004).
Kurosu, T. Quasispecies of dengue virus. Tropical Medicine and Health 39, S29–S36 (2011).
Lewis-Rogers, N. & Crandall, K. A. Evolution of Picornaviridae: An examination of phylogenetic relationships and cophylogeny. Molecular Phylogenetics and Evolution 54, 995–1005 (2010).
Turmelle, A. S. & Olival, K. J. Correlates of Viral Richness in Bats (Order Chiroptera). EcoHealth 6, 522–539 (2009).
Webber, Q. M. R., Fletcher, Q. E. & Willis, C. K. R. Viral Richness is Positively Related to Group Size, but Not Mating System, in Bats. EcoHealth 14, 652–661 (2017).
Anthony, S. J. et al. Coronaviruses in bats from Mexico. Journal of General Virology 94, 1028–1038 (2013).
Yamashita, T. et al. Complete Nucleotide Sequence and Genetic Organization of Aichi Virus, a Distinct Member of the Picornaviridae Associated with Acute Gastroenteritis in Humans. J. VIROL. 72, 5 (1998).
Carmona-Vicente, N. et al. Phylogeny and prevalence of kobuviruses in dogs and cats in the UK. Veterinary Microbiology 164, 246–252 (2013).
Li, L. et al. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. Journal of General Virology 92, 2534–2541 (2011).
Phan, T. G. et al. The Fecal Viral Flora of Wild Rodents. PLoS Pathogens 7, e1002218 (2011).
Cho, Y.-Y. et al. Molecular evolution of kobuviruses in cats. Archives of Virology 160, 537–541 (2015).
Cho, Y.-Y. et al. Molecular Characterization of the Full Kobuvirus Genome in a Cat. Genome Announcements 2 (2014).
Norby, E. E., Jarman, R. G., Keiser, P. B., Binn, L. N. & Hang, J. Genome Sequence of a Novel Canine Picornavirus Isolated from an American Foxhound. Genome Announcements 5 (2017).
IUCN. Miniopterus schreibersii: Hutson, A. M., Aulagnier, S., Benda, P., Karataş, A., Palmeirim, J. & Paunović, M.: The IUCN Red List of Threatened Species 2008: e.T13561A4160556. doi:10.2305/IUCN.UK.2008.RLTS.T13561A4160556.en (2008).
Li, S. et al. Mitochondrial genetic differentiation and morphological difference of Miniopterus fuliginosus and Miniopterus magnater in China and Vietnam. Ecology and Evolution 5, 1214–1223 (2015).
Puechmaille, S. J. et al. A new species of the Miniopterus schreibersii species complex (Chiroptera: Miniopteridae) from the Maghreb Region, North Africa. Zootaxa 3794, 108 (2014).
Simmonds, P. Recombination and Selection in the Evolution of Picornaviruses and Other Mammalian Positive-Stranded RNA Viruses. Journal of Virology 80, 11124–11140 (2006).
Lukashev, A. N. Recombination among picornaviruses. Reviews in Medical Virology 20, 327–337 (2010).
Acknowledgements
This research was supported by the Hungarian Scientific Research Fund (OTKA) K112440 and by the National Research, Development and Innovation Office (grant number NKFIH KH 129599). The project was supported by the European Union, co-financed by the European Social Fund: Comprehensive Development for Implementing Smart Specialization Strategies at the University of Pécs (EFOP-3.6.1.- 16-2016-00004), and by the University of Pécs within the “Viral Pathogenesis” Talent Centre program. The study was financed by the Higher Education Institutional Excellence Program of the Ministry of Innovation and Technology in Hungary, within the framework of the “Innovation for a sustainable life and environment” thematic programme of the University of Pécs (TUDFO/47138/2019-ITM). GK was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Contributions
S.Z. and G.K. conceived the experiments, S.Z., B.Z., K.K., M.M., H.P. and F.F. conducted the experiments, B.S. generated the figures, R.H. and P.U. analyzed the results, F.J. supervised the work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeghbib, S., Herczeg, R., Kemenesi, G. et al. Genetic characterization of a novel picornavirus in Algerian bats: co-evolution analysis of bat-related picornaviruses. Sci Rep 9, 15706 (2019). https://doi.org/10.1038/s41598-019-52209-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52209-2
- Springer Nature Limited
This article is cited by
-
Novel viruses detected in bats in the Republic of Korea
Scientific Reports (2020)